FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
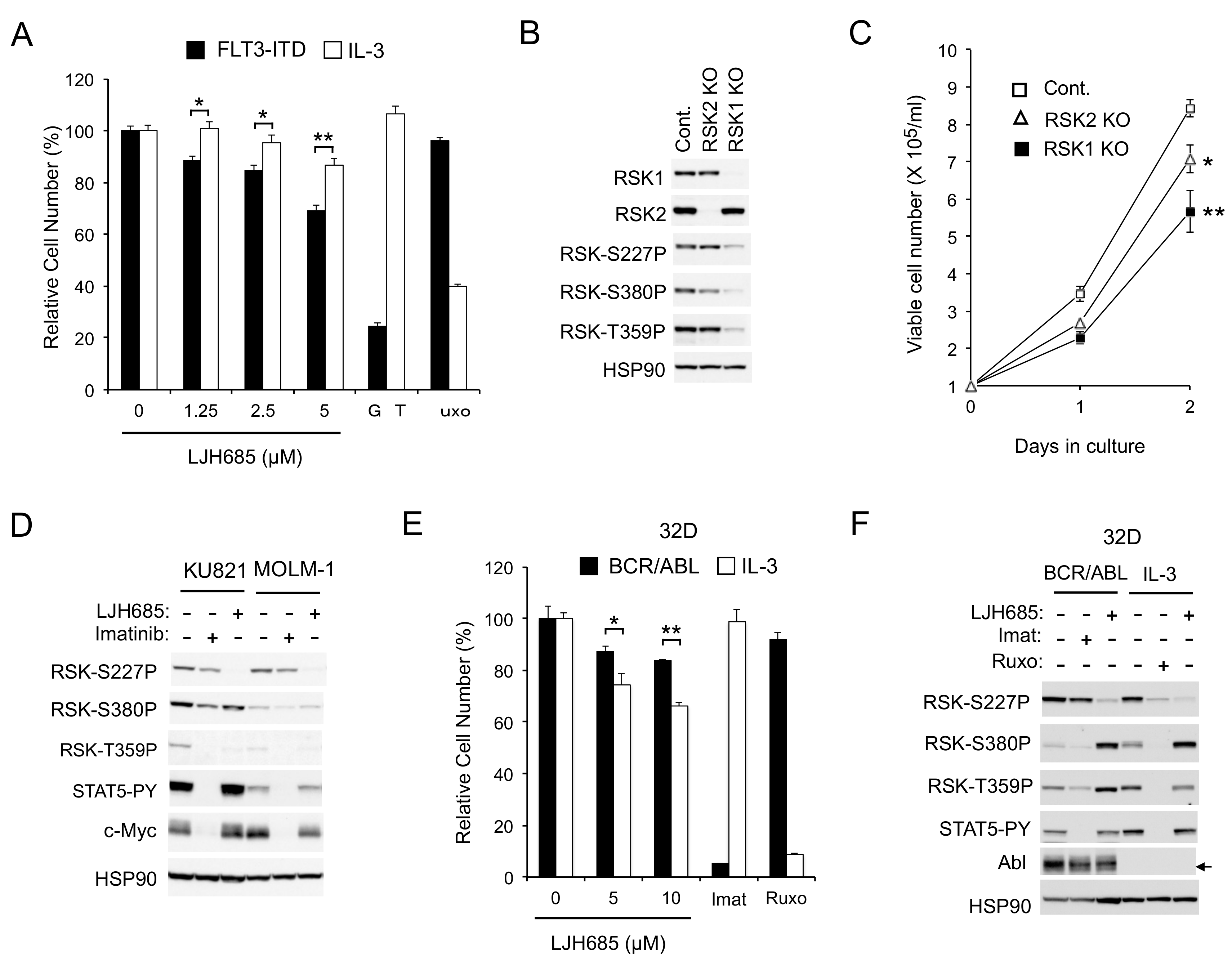
2.1. RSKs Play a Crucial Role in Proliferation of Leukemic Cells Dependent on FLT3-ITD But Not BCR/ABL or JAK2-V617F
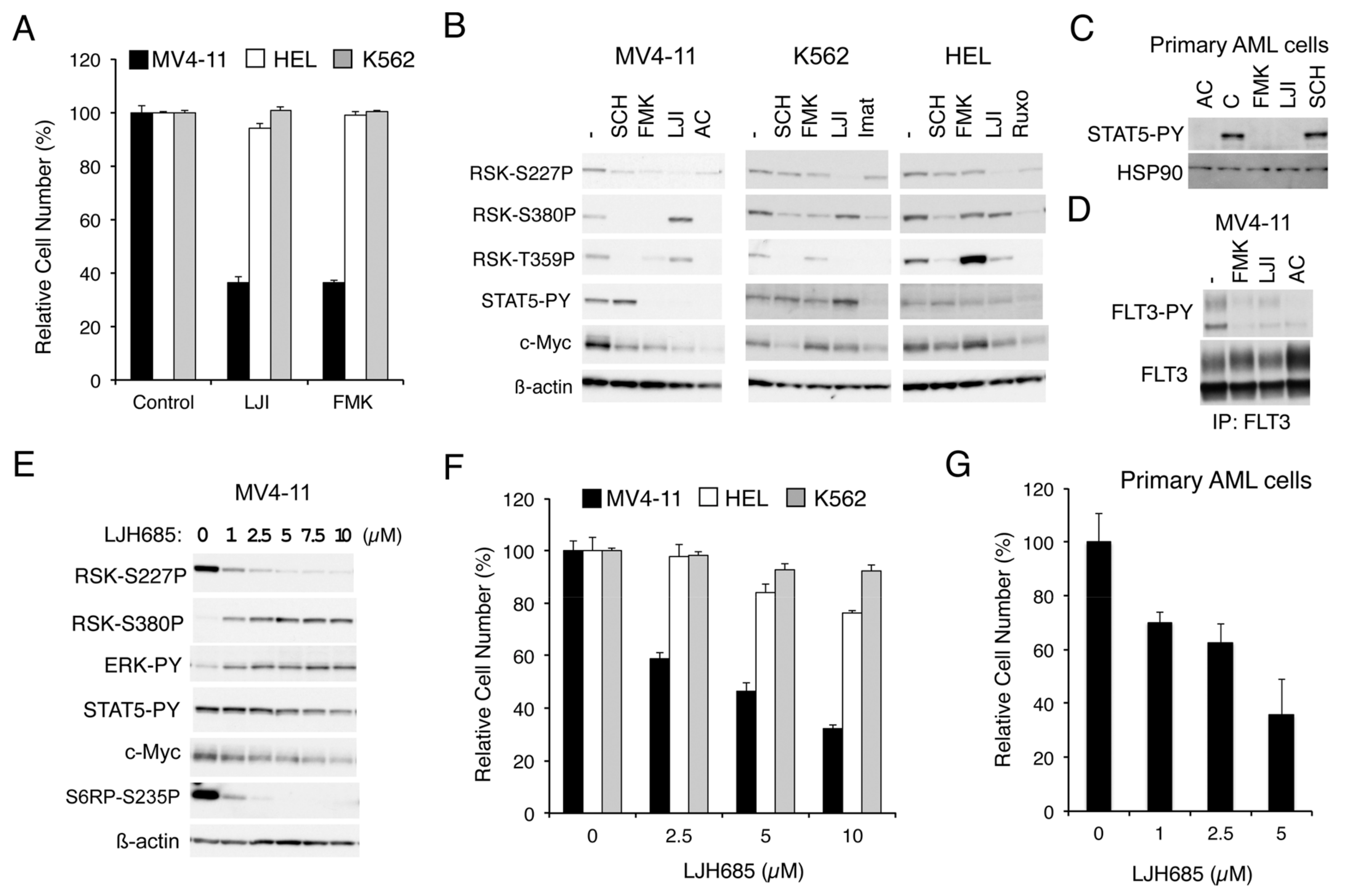
2.1.1. RSK Inhibitors FMK and LJI308 Inhibit Proliferation of Leukemic Cell Lines Dependent on FLT3-ITD But Not BCR/ABL or JAK2-V617F
2.1.2. FMK or LJI308 Inhibits FLT3-ITD through an Off-Target Effect
2.1.3. The Specific RSK Inhibitor LJH685 or Knockout of RSK in MV4-11 Inhibits Proliferation of MV4-11 and a Model Cell Line Transformed by FLT3-ITD
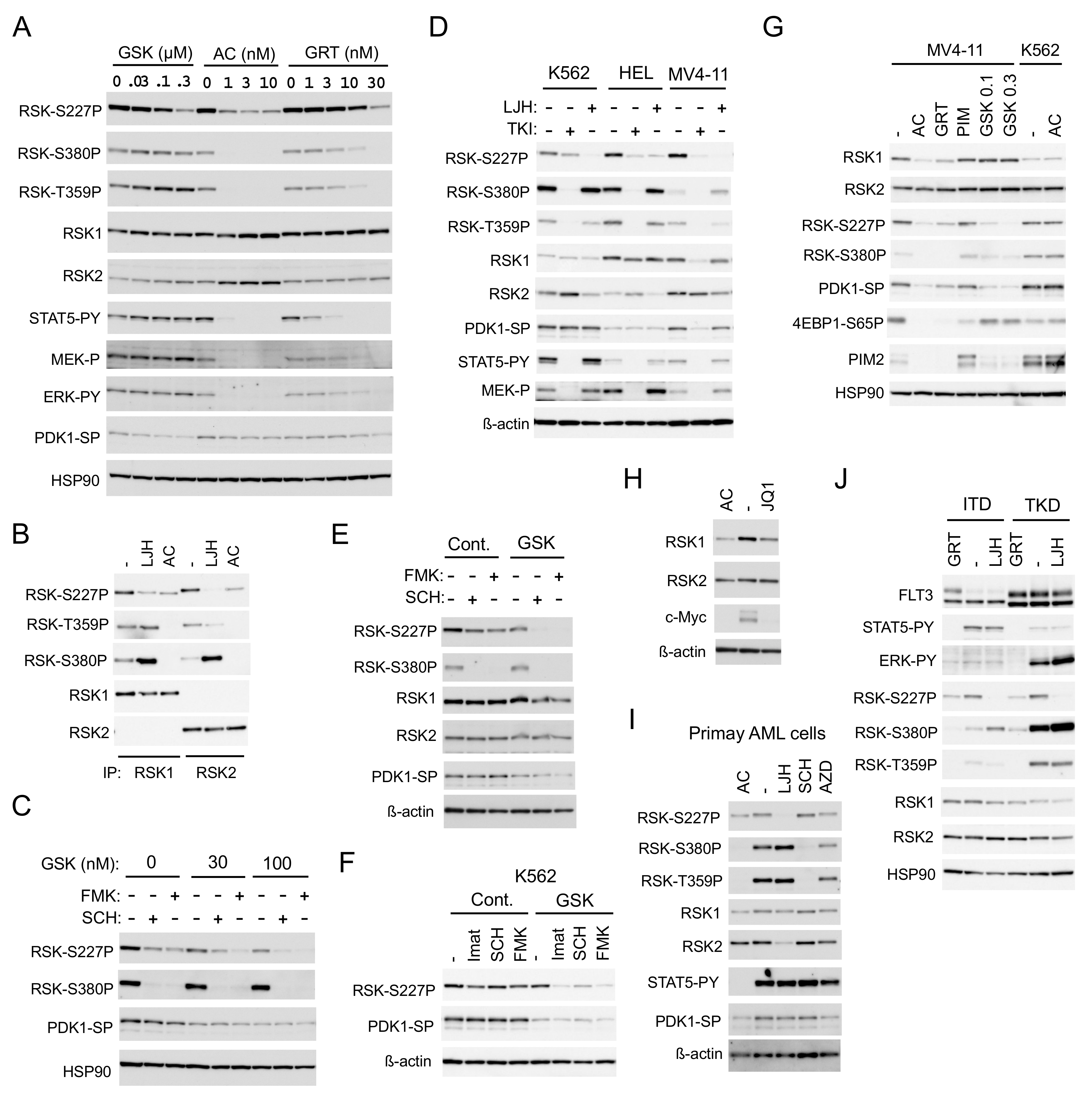
2.2. FLT3-ITD Activates RSKs through Activation of the MEK/ERK Pathway and PDK1
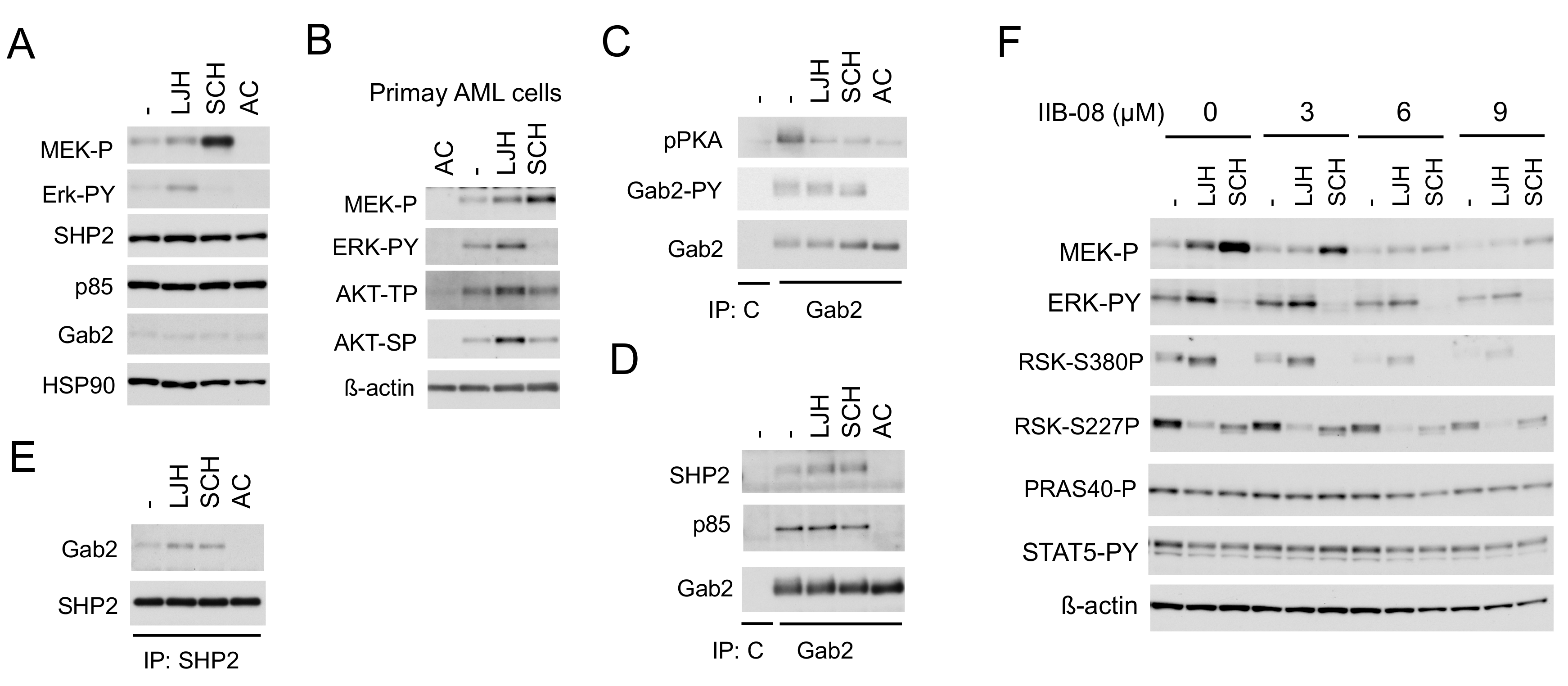
2.3. SHP2 Interacting with Gab2 Mediates Activation of the MEK/ERK Pathway and Its Negative Feedback Regulation by RSKs in FLT3-ITD-Positive AML Cells
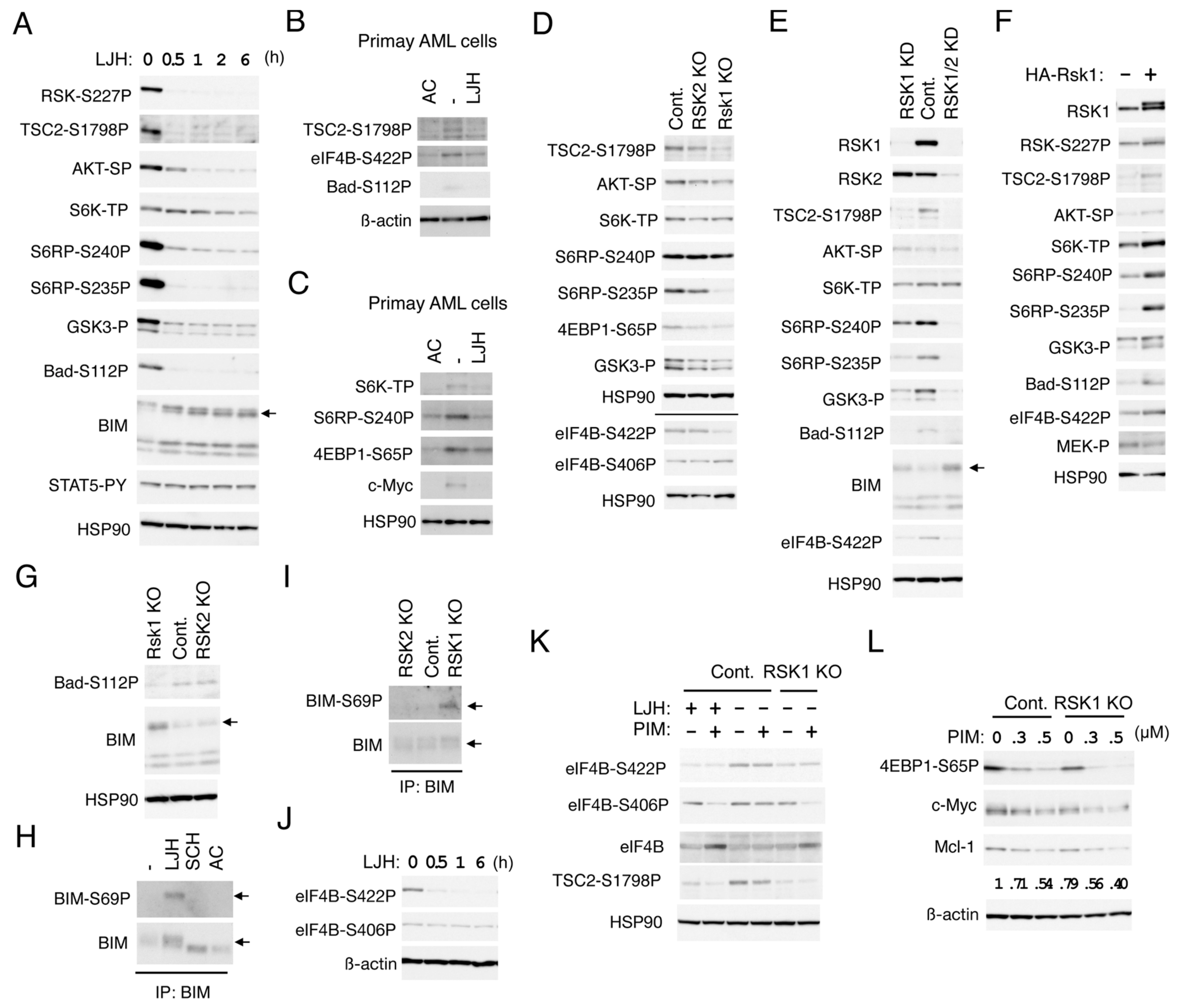
2.4. RSK1 Activates the mTORC1 Pathways in FLT3-ITD-Positive AML Cells
2.5. RSK1 Negatively Regulates Bad and BIM-EL in FLT3-ITD-Positive AML Cells
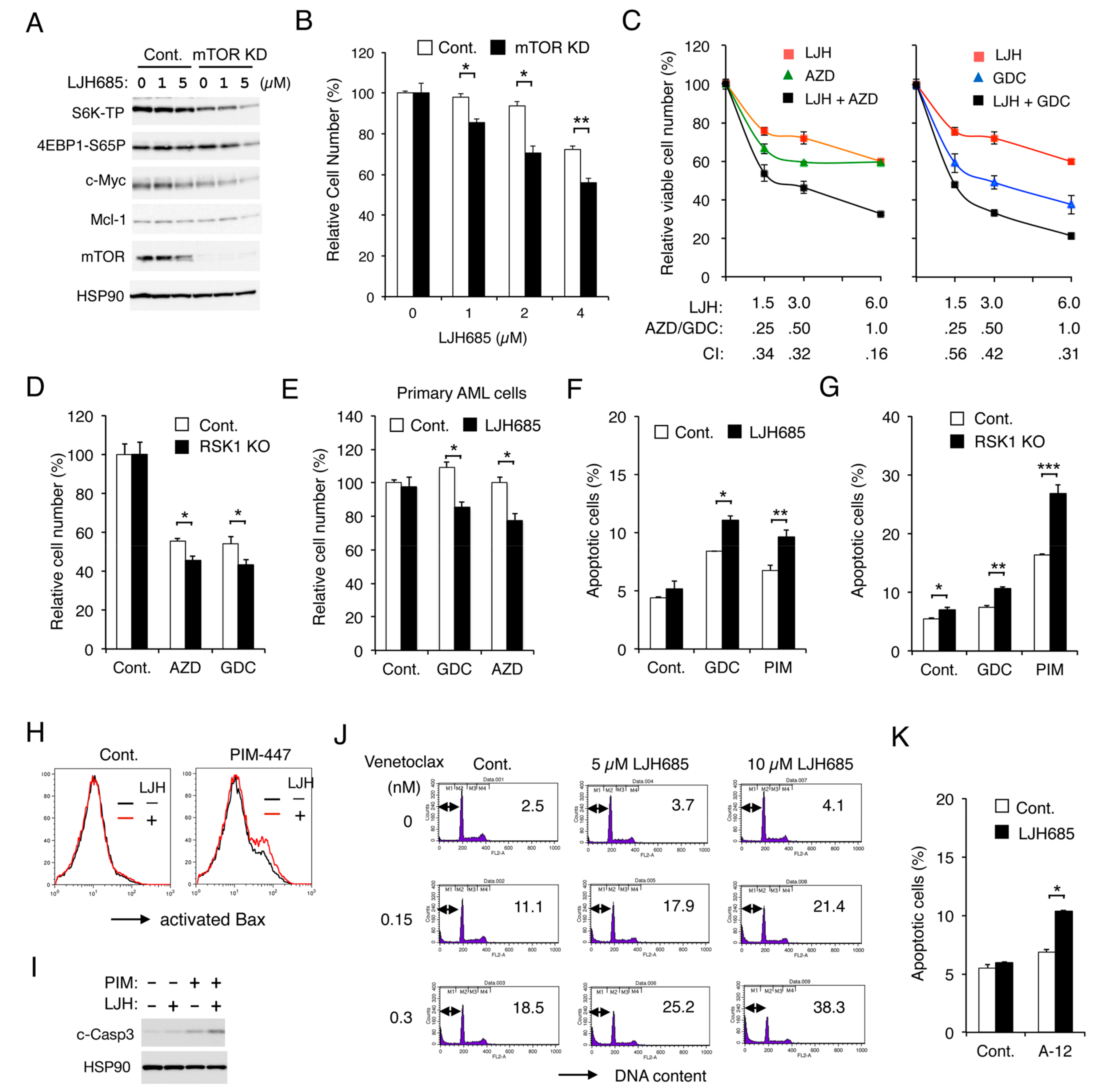
2.6. RSK1 and PIM Play Cooperative Roles Downstream of FLT3-ITD in Enhancing the mTORC1/eIF4F Pathway and Phosphorylation of eIF4B
2.7. Inhibition of RSK and That of PIM or PI3K Cooperatively Inhibit Proliferation and Cooperatively Induce Apoptosis through the Mitochondrial Apoptotic Pathway in FLT3-ITD-Positive AML Cells
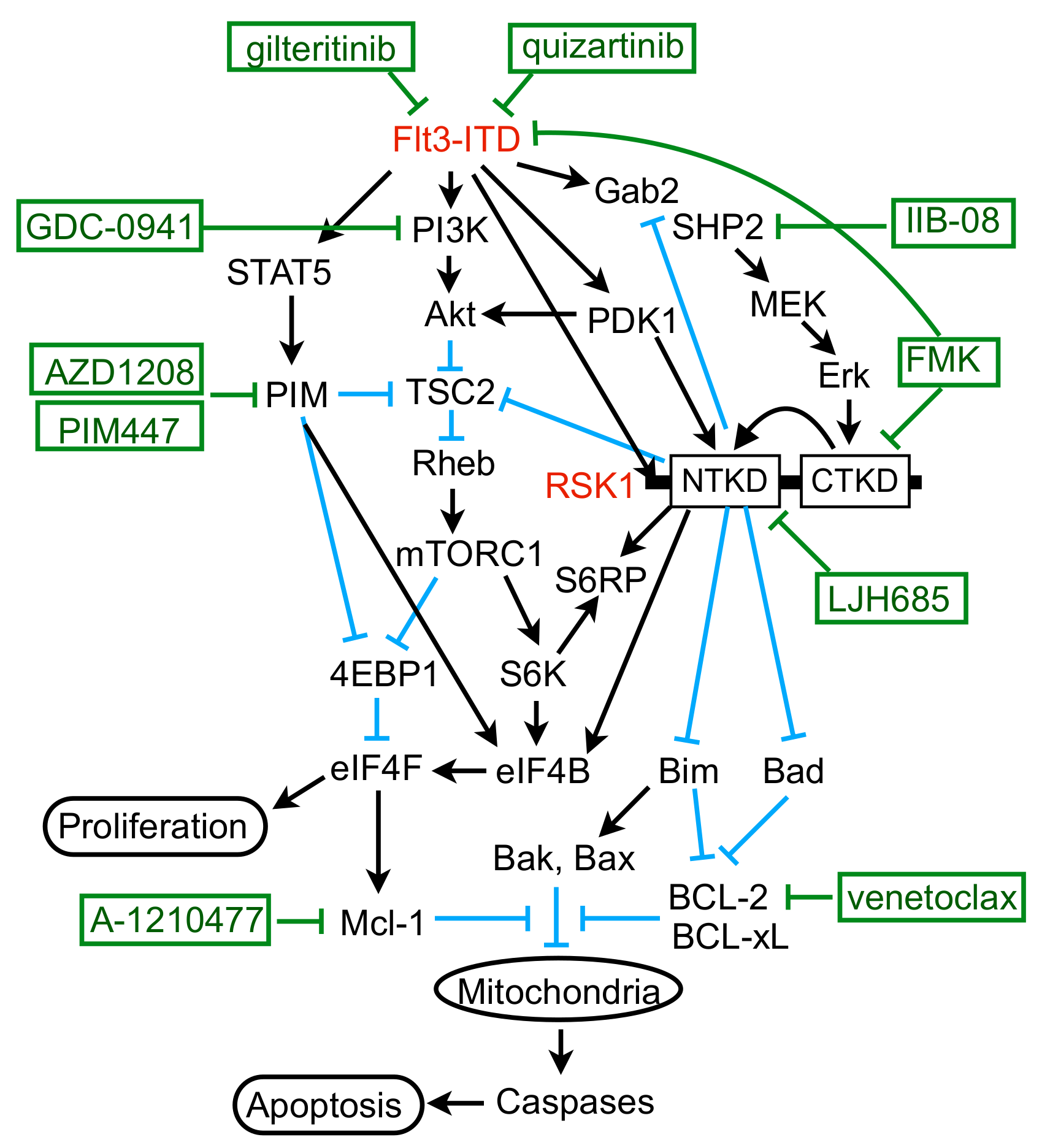
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Expression Plasmids, Transfection, and Infection
4.3. Analyses of Cell Proliferation and Apoptosis
4.4. Immunoprecipitation and Immunoblotting
4.5. Analyses of Primary AML Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hospital, M.A.; Green, A.S.; Maciel, T.T.; Moura, I.C.; Leung, A.Y.; Bouscary, D.; Tamburini, J. Flt3 inhibitors: Clinical potential in acute myeloid leukemia. OncoTargets Ther. 2017, 10, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Appelbaum, F.R. Structural and functional alterations of flt3 in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 4263–4269. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. Molecular pathways: Bcr-abl. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Springuel, L.; Renauld, J.C.; Knoops, L. Jak kinase targeting in hematologic malignancies: A sinuous pathway from identification of genetic alterations towards clinical indications. Haematologica 2015, 100, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Sassano, A.; Platanias, L.C. Targeting mtor for the treatment of aml. New agents and new directions. Oncotarget 2011, 2, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mtorc1 by pi3k signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Topisirovic, I. Signaling pathways involved in the regulation of mrna translation. Mol. Cell Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. Ras/erk signaling promotes site-specific ribosomal protein s6 phosphorylation via rsk and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed]
- Gores, G.J.; Kaufmann, S.H. Selectively targeting mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev. 2012, 26, 305–311. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the bcl-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective bcl-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Nogami, A.; Ishida, S.; Akiyama, H.; Chen, C.; Umezawa, Y.; Miura, O. Flt3-itd induces expression of pim kinases through stat5 to confer resistance to the pi3k/akt pathway inhibitors on leukemic cells by enhancing the mtorc1/mcl-1 pathway. Oncotarget 2018, 9, 8870–8886. [Google Scholar] [CrossRef] [PubMed]
- Nogami, A.; Oshikawa, G.; Okada, K.; Fukutake, S.; Umezawa, Y.; Nagao, T.; Kurosu, T.; Miura, O. Flt3-itd confers resistance to the pi3k/akt pathway inhibitors by protecting the mtor/4ebp1/mcl-1 pathway through stat5 activation in acute myeloid leukemia. Oncotarget 2015, 6, 9189–9205. [Google Scholar] [CrossRef] [PubMed]
- Nogami, A.; Okada, K.; Ishida, S.; Akiyama, H.; Umezawa, Y.; Miura, O. Inhibition of the stat5/pim kinase axis enhances cytotoxic effects of proteasome inhibitors on flt3-itd-positive aml cells by cooperatively inhibiting the mtorc1/4ebp1/s6k/mcl-1 pathway. Transl. Oncol. 2019, 12, 336–349. [Google Scholar] [CrossRef]
- Casalvieri, K.A.; Matheson, C.J.; Backos, D.S.; Reigan, P. Selective targeting of rsk isoforms in cancer. Trends Cancer 2017, 3, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Houles, T.; Roux, P.P. Defining the role of the rsk isoforms in cancer. Semin. Cancer Biol. 2018, 48, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Blevins, D.; Jin, L.; Chung, T.W.; Williams, I.R.; Lee, B.H.; Lin, J.X.; Leonard, W.J.; Taunton, J.; Khoury, H.J.; et al. P90rsk2 is essential for flt3-itd- but dispensable for bcr-abl-induced myeloid leukemia. Blood 2011, 117, 6885–6894. [Google Scholar] [CrossRef]
- Hospital, M.A.; Jacquel, A.; Mazed, F.; Saland, E.; Larrue, C.; Mondesir, J.; Birsen, R.; Green, A.S.; Lambert, M.; Sujobert, P.; et al. Rsk2 is a new pim2 target with pro-survival functions in flt3-itd-positive acute myeloid leukemia. Leukemia 2018, 32, 597–605. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel erk inhibitor with activity in models of acquired resistance to braf and mek inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef]
- Aronchik, I.; Appleton, B.A.; Basham, S.E.; Crawford, K.; Del Rosario, M.; Doyle, L.V.; Estacio, W.F.; Lan, J.; Lindvall, M.K.; Luu, C.A.; et al. Novel potent and selective inhibitors of p90 ribosomal s6 kinase reveal the heterogeneity of rsk function in mapk-driven cancers. Mol. Cancer Res. 2014, 12, 803–812. [Google Scholar] [CrossRef]
- Jain, R.; Mathur, M.; Lan, J.; Costales, A.; Atallah, G.; Ramurthy, S.; Subramanian, S.; Setti, L.; Feucht, P.; Warne, B.; et al. Discovery of potent and selective rsk inhibitors as biological probes. J. Med. Chem. 2015, 58, 6766–6783. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.T.; Nishiguchi, G.; Han, W.; Lan, J.; Simmons, R.; Atallah, G.; Ding, Y.; Tamez, V.; Zhang, Y.; Mathur, M.; et al. Identification of n-(4-((1r,3s,5s)-3-amino-5-methylcyclohexyl)pyridin-3-yl)-6-(2,6-difluorophenyl)-5-fluoropicolinamide (pim447), a potent and selective proviral insertion site of moloney murine leukemia (pim) 1, 2, and 3 kinase inhibitor in clinical trials for hematological malignancies. J. Med. Chem. 2015, 58, 8373–8386. [Google Scholar] [PubMed]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. Azd1208, a potent and selective pan-pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Wohrle, F.U.; Daly, R.J.; Brummer, T. Function, regulation and pathological roles of the gab/dos docking proteins. Cell Commun. Signal 2009, 7, 22. [Google Scholar] [CrossRef]
- Zhang, S.; Broxmeyer, H.E. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and pi3 kinase. Biochem. Biophys. Res. Commun. 2000, 277, 195–199. [Google Scholar] [CrossRef]
- Wang, L.; Iorio, C.; Yan, K.; Yang, H.; Takeshita, S.; Kang, S.; Neel, B.G.; Yang, W. A erk/rsk-mediated negative feedback loop regulates m-csf-evoked pi3k/akt activation in macrophages. FASEB J. 2018, 32, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Vaughan, T.; Bunting, K.D. Gab adapter proteins as therapeutic targets for hematologic disease. Adv. Hematol. 2012, 2012, 380635. [Google Scholar] [CrossRef]
- Zhang, X.; He, Y.; Liu, S.; Yu, Z.; Jiang, Z.X.; Yang, Z.; Dong, Y.; Nabinger, S.C.; Wu, L.; Gunawan, A.M.; et al. Salicylic acid based small molecule inhibitor for the oncogenic src homology-2 domain containing protein tyrosine phosphatase-2 (shp2). J. Med. Chem. 2010, 53, 2482–2493. [Google Scholar] [CrossRef]
- Bui, N.L.; Pandey, V.; Zhu, T.; Ma, L.; Basappa; Lobie, P.E. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018, 415, 177–186. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Yang, X.; Liu, L.; Sternberg, D.; Tang, L.; Galinsky, I.; DeAngelo, D.; Stone, R. The flt3 internal tandem duplication mutation prevents apoptosis in interleukin-3-deprived baf3 cells due to protein kinase a and ribosomal s6 kinase 1-mediated bad phosphorylation at serine 112. Cancer Res. 2005, 65, 7338–7347. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, M.; McLeod, L.E.; Pratt, P.F.; Proud, C.G. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of erk and involves phosphorylation of tuberous sclerosis complex 2 (tsc2). Biochem. J. 2005, 388, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal s6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating tsc2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef]
- Zhang, F.; Beharry, Z.M.; Harris, T.E.; Lilly, M.B.; Smith, C.D.; Mahajan, S.; Kraft, A.S. Pim1 protein kinase regulates pras40 phosphorylation and mtor activity in fdcp1 cells. Cancer Biol. Ther. 2009, 8, 846–853. [Google Scholar] [CrossRef]
- Tamburini, J.; Green, A.S.; Bardet, V.; Chapuis, N.; Park, S.; Willems, L.; Uzunov, M.; Ifrah, N.; Dreyfus, F.; Lacombe, C.; et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [Google Scholar] [CrossRef]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef]
- Carriere, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic mapk signaling stimulates mtorc1 activity by promoting rsk-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef]
- Kuang, E.; Fu, B.; Liang, Q.; Myoung, J.; Zhu, F. Phosphorylation of eukaryotic translation initiation factor 4b (eif4b) by open reading frame 45/p90 ribosomal s6 kinase (orf45/rsk) signaling axis facilitates protein translation during kaposi sarcoma-associated herpesvirus (kshv) lytic replication. J. Biol. Chem. 2011, 286, 41171–41182. [Google Scholar] [CrossRef]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.; Blenis, J.; Pende, M.; Sonenberg, N. The mtor/pi3k and mapk pathways converge on eif4b to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef]
- van Gorp, A.G.; van der Vos, K.E.; Brenkman, A.B.; Bremer, A.; van den Broek, N.; Zwartkruis, F.; Hershey, J.W.; Burgering, B.M.; Calkhoven, C.F.; Coffer, P.J. Agc kinases regulate phosphorylation and activation of eukaryotic translation initiation factor 4b. Oncogene 2009, 28, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Xiong, Y.; Song, J.H.; Mahajan, S.; DuPont, R.; McEachern, K.; DeAngelo, D.J.; Cortes, J.E.; Minden, M.D.; Ebens, A.; et al. The pim-1 protein kinase is an important regulator of met receptor tyrosine kinase levels and signaling. Mol. Cell Biol. 2014, 34, 2517–2532. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, J.; Chen, K.; Guo, G.; Xi, R.; Rothman, P.B.; Whitten, D.; Zhang, L.; Huang, S.; Chen, J.L. Eif4b phosphorylation by pim kinases plays a critical role in cellular transformation by abl oncogenes. Cancer Res. 2013, 73, 4898–4908. [Google Scholar] [CrossRef] [PubMed]
- Masson, K.; Liu, T.; Khan, R.; Sun, J.; Ronnstrand, L. A role of gab2 association in flt3 itd mediated stat5 phosphorylation and cell survival. Br. J. Haematol. 2009, 146, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.; Crouin, C.; Deon, C.; Loyaux, D.; Bertoglio, J. Phosphorylation of grb2-associated binder 2 on serine 623 by erk mapk regulates its association with the phosphatase shp-2 and decreases stat5 activation. J. Immunol. 2004, 173, 3962–3971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lavoie, G.; Fort, L.; Huttlin, E.L.; Tcherkezian, J.; Galan, J.A.; Gu, H.; Gygi, S.P.; Carreno, S.; Roux, P.P. Gab2 phosphorylation by rsk inhibits shp2 recruitment and cell motility. Mol. Cell Biol. 2013, 33, 1657–1670. [Google Scholar] [CrossRef]
- Zhang, X.; Lavoie, G.; Meant, A.; Aubert, L.; Cargnello, M.; Haman, A.; Hoang, T.; Roux, P.P. Extracellular signal-regulated kinases 1 and 2 phosphorylate gab2 to promote a negative-feedback loop that attenuates phosphoinositide 3-kinase/akt signaling. Mol. Cell Biol. 2017, 37, e00357-16. [Google Scholar] [CrossRef]
- Shimamura, A.; Ballif, B.A.; Richards, S.A.; Blenis, J. Rsk1 mediates a mek-map kinase cell survival signal. Curr. Biol. 2000, 10, 127–135. [Google Scholar] [CrossRef]
- Minami, Y.; Yamamoto, K.; Kiyoi, H.; Ueda, R.; Saito, H.; Naoe, T. Different antiapoptotic pathways between wild-type and mutated flt3: Insights into therapeutic targets in leukemia. Blood 2003, 102, 2969–2975. [Google Scholar] [CrossRef]
- Dehan, E.; Bassermann, F.; Guardavaccaro, D.; Vasiliver-Shamis, G.; Cohen, M.; Lowes, K.N.; Dustin, M.; Huang, D.C.; Taunton, J.; Pagano, M. Betatrcp- and rsk1/2-mediated degradation of bimel inhibits apoptosis. Mol. Cell 2009, 33, 109–116. [Google Scholar] [CrossRef]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of bim in health and disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [PubMed]
- Nordigarden, A.; Kraft, M.; Eliasson, P.; Labi, V.; Lam, E.W.; Villunger, A.; Jonsson, J.I. Bh3-only protein bim more critical than puma in tyrosine kinase inhibitor-induced apoptosis of human leukemic cells and transduced hematopoietic progenitors carrying oncogenic flt3. Blood 2009, 113, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. Flt3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through foxo proteins. Oncogene 2004, 23, 3338–3349. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. Bid preferentially activates bak while bim preferentially activates bax, affecting chemotherapy response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Umezawa, Y.; Ishida, S.; Okada, K.; Nogami, A.; Miura, O. Inhibition of usp9x induces apoptosis in flt3-itd-positive aml cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. 2019, 453, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, G.; Nagao, T.; Wu, N.; Kurosu, T.; Miura, O. C-cbl and cbl-b ligases mediate 17-allylaminodemethoxygeldanamycin-induced degradation of autophosphorylated flt3 kinase with internal tandem duplication through the ubiquitin proteasome pathway. J. Biol. Chem. 2011, 286, 30263–30273. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, T.; Tsuji, K.; Kida, A.; Koyama, T.; Yamamoto, M.; Miura, O. Rottlerin synergistically enhances imatinib-induced apoptosis of bcr/abl-expressing cells through its mitochondrial uncoupling effect independent of protein kinase c-delta. Oncogene 2007, 26, 2975–2987. [Google Scholar] [CrossRef]
- Klucher, K.M.; Lopez, D.V.; Daley, G.Q. Secondary mutation maintains the transformed state in baf3 cells with inducible bcr/abl expression. Blood 1998, 91, 3927–3934. [Google Scholar] [CrossRef]
- Nagao, T.; Kurosu, T.; Umezawa, Y.; Nogami, A.; Oshikawa, G.; Tohda, S.; Yamamoto, M.; Miura, O. Proliferation and survival signaling from both jak2-v617f and lyn involving gsk3 and mtor/p70s6k/4ebp1 in pvtl-1 cell line newly established from acute myeloid leukemia transformed from polycythemia vera. PLoS ONE 2014, 9, e84746. [Google Scholar] [CrossRef]
- Ishida, S.; Akiyama, H.; Umezawa, Y.; Okada, K.; Nogami, A.; Oshikawa, G.; Nagao, T.; Miura, O. Mechanisms for mtorc1 activation and synergistic induction of apoptosis by ruxolitinib and bh3 mimetics or autophagy inhibitors in jak2-v617f-expressing leukemic cells including newly established pvtl-2. Oncotarget 2018, 9, 26834–26851. [Google Scholar] [CrossRef][Green Version]
- Richards, S.A.; Dreisbach, V.C.; Murphy, L.O.; Blenis, J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal s6 kinase 1. Mol. Cell Biol. 2001, 21, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide crispr screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgrna design to maximize activity and minimize off-target effects of crispr-cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Kurosu, T.; Ohki, M.; Wu, N.; Kagechika, H.; Miura, O. Sorafenib induces apoptosis specifically in cells expressing bcr/abl by inhibiting its kinase activity to activate the intrinsic mitochondrial pathway. Cancer Res. 2009, 69, 3927–3936. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827. https://doi.org/10.3390/cancers11121827
Watanabe D, Nogami A, Okada K, Akiyama H, Umezawa Y, Miura O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers. 2019; 11(12):1827. https://doi.org/10.3390/cancers11121827
Chicago/Turabian StyleWatanabe, Daisuke, Ayako Nogami, Keigo Okada, Hiroki Akiyama, Yoshihiro Umezawa, and Osamu Miura. 2019. "FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM" Cancers 11, no. 12: 1827. https://doi.org/10.3390/cancers11121827
APA StyleWatanabe, D., Nogami, A., Okada, K., Akiyama, H., Umezawa, Y., & Miura, O. (2019). FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers, 11(12), 1827. https://doi.org/10.3390/cancers11121827