DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression

 ,
,  ,
,  and
and
Abstract
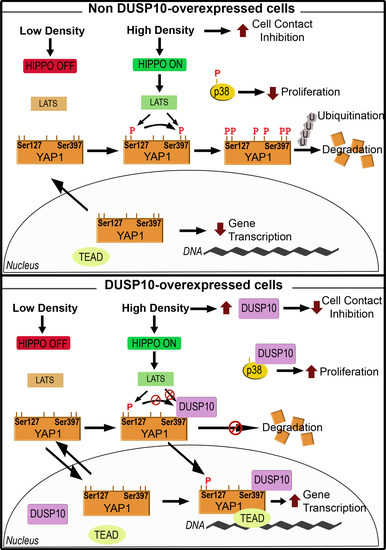
1. Introduction
2. Results
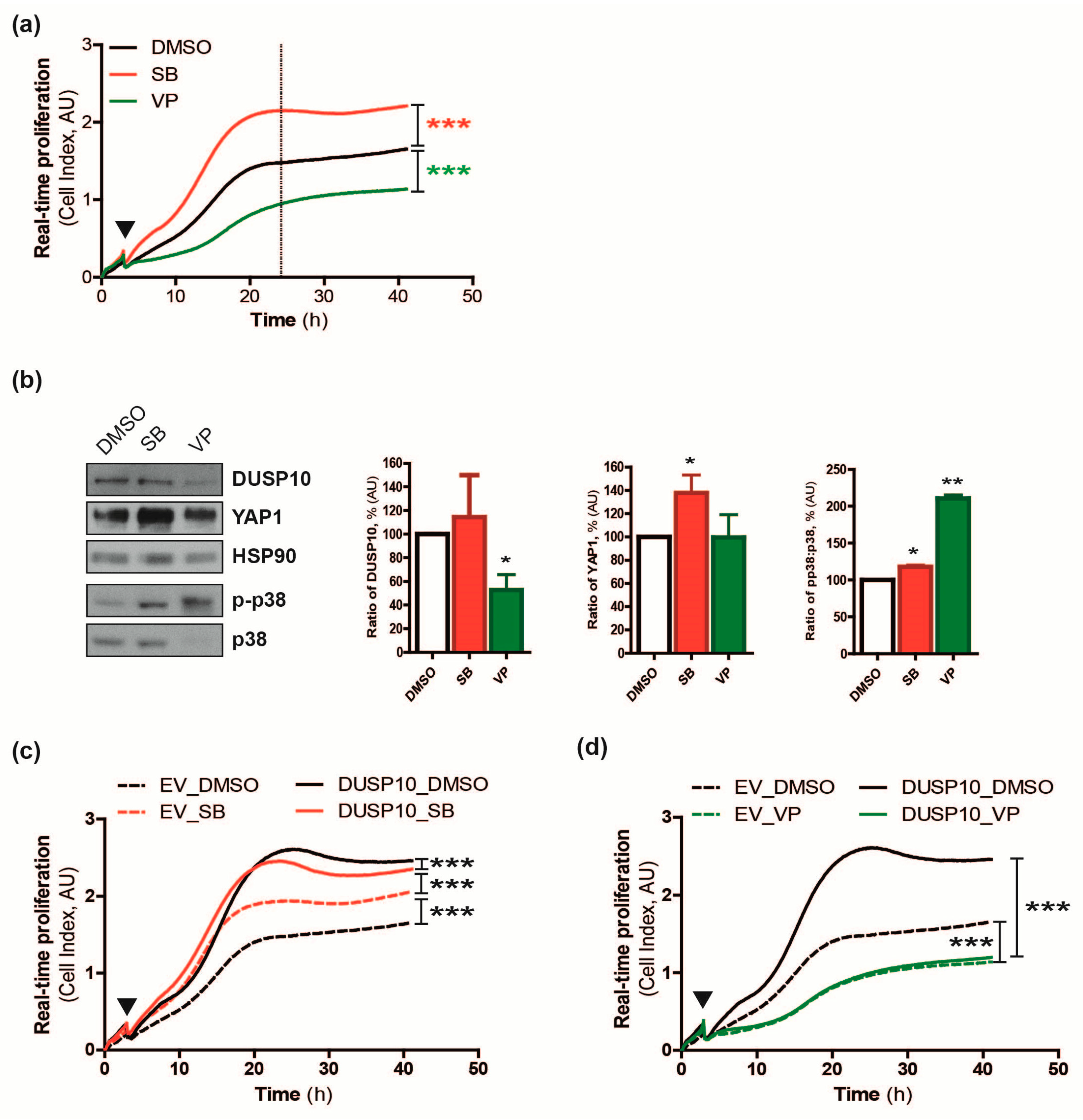
2.1. DUSP10 Regulates Cell Proliferation of CRC Cell Lines In Vitro and In Vivo
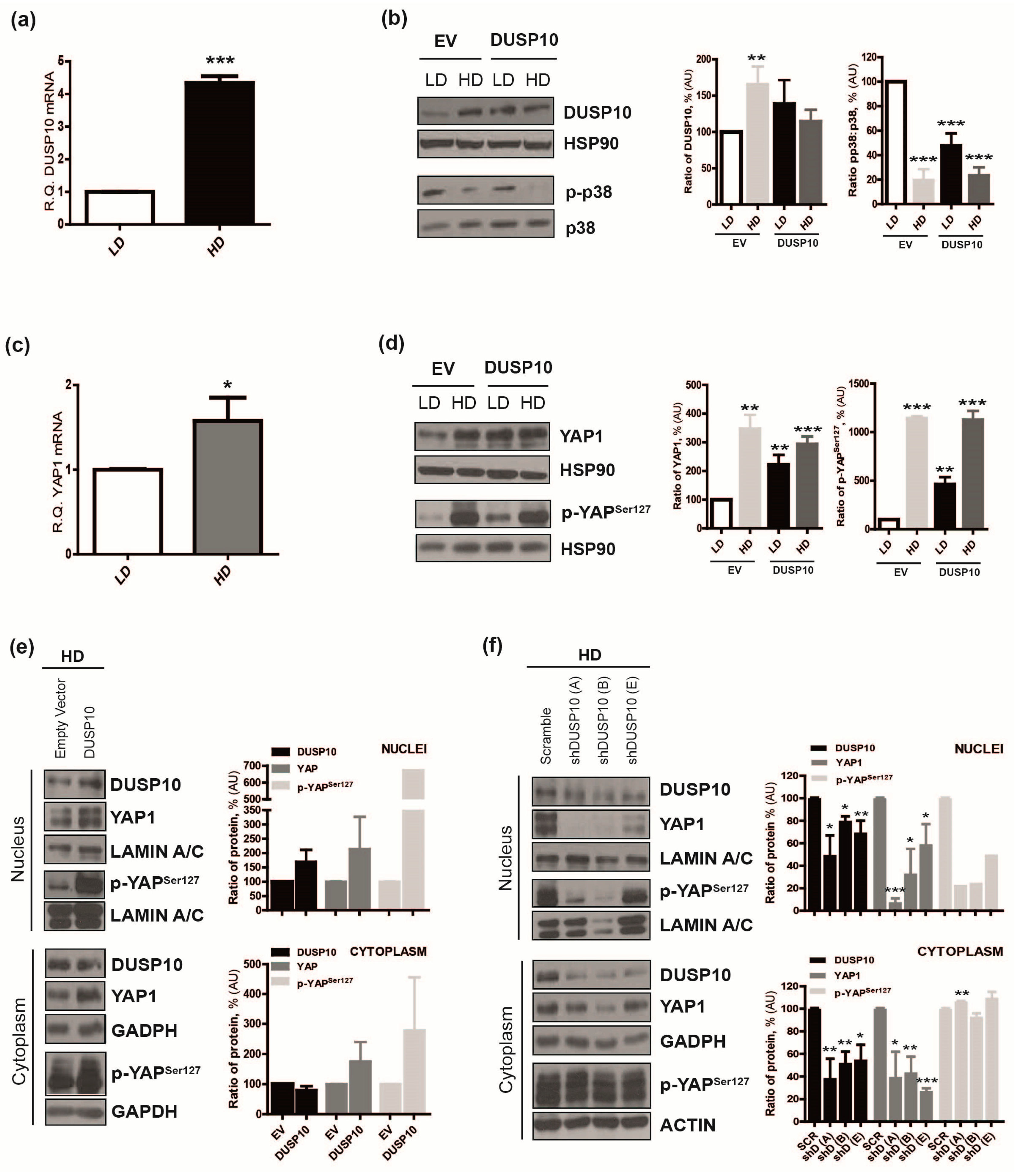
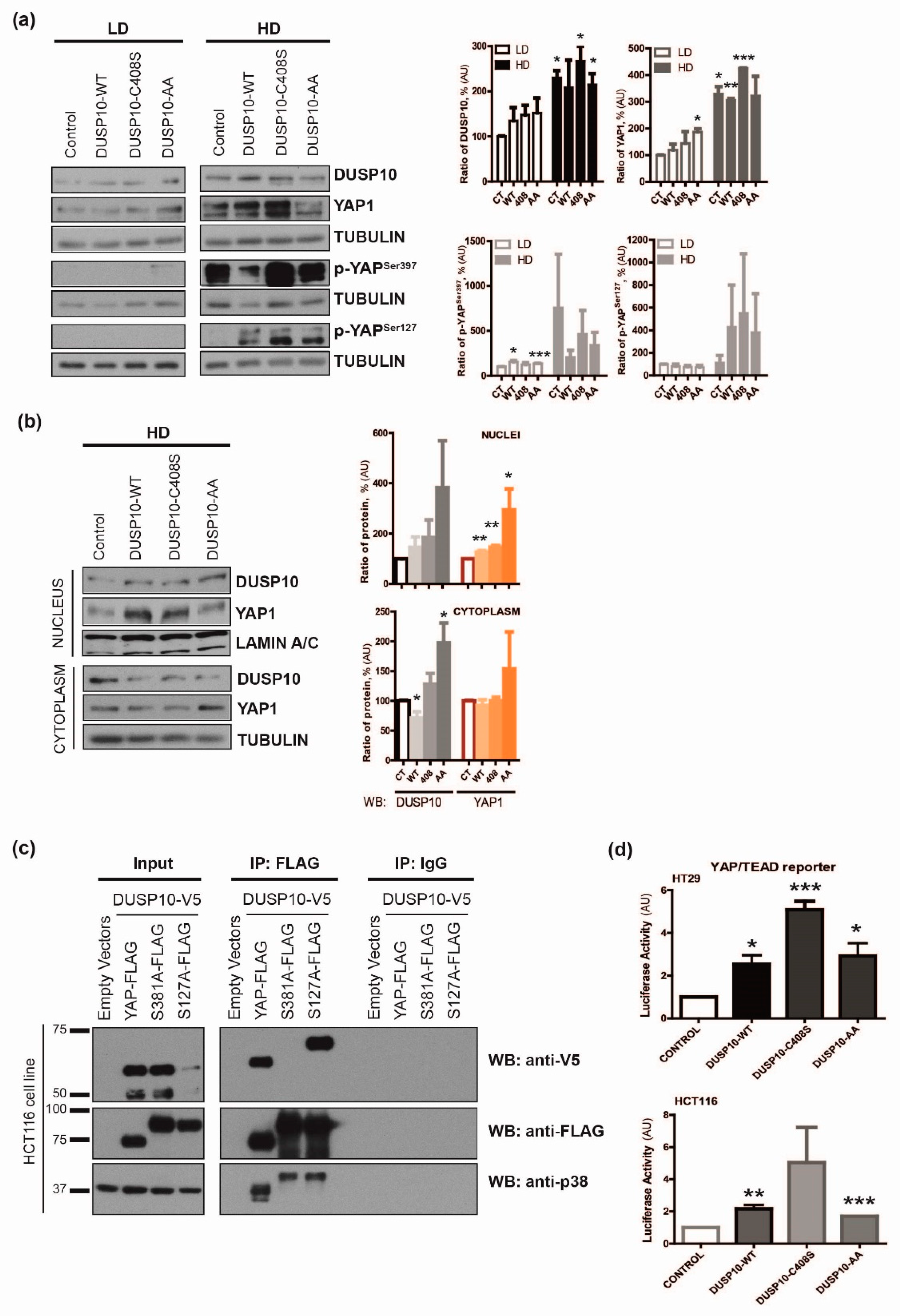
2.2. DUSP10 Is Increased in HD and Correlates with YAP1 Expression in CRC Cell Lines
2.3. DUSP10 Interacts with YAP1 through Ser397 Residue
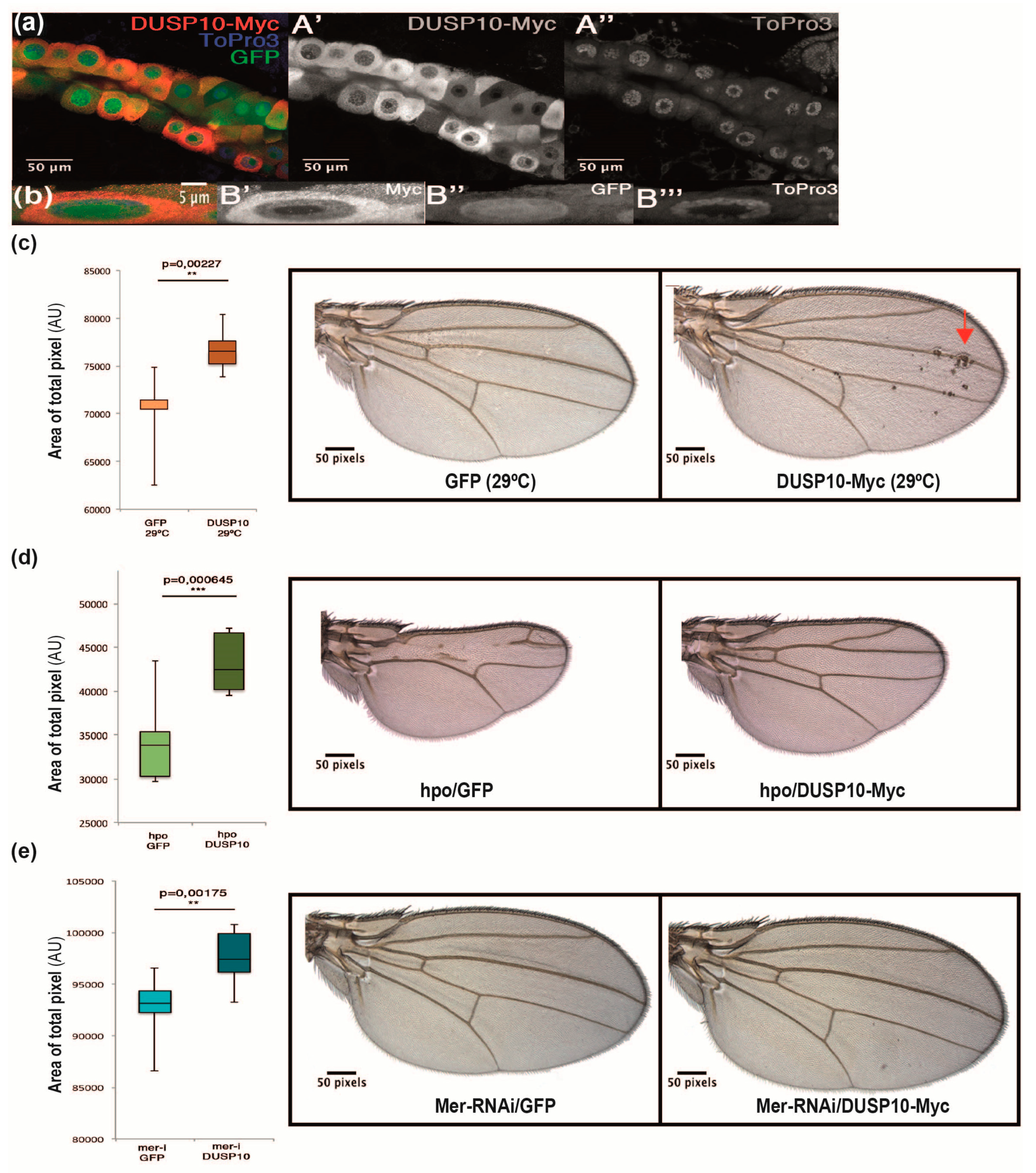
2.4. DUSP10 Regulates the Hippo Pathway in Drosophila
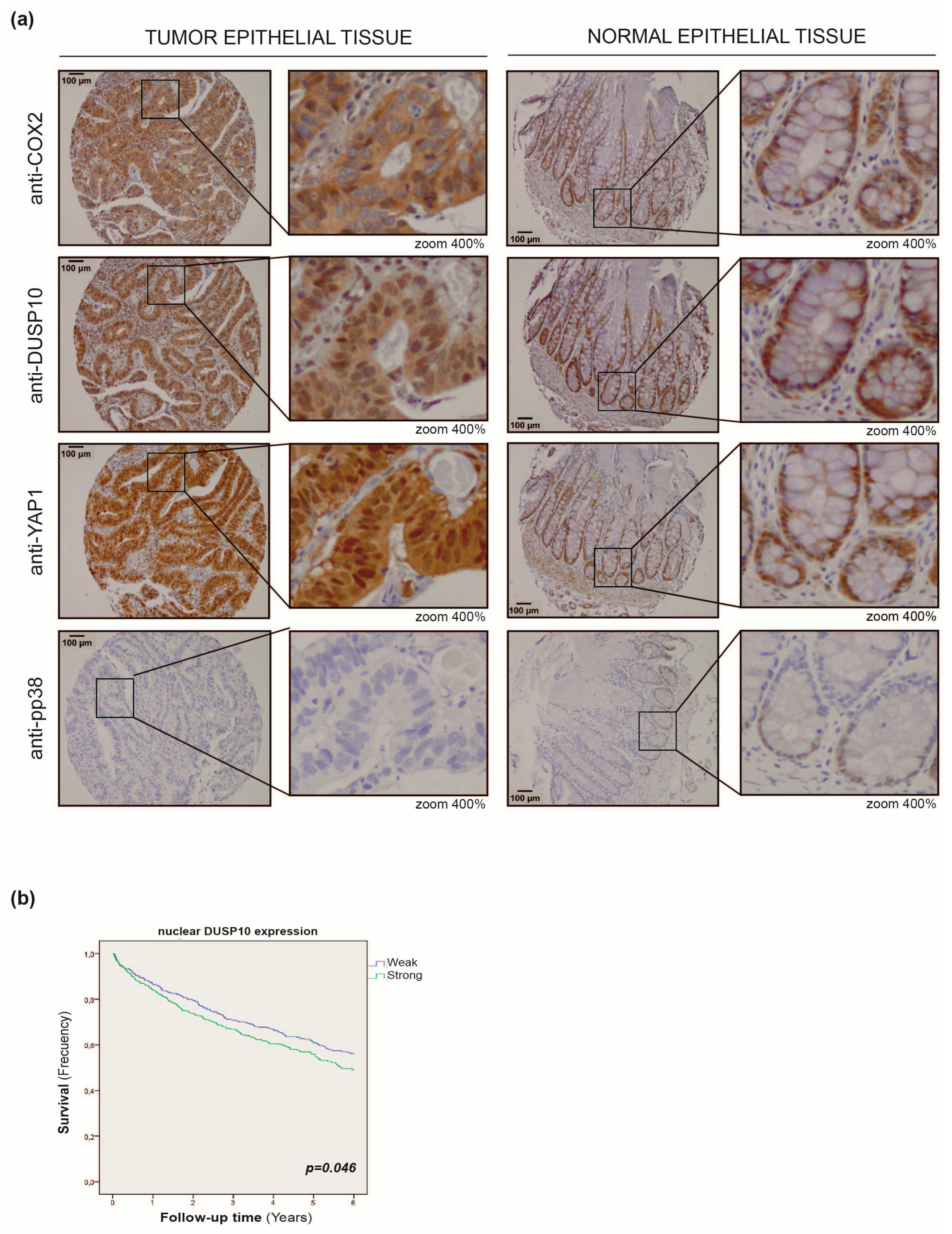
2.5. DUSP10 Is Increased in CRC Patient Samples
2.6. Nuclear DUSP10 Expression Correlates with Bad Prognosis in CRC Patients
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Transient Expression Constructs
4.3. Stable Expression Constructs
4.4. Cell Proliferation Assays
4.5. Cell Density Assay
4.6. Luciferase Reporter Activity Assay
4.7. Protein Extraction and Immunoblot Analysis
4.8. Immunoprecipitation
4.9. Immunofluorescence and Confocal Fluorescence Microscopy
4.10. RNA Isolation and qPCR Analysis
4.11. Drosophila Studies
4.12. Mouse Studies
4.13. Human Studies
4.14. Study Approval
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Okamoto, K.; Kasi, P.M.; Kawakami, H. Molecular Biomarkers in the Personalized Treatment of Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2016, 14, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M. Contact inhibition and malignancy. Nature 1979, 281, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Jiao, H.; Teh, H.Y.; Takahashi, H.; Png, C.W.; Phoon, M.C.; Suzuki, Y.; Sawasaki, T.; Xiao, H.; Chow, V.T.K.; et al. MAPK Phosphatase 5 Expression Induced by Influenza and Other RNA Virus Infection Negatively Regulates IRF3 Activation and Type I Interferon Response. Cell Rep. 2015, 10, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Shiiba, K.; Katagiri, C.; Kasugai, I.; Masuda, K.; Sato, I.; Sato, M.; Kakugawa, Y.; Nomura, E.; Hayashi, K.; et al. Novel function of MKP-5/DUSP10, a phosphatase of stress-activated kinases, on ERK-dependent gene expression, and upregulation of its gene expression in colon carcinomas. Oncol Rep. 2012, 28, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Martinez, M.; Stamatakis, K.; Fresno, M. The Dual-Specificity Phosphatase 10 (DUSP10): Its Role in Cancer, Inflammation, and Immunity. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3sigma sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2016, 6, 27–37. [Google Scholar]
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; White, R.F.; McVey, M.J.; Legos, J.J.; et al. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J. Pharm. Exp. 2001, 296, 312–321. [Google Scholar]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Bermudez, O.; Pages, G.; Gimond, C. The dual-specificity MAP kinase phosphatases: Critical roles in development and cancer. Am. J. Physiol. Cell. Physiol 2010, 299, 189–202. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Puliafito, A.; Hufnagel, L.; Neveu, P.; Streichan, S.; Sigal, A.; Fygenson, D.K.; Shraiman, B.I. Collective and single cell behavior in epithelial contact inhibition. Proc. Natl. Acad Sci. USA 2012, 109, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Eisenhoffer, G.T.; Rosenblatt, J. Bringing balance by force: Live cell extrusion controls epithelial cell numbers. Trends Cell Biol. 2013, 23, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Dev. (Camb. Engl.) 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, L.; Weng, W.; Qiao, Y.; Zhang, Y.; He, J.; Wang, H.; Xiao, W.; Li, L.; Chu, Q.; et al. Mutual interaction between YAP and CREB promotes tumorigenesis in liver cancer. Hepatology 2013, 58, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Mei, Z.Q.; Wu, J.W.; Wang, Z.X. Enzymatic activity and substrate specificity of mitogen-activated protein kinase p38alpha in different phosphorylation states. J. Biol. Chem. 2008, 283, 26591–26601. [Google Scholar] [CrossRef] [PubMed]
- Perrimon, N. Signalling pathways initiated by receptor protein tyrosine kinases in Drosophila. Curr. Opin. Cell Biol. 1994, 6, 260–266. [Google Scholar] [CrossRef]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, S.H.; Lee, O.J.; Huang, S.M.; Kwon, J.L.; Kim, J.M.; Kim, J.Y.; Seong, I.O.; Song, K.S.; Kim, K.H. Differential expression of Yes-associated protein and phosphorylated Yes-associated protein is correlated with expression of Ki-67 and phospho-ERK in colorectal adenocarcinoma. Histol. Histopathol. 2013, 28, 1483–1490. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, C.; Li, Q.; Xu, K.; Wang, E. Clinical and prognostic significance of Yes-associated protein in colorectal cancer. Tumour Biol. 2013, 34, 2169–2174. [Google Scholar] [CrossRef]
- Ou, C.; Sun, Z.; Li, S.; Li, G.; Li, X.; Ma, J. Dual roles of yes-associated protein (YAP) in colorectal cancer. Oncotarget 2017, 8, 75727–75741. [Google Scholar] [CrossRef]
- Hardwick, J.C.; van den Brink, G.R.; Offerhaus, G.J.; van Deventer, S.J.; Peppelenbosch, M.P. NF-kappaB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene 2001, 20, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Wakeman, D.; Schneider, J.E.; Liu, J.; Wandu, W.S.; Erwin, C.R.; Guo, J.; Stappenbeck, T.S.; Warner, B.W. Deletion of p38-alpha mitogen-activated protein kinase within the intestinal epithelium promotes colon tumorigenesis. Surgery 2012, 152, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, K.; Jimenez-Martinez, M.; Jimenez-Segovia, A.; Chico-Calero, I.; Conde, E.; Galan-Martinez, J.; Ruiz, J.; Pascual, A.; Barrocal, B.; Lopez-Perez, R.; et al. Prostaglandins induce early growth response 1 transcription factor mediated microsomal prostaglandin E2 synthase up-regulation for colorectal cancer progression. Oncotarget 2015, 6, 39941–39959. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Rubin, G.M.; Spradling, A.C. Genetic transformation of Drosophila with transposable element vectors. Science 1982, 218, 348–353. [Google Scholar] [CrossRef]
- Cruz, C.; Glavic, A.; Casado, M.; de Celis, J.F. A gain-of-function screen identifying genes required for growth and pattern formation of the Drosophila melanogaster wing. Genetics 2009, 183, 1005–1026. [Google Scholar] [CrossRef]
- Buskens, C.J.; Van Rees, B.P.; Sivula, A.; Reitsma, J.B.; Haglund, C.; Bosma, P.J.; Offerhaus, G.J.; Van Lanschot, J.J.; Ristimaki, A. Prognostic significance of elevated cyclooxygenase 2 expression in patients with adenocarcinoma of the esophagus. Gastroenterology 2002, 122, 1800–1807. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Campeau, E.; Ruhl, V.E.; Rodier, F.; Smith, C.L.; Rahmberg, B.L.; Fuss, J.O.; Campisi, J.; Yaswen, P.; Cooper, P.K.; Kaufman, P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 2009, 4, e6529. [Google Scholar] [CrossRef]
- Yang, X.; Boehm, J.S.; Yang, X.; Salehi-Ashtiani, K.; Hao, T.; Shen, Y.; Lubonja, R.; Thomas, S.R.; Alkan, O.; Bhimdi, T.; et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 2011, 8, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DUSP10 Cytoplasmic (n= 67) | DUSP10 Nuclei (n= 67) | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristic | No. of Patients a | Weak | Strong | p-Value b | Weak | Strong | p-Value b | |
| COX2 cytoplasmic, No. (%) | ||||||||
| Weak | 22 (32.8) | 8 (61.5) | 14 (25.9) | 0.022 | - | - | - | |
| Strong | 45 (67.2) | 5 (38.5) | 40 (74.1) | - | - | - | ||
| YAP1nuclei, No. (%) | ||||||||
| Weak | 18 (28.6) | - | - | - | 8 (61.5) | 10 (20.0) | 0.006 | |
| Strong | 45 (71.4) | - | - | - | 5 (38.5) | 40 (80.0) | ||
| p-p38 staining, No. (%) | ||||||||
| Positive | 2 (3.1) | 0 (0.0) | 2 (3.7) | 1.0 | 0 (0.0) | 2 (3.9) | 1.0 | |
| Negative | 62 (96.9) | 10 (100.0) | 52 (96.3) | 13 (100.0) | 49 (96.1) | |||
| Cytoplasmic DUSP10 (n = 961 ) | Nuclear DUSP10 (n = 961) | |||||||
|---|---|---|---|---|---|---|---|---|
| Categories | No. of Patients a | Weak | Strong | p-Value b | Weak | Strong | p-Value b | |
| Sex, No. (%) | ||||||||
| Female | 479 (49.8) | 293 (51.7) | 186 (47.2) | 0.190 | 235 (47.7) | 244 (52.1) | 0.176 | |
| Male | 482 (50.2) | 274 (48.3) | 208 (52.8) | 258 (52.3) | 224 (47.9) | |||
| Age, No. (%) | ||||||||
| ≤65 | 329 (34.2) | 199 (35.1) | 130 (33.0) | 0.457 | 155 (31.4) | 174 (37.2) | 0.156 | |
| 66-74 | 292 (30.4) | 172 (30.3) | 120 (30.5) | 159 (32.3) | 133 (28.4) | |||
| ≥75 | 340 (35.4) | 196 (34.6) | 144 (36.5) | 179 (36.3) | 161 (34.4) | |||
| Stage, No. (%) | ||||||||
| I | 136 (14.2) | 88 (15.6) | 48 (12.2) | 0.391 | 80 (16.2) | 56 (12.0) | 0.043 | |
| II | 392 (40.9) | 232 (41.1) | 160 (40.7) | 201 (40.8) | 191 (41.1) | |||
| III | 271 (28.3) | 147 (26.0) | 124 (31.6) | 140 (28.4) | 131 (28.2) | |||
| IV | 159 (16.6) | 98 (17.3) | 61 (16.6) | 72 (14.6) | 87 (18.7) | |||
| Unknown | 3 (0.3) | |||||||
| Grade, No. (%) | ||||||||
| Well differentiated | 96 (10.0) | 50 (8.8) | 46 (11.7) | 0.027 | 53 (10.8) | 43 (9.2) | 0.185 | |
| Moderately differentiated | 607 (63.2) | 348 (61.4) | 259 (65.7) | 293 (59.4) | 314 (67.1) | |||
| Poorly differentiated | 182 (18.9) | 118 (20.8) | 64 (16.2) | 108 (21.9) | 74 (15.8) | |||
| Unknown | 76 (7.9) | |||||||
| Chemotherapy, No. (%) | ||||||||
| No | 673 (70.0) | 412 (72.7) | 261 (66.2) | 0.038 | 352 (71.4) | 321 (68.6) | 0.360 | |
| Yes | 288 (30.0) | 155 (27.3) | 133 (33.8) | 141 (28.6) | 147 (31.4) | |||
| Radiotherapy, No. (%) | ||||||||
| No | 948 (98.6) | 557 (98.2) | 391 (99.2) | 0.259 | 485 (98.4) | 463 (98.9) | 0.580 | |
| Yes | 13 (1.4) | 10 (1.8) | 3 (0.8) | 8 (1.6) | 5 (1.1) | |||
| Nuclear DUSP10 | No. of Patients | No. of Death | RR Univariate (95% CI) | p-Value a | RR Adjusted 1 (95% CI) | p-Value a | RR Adjusted 2 (95% CI) | p-Value a | |
|---|---|---|---|---|---|---|---|---|---|
| Weak | 481 | 211 | 1 [Reference] | 0.047 | 1 [Reference] | 0.033 | 1 [Reference] | 0.189 | |
| Strong | 459 | 213 | 1.22 (1.00–1.49) | 1.24 (1.02–1.51) | 1.14 (0.94–1.39) | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Martínez, M.; Ostalé, C.M.; van der Burg, L.R.; Galán-Martínez, J.; Hardwick, J.C.H.; López-Pérez, R.; Hawinkels, L.J.A.C.; Stamatakis, K.; Fresno, M. DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression. Cancers 2019, 11, 1767. https://doi.org/10.3390/cancers11111767
Jiménez-Martínez M, Ostalé CM, van der Burg LR, Galán-Martínez J, Hardwick JCH, López-Pérez R, Hawinkels LJAC, Stamatakis K, Fresno M. DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression. Cancers. 2019; 11(11):1767. https://doi.org/10.3390/cancers11111767
Chicago/Turabian StyleJiménez-Martínez, Marta, Cristina M. Ostalé, Lennart R. van der Burg, Javier Galán-Martínez, James C. H. Hardwick, Ricardo López-Pérez, Lukas J. A. C. Hawinkels, Konstantinos Stamatakis, and Manuel Fresno. 2019. "DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression" Cancers 11, no. 11: 1767. https://doi.org/10.3390/cancers11111767
APA StyleJiménez-Martínez, M., Ostalé, C. M., van der Burg, L. R., Galán-Martínez, J., Hardwick, J. C. H., López-Pérez, R., Hawinkels, L. J. A. C., Stamatakis, K., & Fresno, M. (2019). DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression. Cancers, 11(11), 1767. https://doi.org/10.3390/cancers11111767