2. Results
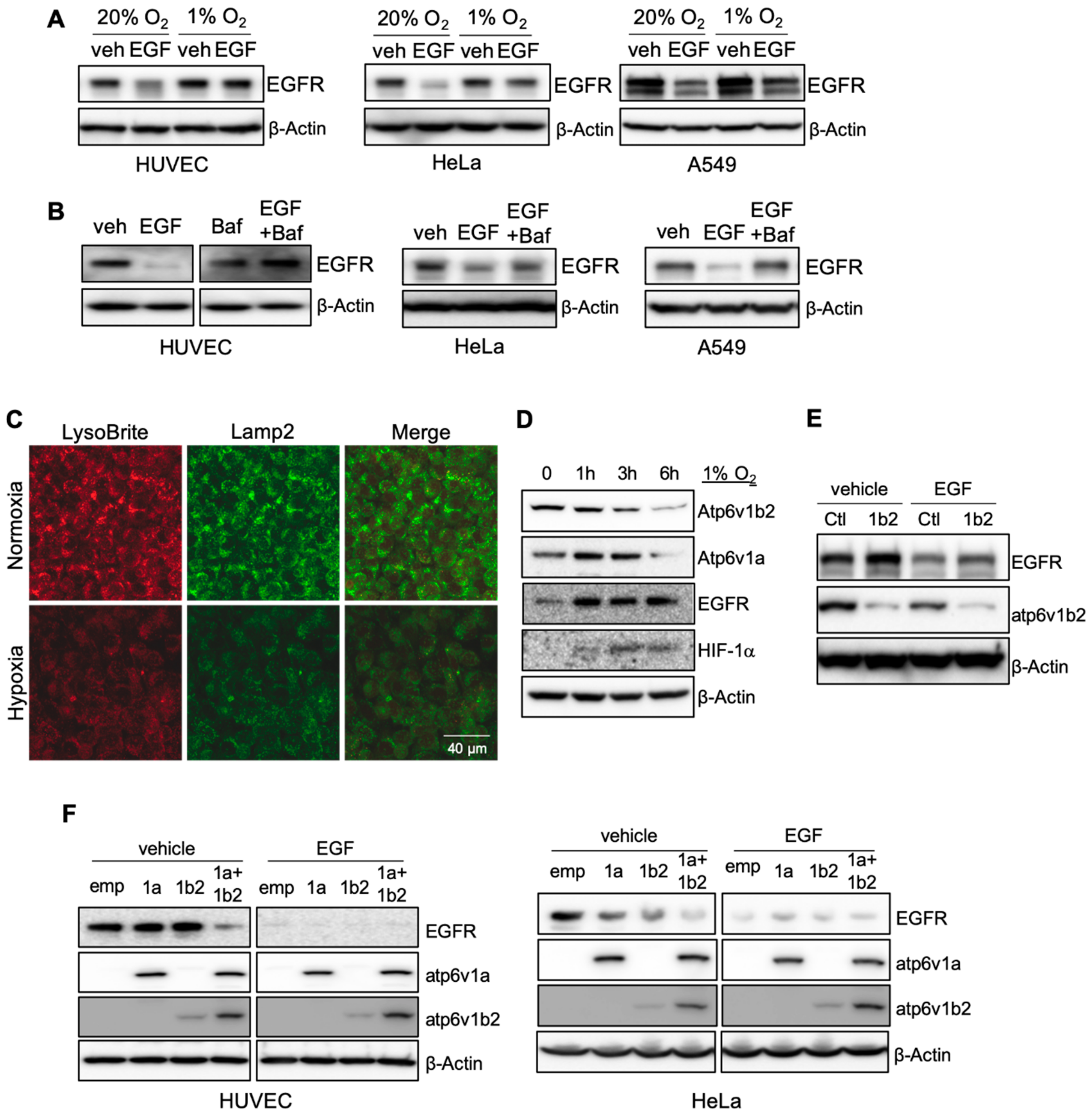
In studying the effect of hypoxia on RTK activity, we identified a link between hypoxia and receptor signaling. Human endothelial cells were cultured either in normoxic (20% O
2) or hypoxic (1% O
2) conditions in the presence or absence of growth factors. Ligand stimulation with recombinant epidermal growth factor (EGF) resulted in a reduction of EGF receptor (EGFR) levels in cells cultured in normoxia. Ligand stimulation failed to induce EGFR degradation in hypoxia (
Figure 1A). We observed similar results in two epithelial tumor cell lines, HeLa and A549 (
Figure 1A). Elevated EGFR levels in hypoxic conditions resulted in increased receptor phosphorylation and activation of downstream signaling in both types of cells (
Figure S1A). Hypoxia also blocked VEGF-induced VEGFR2 reduction in endothelial cells (
Figure S1B). These findings reveal a general inhibitory role of hypoxia in RTK degradation.
The failure of receptor degradation under hypoxic conditions suggested that hypoxia may play a role in lysosomal function. We used Bafilomycin A1, a specific inhibitor of V-ATPase, to block lysosomal acidification/activation under normoxic conditions and observed that EGF-induced EGFR degradation was reduced in cultured endothelial cells, as well as epithelial tumor cell lines after ligand stimulation, as seen in hypoxic conditions (
Figure 1B). The addition of chloroquine (CQ), another lysosomal specific inhibitor, also blocked ligand-stimulation induced VEGFR2 reduction in endothelial cells (
Figure S1C). These results suggest that hypoxia may suppress receptor degradation through the inhibition of lysosomal activity.
We examined if hypoxia affects lysosome acidification. Using LysoBrite, a dye that is specific for acidic organelles, we observed a significant reduction in the LysoBrite signal in cells cultured in hypoxic conditions. The hypoxic treatment also led to a reduction of Lamp2, a marker for lysosomes (
Figure 1C). The acidification of lysosomes is generated by the action of V-ATPase [
9,
10]. We measured two catalytic components of V-ATPase, Atp6v1a and Atp6v1b2, in cells incubated under hypoxic conditions and observed a gradual reduction of both subunits, which correlated with an increase of EGFR (
Figure 1D). Thus, the inhibitory effect of hypoxia on lysosomal acidification is caused by the reduction of V-ATPase.
To further establish the connection between the acidification of lysosomes by V-ATPase with ligand-induced receptor degradation, we knocked down Atp6v1b2 expression in cells. Reduction of V-ATPase inhibited ligand-mediated receptor degradation, resulting in increased levels of EGFR (
Figure 1E). Modest ectopic over-expression of Atp6v1a or Atp6v1b2 in endothelial cells did not result in a significant change in EGFR levels. Over-expression of both components led to a substantial reduction of EGFR (
Figure 1F). In epithelial cells, ectopic expression of either one of these V-ATPase subunits led to a clear reduction of EGFR, and over-expression of both produced a more pronounced response than either one alone (
Figure 1F). The difference observed between endothelial and epithelial cells may reflect the difference of endogenous V-ATPase subunit levels in different types of cells.
Hypoxia inhibits mTOR activity through the TSC1/2 complex and regulated in development and DNA damage response 1 (REDD1) [
11,
12,
13], and the mTOR pathway is involved in controlling V-ATPase assembly during dendritic cell maturation [
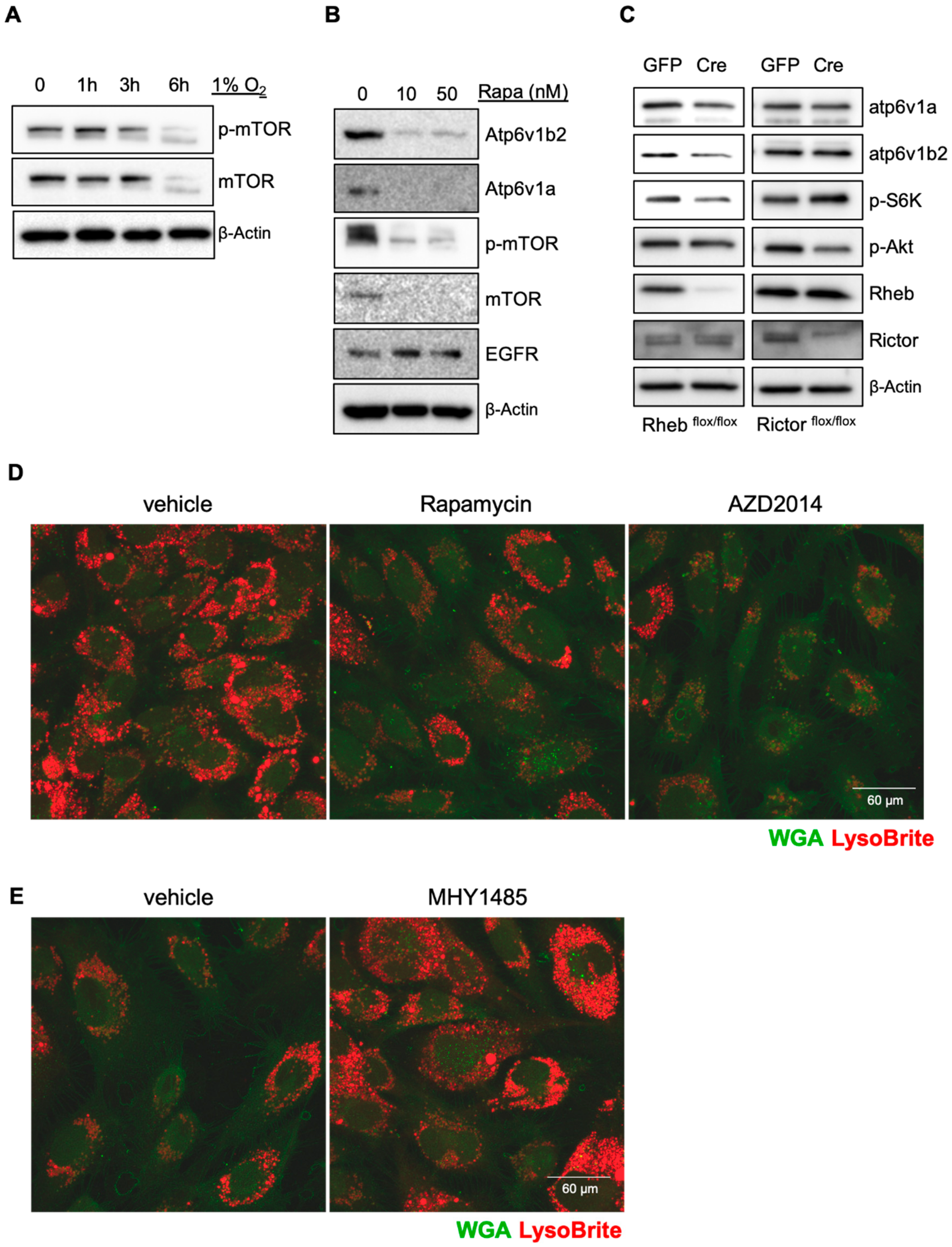
14]. We thus investigated if mTOR is responsible for hypoxia-mediated inhibition of V-ATPase expression. Hypoxia led to a reduction of both total mTOR and phosphorylated mTOR (
Figure 2A). Blocking mTOR activity with rapamycin significantly reduced the levels of V-ATPase subunits, which correlated with increased EGFR levels (
Figure 2B). To determine which mTOR complex regulates the expression of V-ATPase, we purified pulmonary microvascular endothelial cells from mTORc1 (Rheb
flox/flox) or mTORc2 (Rictor
flox/flox) floxed mice, and infected them with adenoviral vectors for the Cre recombinase. Deletion of Rheb (mTORc1) but not Rictor (mTORc2) resulted in a reduction of Atp6v1a and Atp6v1b2 (
Figure 2C). Treating endothelial cells with the mTORc1-specific inhibitors rapamycin and AZD2014 led to reduction of the acidification of lysosomes (
Figure 2D). Activation of mTORc1 using MHY1485 increased acidification of lysosomes (
Figure 2E). These results suggest that hypoxia inhibits lysosomal acidification/activation through suppression of mTORc1 mediated V-ATPase expression.
V-ATPase regulation by mTORc1 involves TFEB [
15], which binds to the regulatory regions of many lysosomal genes, activating their expression [
16]. TFEB shuttles between the cytosol and nucleus, and nuclear translocation is required for its function. In normoxic cells, immunostaining for TFEB protein revealed the majority was in the nucleus, whereas it was mainly in the cytosol under hypoxic conditions, as was seen when cells were cultured in normoxia in the presence of an mTORc1 specific inhibitor (
Figure S2A). Addition of the mTORc1 activator, MHY1485, reversed the hypoxia-mediated blockade of TFEB nuclear translocation (
Figure S2B). These findings suggest that hypoxia inhibits TFEB nuclear translocation via inhibition of mTOR, thereby preventing V-ATPase transcription.
TFEB phosphorylation regulates nuclear translocation. Both Serine 142 (S142) [
17], and a serine-rich region at the C-terminus (
462SSRRSSFS
469) [
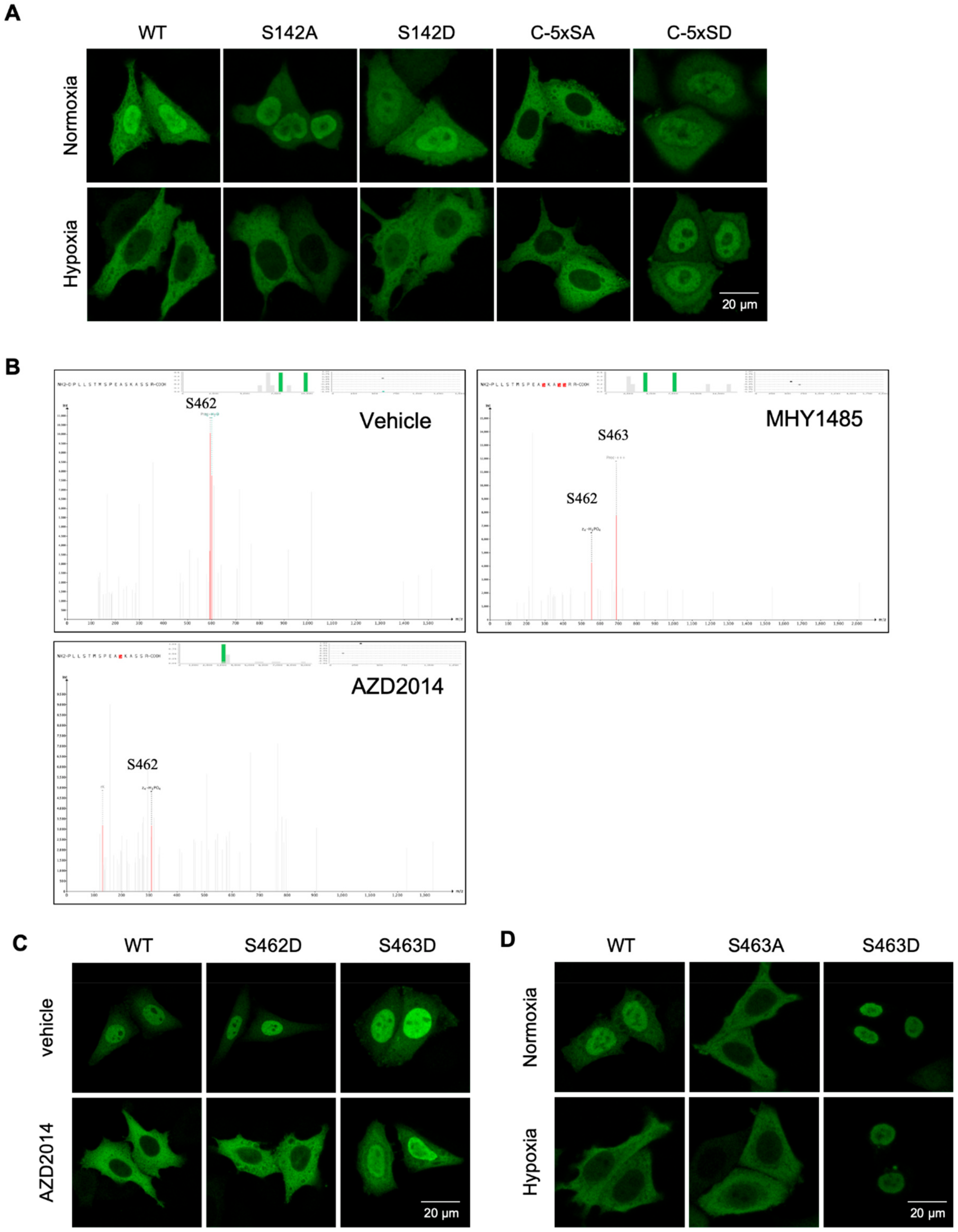
15] have been implicated in TFEB nuclear translocation. To determine which site(s) is(are) responsible for hypoxia-mediated translocation, we transfected cells with phospho-mimic or inhibitory mutants of TFEB, and subjected them to normoxia or hypoxia. Mutating S142 to either alanine or aspartic acid had no effect on TFEB nuclear translocation in response to hypoxia. Mutating all five serine residues at the C-terminus to alanines blocked TFEB nuclear translocation. Mutating them to aspartic acids led to spontaneous nuclear translocation of TEFB regardless the levels of oxygen (
Figure 3A). To determine which serine residue(s) in this cluster is phosphorylated by mTORc1, we over-expressed Myc-tag TFEB in HeLa cells, followed by treatment with either vehicle, or MHY1485, or AZD2014. TFEB protein was pulled down with an anti-Myc antibody and subjected to mass-spectrometry. Phosphorylation at S462 was detected in all three treatment groups, while phosphorylation at S463 only occurred after a stimulation with mTORc1 activator MHY1485 (
Figure 3B). There was no detectable phosphorylation signal on any other serine residue in this cluster. S142 in TFEB was phosphorylated regardless the activation status of mTORc1 (
Figure S3). Thus, mTORc1 specifically phosphorylates TFEB at the Serine 463 site.
To evaluate the role of phosphorylation at S463 on TFEB nuclear translocation, we mutated this serine to either alanine (S463A) or aspartic acid (S463D). As a control, we also mutated S462 to aspartic acid (S462D). The mutant constructs were transfected into HeLa cells followed by treatment with AZD2014. Blocking mTORc1 inhibited the nuclear translocation of wild-type TFEB, as well as the S462D mutant. However, AZD2014 failed to block S463D nuclear translocation (
Figure 3C). Transfection of the S463A and S463D mutants into HeLa cells, and their exposure to either normoxic or hypoxic conditions, revealed the S463A mutation blocked TFEB nuclear translocation, and the S463D mutant spontaneously translocated to the nucleus, independent of oxygen tension (
Figure 3D). Activation of mTORc1 with MHY1485 reversed the hypoxia-induced nuclear blockade of wild-type TFEB. The S463D mutant spontaneously translocated to the nucleus and the S463A mutant failed to translocate to the nucleus regardless of oxygen tension and mTOR activity (
Figure S4). Thus, mTORc1 phosphorylates TFEB at S463, and this phosphorylation is necessary and sufficient for hypoxia-mediated TFEB nuclear translocation.
Hypoxia is a hallmark of solid tumors and is positively correlated with invasive phenotypes, resistance to therapy and poor prognosis of cancer patients [
2,
18,
19,
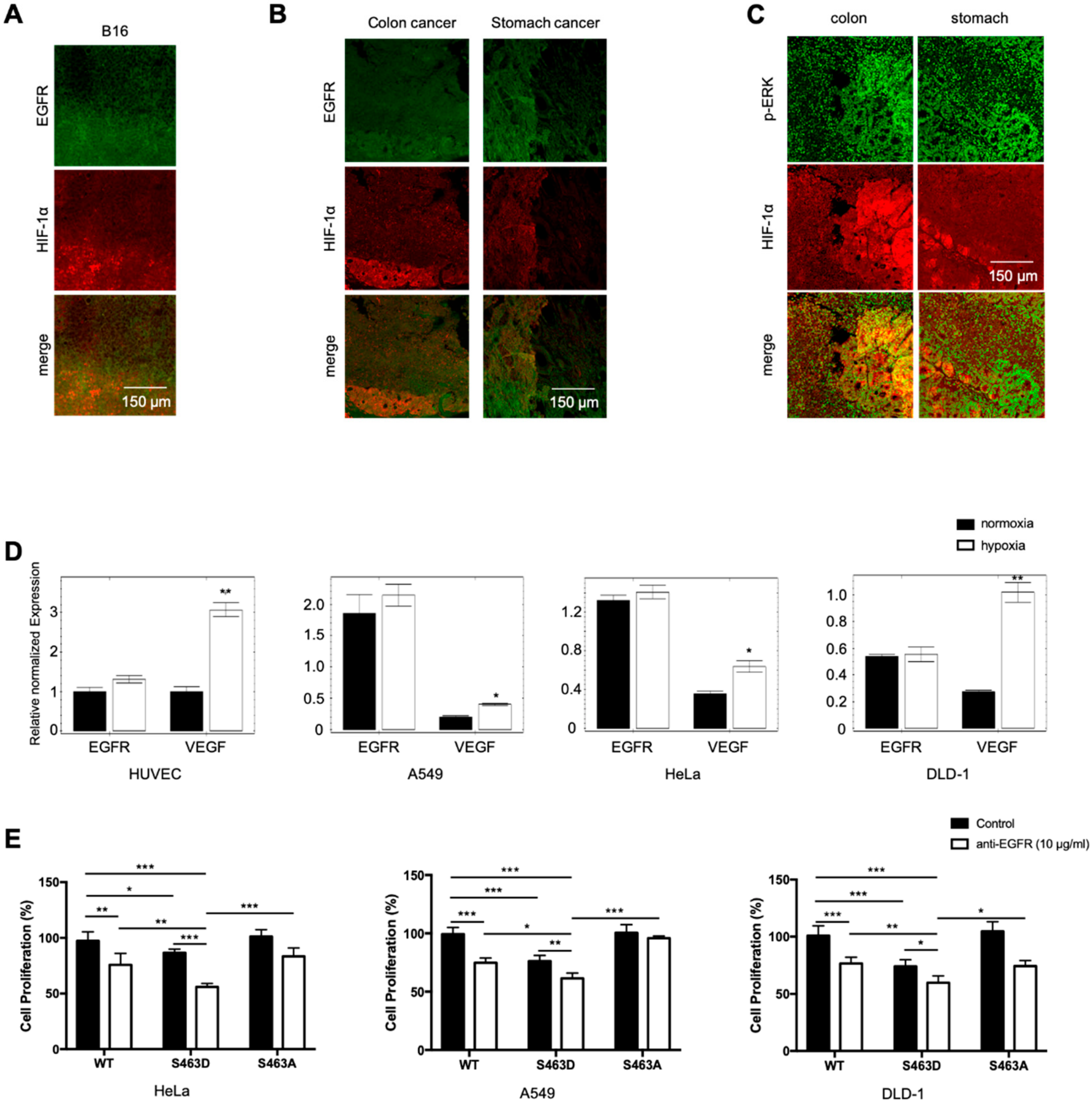
20]. RTKs are major drivers in cancer growth and progression, and RTK inhibitors are commonly used in cancer therapy. Since hypoxia blocks RTK degradation, hypoxic tumors may have elevated RTK levels that contribute to the phenotypes associated with poor prognosis. Analyzing murine B16 melanoma tumor tissues, we found a positive correlation between hypoxia, measured by HIF-1α accumulation, and EGFR levels. The hypoxic region had significantly higher levels of EGFR than the less hypoxic region of the tumor (
Figure 4A). Analysis of human cancer tissues also revealed a positive correlation between EGFR levels and hypoxia in both colon and stomach cancer samples (
Figure 4B). In addition, there is a positive correlation of hypoxia with phosphorylation of ERK, a downstream mediator of EGFR signaling (
Figure 4C). To determine what drives elevated EGFR levels in hypoxic tumors, we incubated three different tumor cell lines plus one endothelial cell line in hypoxia or normoxia and measured transcription by RT-qPCR. There was no statistical increase in EGFR transcript levels in the four cell lines, regardless of the hypoxic or nontoxic conditions.
VEGF, a gene known to be regulated by hypoxia, was increased in all four cell lines subjected to hypoxia (
Figure 4D). These findings suggest that increased RTK levels in hypoxic tumors are likely due to inhibited receptor degradation in lysosomes.
EGF signaling is involved in cell proliferation and cancer progression, and EGF inhibitors are widely used in cancer therapy. Given that hypoxia blocked lysosome-mediated EGFR degradation, resulting in increased signaling, lysosomal activators should increase receptor degradation and potentially provide therapeutic benefit in anti-RTK cancer therapy. To evaluate this idea, we ectopically expressed the wild-type and S463D mutant of TFEB in three human cancer cell lines to modulate lysosomal acidification/activation and treated the cells with EGFR inhibitors under hypoxia. S463A mutant was included as a control. Activation of lysosomes with the S463D significantly reduced cell proliferation in all three tumor lines. A combination of lysosomal activation and EGFR inhibitors produced the strongest inhibition of all tumor cells (
Figure 4E). In contrast, there is no significant difference between cells transfected with the WT or the S463A mutant, consistent with the finding that hypoxia blocks TFEB phosphorylation and nuclear translocation.
4. Materials and Methods
4.1. Experimental Animals
The mice were maintained in pathogen-free facilities in the National Cancer Institute (Frederick, MD, USA). The study was approved by the NCI Animal Care and Use Committee (protocol number 17-009, 17-010 and 17-048), and in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Rheb- and Rictor-flox mice are on a C57BL/6 background. Age and sex matched mice were used in isolation of endothelial cells, and pooled cells from 3–5 mice per group were used. 5 × 105 of B16 cells were injected to the right flank of C57BL/6 mice. The tumor tissue was harvested between 3–5 weeks post-injection.
4.2. Cell Culture and Bioassays
Human umbilical vein endothelial cells (HUVECs) and human epithelial cell lines (HeLa, A549, and DLD-1) were obtained from Lonza (Walkersville, MD, USA) and ATCC (Manassas, VA, USA), respectively. Lung endothelial cells were isolated from Rheb- and Rictor-floxed mice as described [
24]. The cells were cultured according to the manufacture’s protocols. Recombinant EGF and VEGF proteins were purchased from ProSpec (East Brunswick, NJ, USA). For EGFR and VEGFR2 degradation assays, serum starved cells were stimulated with 50 ng/mL of EGF or 100 ng/mL of VEGF for 5 h. For hypoxia, cells were incubated in 1% O
2 for 1–6 h. mTORc1 inhibitor, Rapamycin (Selleckchem, Houston, TX, USA) or AZD2014 (Selleckchem) at 10 nM, and mTORc1 activator, MHY1485 at 2 μM, were used. Cell transfection of Myc tagged TFEB and V-ATPase components, pcDNA-ATP6V1A plus pcDNA-ATP6V1B2 were carried out by using Fugene HD (Promega, Madison, WI, USA). For the anti-EGFR assay, cells were treated with 10 μg/mL of anti-EGFR monoclonal antibody (Sigma, clone LA1, St. Louise, MO, USA) or control mouse IgG for 16 h. XTT cell proliferation assay was conducted following the description.
4.3. Immunofluorescent Staining and Microscopy
Cells were incubated with LysoBrite-NIR (AAT Bio, Sunyvale, CA, USA) following the manufacturers protocol. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with a primary antibody followed by fluorescent dye-conjugated secondary antibody. Antibodies against Lamp2 and Myc tag for TFEB were obtained from DSHB and Cell Signaling, respectively. Alexa 488-conjugated wheat germ agglutinin (Thermo Fisher Scientific, Waltham, MA, USA) was used to stain the membranous structure. The tumor tissue was stained with antibodies against EGFR (Sigma-Aldrich, St. Louise, MO, USA), HIF-1α (Abcam, Cambridge, UK) and p-ERK (Cell Signaling, Danvers, MA, USA). Human tumor tissue slides were purchased from Genetex (Irvine, CA, USA).
Confocal microscopy images were analyzed with Zen software (Carl Zeiss, Oberkochen, Germany) and imageJ (NIH, Bethesda, MD, USA).
4.4. Western Blot and RT-qPCR
Cells were lysed with sample buffer and briefly ultrasonicated. SDS-PAGE and Western blot was carried out on 5 μg of total protein. Specific antibodies for EGFR (#2085), VEGFR2 (#9698), p-mTOR (#5536), mTOR (#2983), p-S6K (#9204), p-Akt (#4060), Rheb (#13879), Rictor (#9476) were purchased from Cell Signaling Technology (Danvers, MA) and antibodies against Atp6v1a (#GT3846) and Apt6v1b2 (#GTX110783) from GeneTex (Irvine, CA, USA). Isolated by RNeasy mini (Qiagen, Hilden, Germany), 100 μg of total RNA was propagated to RT-qPCR from cell lines. Relative mRNA level was measured with primers purchased against EGFR (#KSPQ12012, Sigma-Aldrich) and VEGF (#KSPQ12012, Sigma-Aldrich).
4.5. Statistics
Prism (Graphpad) was used for all statistical analyses. For analysis, two-way ANOVA was used followed by Bonferroni Multiple Comparisons test.


{kind=link}
{kind=link}
{kind=link}
{kind=link}



