Tumor Endothelial Heterogeneity in Cancer Progression


Abstract
1. Introduction
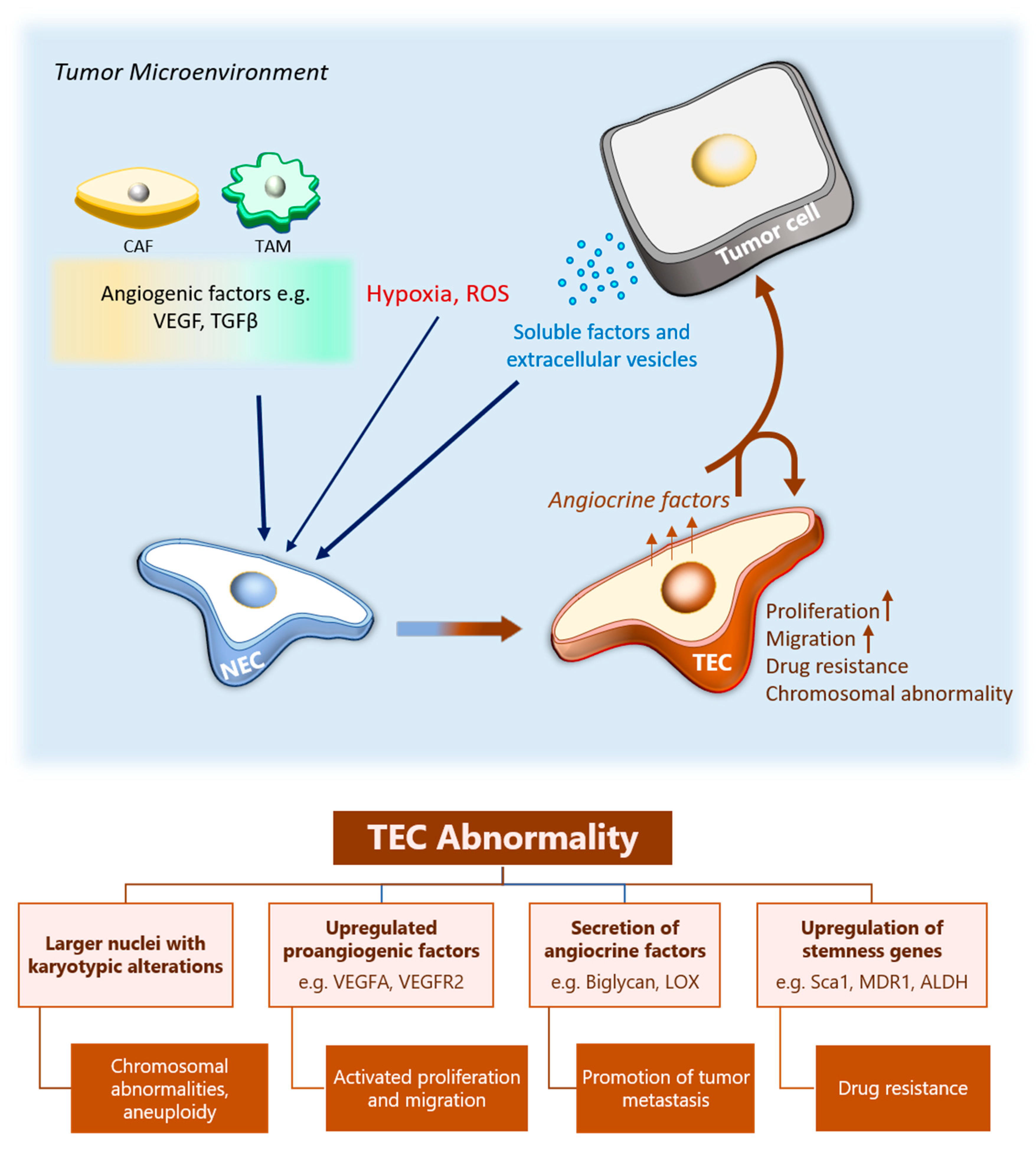
2. Abnormalities of TECs
2.1. Tumor Blood Vessels and Normal Blood Vessels
2.2. Differential Characteristics of Tumor and Normal Endothelial Cells
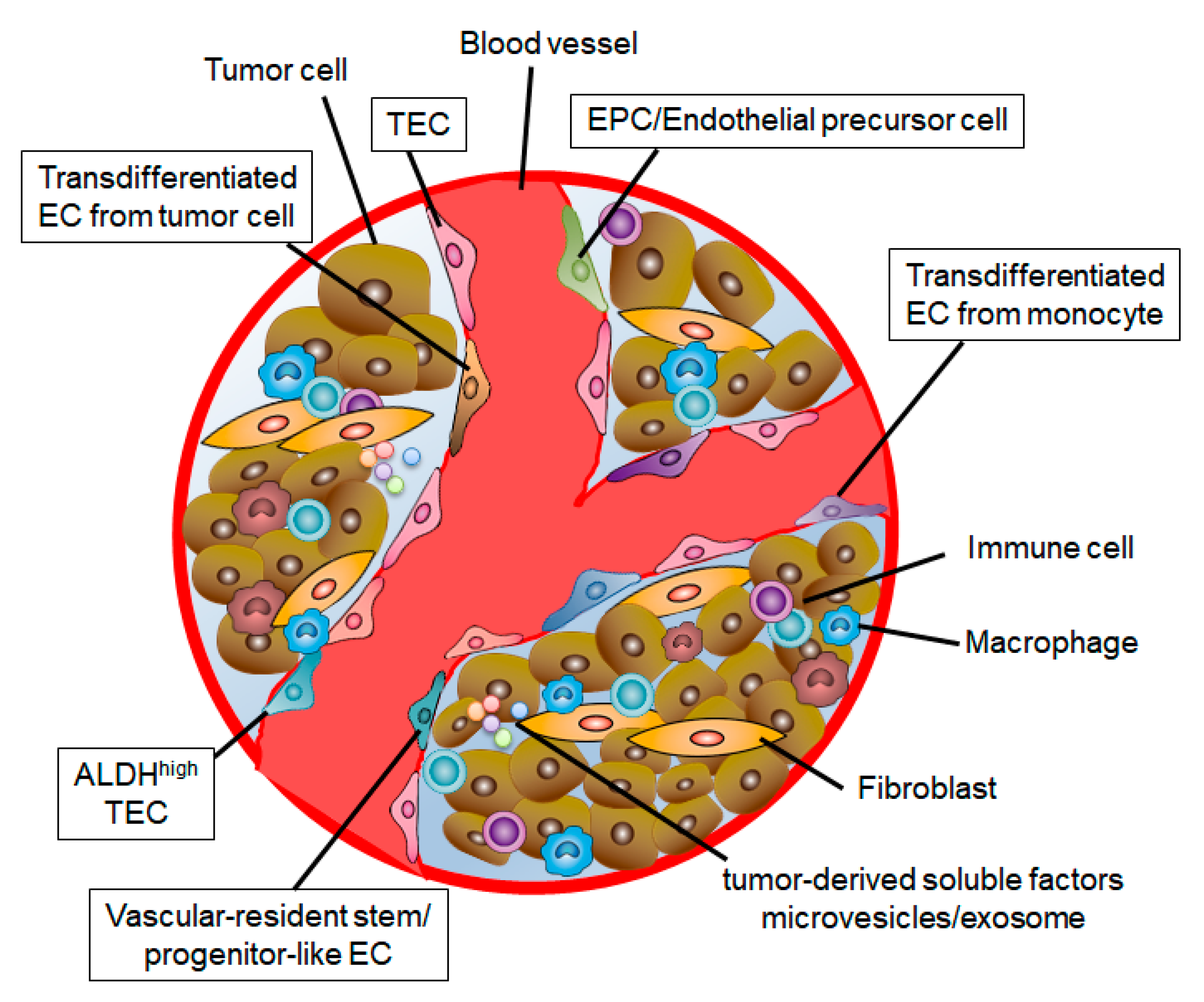
3. Heterogeneity of TECs
3.1. Different Roles in ECs during Angiogenesis
3.2. Origin of TECs
3.3. Stem Cell Population in TECs
3.4. The Effect of Tumor Microenvironment on TEC Heterogeneity
4. The Role of TECs in Cancer Progression
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Culy, C. Bevacizumab: Antiangiogenic cancer therapy. Drugs Today (Barc) 2005, 41, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.A.; Megyesi, J.F.; Henzel, W.J.; Ferrara, N.; Folkman, J. Conditioned medium from mouse sarcoma 180 cells contains vascular endothelial growth factor. Growth Factors 1990, 4, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [Google Scholar] [CrossRef] [PubMed]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef]
- Endrich, B.; Reinhold, H.S.; Gross, J.F.; Intaglietta, M. Tissue perfusion inhomogeneity during early tumor growth in rats. J. Natl. Cancer Inst. 1979, 62, 387–395. [Google Scholar]
- Zhang, X.; Nie, D.; Chakrabarty, S. Growth factors in tumor microenvironment. Front. Biosci. (Landmark Ed) 2010, 15, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am. J. Pathol. 2003, 162, 1747–1757. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: Mechanism of low pH-induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef] [PubMed]
- Konerding, M.A.; Malkusch, W.; Klapthor, B.; van Ackern, C.; Fait, E.; Hill, S.A.; Parkins, C.; Chaplin, D.J.; Presta, M.; Denekamp, J. Evidence for characteristic vascular patterns in solid tumours: Quantitative studies using corrosion casts. Br. J. Cancer 1999, 80, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Sund, M.; Zeisberg, M.; Kalluri, R. Endogenous Stimulators and Inhibitors of Angiogenesis in Gastrointestinal Cancers: Basic Science to Clinical Application. Gastroenterology 2005, 129, 2076–2091. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure — an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.J.; Olive, P.L.; Durand, R.E. Intermittent Blood Flow in a Murine Tumor: Radiobiological Effects. Cancer Res. 1987, 47, 597–601. [Google Scholar] [PubMed]
- Jiang, J.; Tang, Y.L.; Liang, X.H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar]
- Akino, T.; Hida, K.; Hida, Y.; Tsuchiya, K.; Freedman, D.; Muraki, C.; Ohga, N.; Matsuda, K.; Akiyama, K.; Harabayashi, T.; et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am. J. Pathol. 2009, 175, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004, 64, 8249–8255. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Deambrosis, I.; Russo, S.; Deregibus, M.C.; Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003, 17, 1159–1161. [Google Scholar] [CrossRef]
- Alessandri, G.; Chirivi, R.G.; Fiorentini, S.; Dossi, R.; Bonardelli, S.; Giulini, S.M.; Zanetta, G.; Landoni, F.; Graziotti, P.P.; Turano, A.; et al. Phenotypic and functional characteristics of tumour-derived microvascular endothelial cells. Clin. Exp. Metastasis 1999, 17, 655–662. [Google Scholar]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ohga, N.; Hida, Y.; Maishi, N.; Kawamoto, T.; Kitayama, K.; Akiyama, K.; Osawa, T.; Kondoh, M.; Matsuda, K.; et al. Biglycan is a specific marker and an autocrine angiogenic factor of tumour endothelial cells. Br. J. Cancer. 2012, 106, 1214–1223. [Google Scholar] [CrossRef]
- Osawa, T.; Ohga, N.; Akiyama, K.; Hida, Y.; Kitayama, K.; Kawamoto, T.; Yamamoto, K.; Maishi, N.; Kondoh, M.; Onodera, Y.; et al. Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br. J. Cancer 2013, 109, 2237–2247. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Kawamoto, T.; Akiyama, K.; Ohga, N.; Hida, Y.; Yamada, K.; Hojo, T.; Kikuchi, H.; Sato, M.; et al. Tumor endothelial cells express high pentraxin 3 levels. Pathol. Int. 2016, 66, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Otsubo, T.; Hida, Y.; Ohga, N.; Sato, H.; Kai, T.; Matsuki, Y.; Takasu, H.; Akiyama, K.; Maishi, N.; Kawamoto, T.; et al. Identification of novel targets for antiangiogenic therapy by comparing the gene expressions of tumor and normal endothelial cells. Cancer Sci. 2014, 105, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of tumor endothelial cells: Comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays 2016, 38 (Suppl. 1), S65–S74. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Akiyama, K.; Ohmura-Kakutani, H.; Torii, C.; Ohga, N.; Osawa, T.; Kikuchi, H.; Morimoto, H.; Morimoto, M.; et al. Tumor endothelial cells with high aldehyde dehydrogenase activity show drug resistance. Cancer Sci. 2017, 108, 2195–2203. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Sun, H.C.; Zhang, W.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Wang, L.; Wu, W.Z.; Qin, L.X.; Tang, Z.Y. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef]
- Geudens, I.; Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development 2011, 138, 4569–4583. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Asahara, T.; Masuda, H.; Takahashi, T.; Kalka, C.; Pastore, C.; Silver, M.; Kearne, M.; Magner, M.; Isner, J.M. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 1999, 85, 221–228. [Google Scholar] [CrossRef]
- Asahara, T.; Kawamoto, A. Endothelial progenitor cells for postnatal vasculogenesis. Am. J. Physiol. Cell Physiol. 2004, 287, C572–C579. [Google Scholar] [CrossRef]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef]
- Rafii, S.; Lyden, D.; Benezra, R.; Hattori, K.; Heissig, B. Vascular and haematopoietic stem cells: Novel targets for anti-angiogenesis therapy? Nat. Rev. Cancer 2002, 2, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.J.; Ciarrocchi, A.; Mellick, A.S.; Jaggi, J.S.; Bambino, K.; Gupta, S.; Heikamp, E.; McDevitt, M.R.; Scheinberg, D.A.; Benezra, R.; et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007, 21, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Marcola, M.; Rodrigues, C.E. Endothelial progenitor cells in tumor angiogenesis: Another brick in the wall. Stem Cells Int. 2015, 2015, 832649. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Differentiation of endothelium. FASEB J. 1995, 9, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; West, D.C.; Ager, A. Heterogeneity in endothelial cells from large vessels and microvessels. Differentiation 1987, 36, 57–70. [Google Scholar] [CrossRef]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef]
- Zhao, Q.; Eichten, A.; Parveen, A.; Adler, C.; Huang, Y.; Wang, W.; Ding, Y.; Adler, A.; Nevins, T.; Ni, M.; et al. Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res. 2018, 78, 2370–2382. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Streubel, B.; Chott, A.; Huber, D.; Exner, M.; Jager, U.; Wagner, O.; Schwarzinger, I. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N. Engl. J. Med. 2004, 351, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Kulla, A.; Burkhardt, K.; Meyer-Puttlitz, B.; Teesalu, T.; Asser, T.; Wiestler, O.D.; Becker, A.J. Analysis of the TP53 gene in laser-microdissected glioblastoma vasculature. Acta Neuropathol. 2003, 105, 328–332. [Google Scholar]
- Rodriguez, F.J.; Orr, B.A.; Ligon, K.L.; Eberhart, C.G. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 2012, 3, 98–106. [Google Scholar] [CrossRef]
- Chen, H.; Campbell, R.A.; Chang, Y.; Li, M.; Wang, C.S.; Li, J.; Sanchez, E.; Share, M.; Steinberg, J.; Berenson, A.; et al. Pleiotrophin produced by multiple myeloma induces transdifferentiation of monocytes into vascular endothelial cells: A novel mechanism of tumor-induced vasculogenesis. Blood 2009, 113, 1992–2002. [Google Scholar] [CrossRef]
- Fernandez Pujol, B.; Lucibello, F.C.; Gehling, U.M.; Lindemann, K.; Weidner, N.; Zuzarte, M.L.; Adamkiewicz, J.; Elsasser, H.P.; Muller, R.; Havemann, K. Endothelial-like cells derived from human CD14 positive monocytes. Differentiation 2000, 65, 287–300. [Google Scholar] [CrossRef]
- Fernandez Pujol, B.; Lucibello, F.C.; Zuzarte, M.; Lutjens, P.; Muller, R.; Havemann, K. Dendritic cells derived from peripheral monocytes express endothelial markers and in the presence of angiogenic growth factors differentiate into endothelial-like cells. Eur. J. Cell Biol. 2001, 80, 99–110. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Wakabayashi, T.; Kidoya, H.; Muramatsu, F.; Takara, K.; Eino, D.; Yamane, K.; Iba, T.; Takakura, N. Endothelial Side Population Cells Contribute to Tumor Angiogenesis and Antiangiogenic Drug Resistance. Cancer Res. 2016, 76, 3200–3210. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.E.; Rockwell, S. Tumor hypoxia: Its impact on cancer therapy. Cancer Metastasis Rev. 1987, 5, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Hojo, T.; Maishi, N.; Towfik, A.M.; Akiyama, K.; Ohga, N.; Shindoh, M.; Hida, Y.; Minowa, K.; Fujisawa, T.; Hida, K. ROS enhance angiogenic properties via regulation of NRF2 in tumor endothelial cells. Oncotarget 2017, 8, 45484–45495. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, S.M.; Nevis, K.R.; Park, H.L.; Rogers, G.C.; Rogers, S.L.; Cook, J.G.; Bautch, V.L. Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood 2010, 116, 3108–3117. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef]
- Maishi, N.; Ohga, N.; Hida, Y.; Akiyama, K.; Kitayama, K.; Osawa, T.; Onodera, Y.; Shinohara, N.; Nonomura, K.; Shindoh, M.; et al. CXCR7: A novel tumor endothelial marker in renal cell carcinoma. Pathol. Int. 2012, 62, 309–317. [Google Scholar] [CrossRef]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ohga, N.; Akiyama, K.; Hirata, N.; Kitahara, S.; Maishi, N.; Osawa, T.; Yamamoto, K.; Kondoh, M.; Shindoh, M.; et al. Tumor-derived microvesicles induce proangiogenic phenotype in endothelial cells via endocytosis. PLoS ONE 2012, 7, e34045. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra234. [Google Scholar] [CrossRef]
- Xiao, L.; Kim, D.J.; Davis, C.L.; McCann, J.V.; Dunleavey, J.M.; Vanderlinden, A.K.; Xu, N.; Pattenden, S.G.; Frye, S.V.; Xu, X.; et al. Tumor Endothelial Cells with Distinct Patterns of TGFbeta-Driven Endothelial-to-Mesenchymal Transition. Cancer Res. 2015, 75, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Akatsu, Y.; Takahashi, N.; Yoshimatsu, Y.; Kimuro, S.; Muramatsu, T.; Katsura, A.; Maishi, N.; Suzuki, H.I.; Inazawa, J.; Hida, K.; et al. Fibroblast growth factor signals regulate transforming growth factor-beta-induced endothelial-to-myofibroblast transition of tumor endothelial cells via Elk1. Mol. Oncol. 2019, 13, 1706–1724. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Sokeland, G.; Schumacher, U. The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol. Cancer 2019, 18, 12. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Pirtskhalaishvili, G.; Nelson, J.B. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate 2000, 44, 77–87. [Google Scholar] [CrossRef]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef]
- Yamada, K.; Maishi, N.; Akiyama, K.; Towfik Alam, M.; Ohga, N.; Kawamoto, T.; Shindoh, M.; Takahashi, N.; Kamiyama, T.; Hida, Y.; et al. CXCL12-CXCR7 axis is important for tumor endothelial cell angiogenic property. Int. J. Cancer 2015, 137, 2825–2836. [Google Scholar] [CrossRef]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Wang, Y.; Lewen, S.; Berahovich, R.D.; Penfold, M.E.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B.; et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.M.; Kim, S.H.; Jin, X.; Park, J.B.; Kim, S.H.; Joshi, K.; Nakano, I.; Kim, H. Crosstalk between glioma-initiating cells and endothelial cells drives tumor progression. Cancer Res. 2014, 74, 4482–4492. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Dunaway, C.M.; Rao, M.; Short, S.; Hwang, Y.; Gao, Y.; Li, D.; Jiang, A.; Shyr, Y.; Wu, J.Y.; et al. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res. 2011, 71, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bahr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Mathieu, C.; Sii-Felice, K.; Fouchet, P.; Etienne, O.; Haton, C.; Mabondzo, A.; Boussin, F.D.; Mouthon, M.A. Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol. Cell Neurosci. 2008, 38, 569–577. [Google Scholar] [CrossRef]
- Hendrikx, S.; Coso, S.; Prat-Luri, B.; Wetterwald, L.; Sabine, A.; Franco, C.A.; Nassiri, S.; Zangger, N.; Gerhardt, H.; Delorenzi, M.; et al. Endothelial Calcineurin Signaling Restrains Metastatic Outgrowth by Regulating Bmp2. Cell Rep. 2019, 26, 1227–1241. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.B.; Chan-Tack, K.; Hedican, S.P.; Magnuson, S.R.; Opgenorth, T.J.; Bova, G.S.; Simons, J.W. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996, 56, 663–668. [Google Scholar] [PubMed]
- Hatfield, K.; Ryningen, A.; Corbascio, M.; Bruserud, O. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int. J. Cancer 2006, 119, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. Melanoma exosome induction of endothelial cell GM-CSF in pre-metastatic lymph nodes may result in different M1 and M2 macrophage mediated angiogenic processes. Med. Hypotheses 2016, 94, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.S.; Rafii, S. Molecular Checkpoint Decisions Made by Subverted Vascular Niche Transform Indolent Tumor Cells into Chemoresistant Cancer Stem Cells. Cancer Cell 2017, 31, 110–126. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sims, A.H.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P.; Clarke, R.B. GPER mediates the angiocrine actions induced by IGF1 through the HIF-1alpha/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res. 2017, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.G.; Sun, H.C.; Zhu, X.D.; Chai, Z.T.; Zhang, Y.Y.; Ao, J.Y.; Cai, H.; Ma, D.N.; Wang, C.H.; Qin, C.D.; et al. Reduced expression of CD109 in tumor-associated endothelial cells promotes tumor progression by paracrine interleukin-8 in hepatocellular carcinoma. Oncotarget 2016, 7, 29333–29345. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, G.; Strilic, B.; Lammert, E. The vascular niche and its basement membrane. Trends Cell Biol. 2007, 17, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Nagao-Kitamoto, H.; Ohga, N.; Akiyama, K.; Maishi, N.; Kawamoto, T.; Shinohara, N.; Taketomi, A.; Shindoh, M.; Hida, Y.; et al. Suprabasin as a novel tumor endothelial cell marker. Cancer Sci. 2014, 105, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chen, T.T.; Barber, C.L.; Jordan, M.C.; Murdock, J.; Desai, S.; Ferrara, N.; Nagy, A.; Roos, K.P.; Iruela-Arispe, M.L. Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007, 130, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.I.; Bocci, M.; Lovrot, J.; Eleftheriou, N.; Roswall, P.; Cordero, E.; Lindstrom, L.; Bartoschek, M.; Haller, B.K.; Pearsall, R.S.; et al. Endothelial ALK1 Is a Therapeutic Target to Block Metastatic Dissemination of Breast Cancer. Cancer Res. 2015, 75, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Johnstone, R.W.; Zalcberg, J.R.; El-Osta, A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 2005, 24, 8061–8075. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Ohba, Y.; Alam, M.T.; Kawamoto, T.; Ohmura, H.; Yamada, K.; Torii, C.; et al. Inhibition of multidrug transporter in tumor endothelial cells enhances antiangiogenic effects of low-dose metronomic paclitaxel. Am. J. Pathol. 2015, 185, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Percival, A.; He, B.; Ganss, R. Immunomodulation of Tumor Vessels: It Takes Two to Tango. Trends Immunol. 2018, 39, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Turcotte, M.; Spring, K.; Pommey, S.; Royal, I.; Stagg, J. Anti-CD73 therapy impairs tumor angiogenesis. Int. J. Cancer 2014, 134, 1466–1473. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted TNFalpha stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef]
- Johansson-Percival, A.; Li, Z.J.; Lakhiani, D.D.; He, B.; Wang, X.; Hamzah, J.; Ganss, R. Intratumoral LIGHT Restores Pericyte Contractile Properties and Vessel Integrity. Cell Rep. 2015, 13, 2687–2698. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Angiocrine Factors | Functions | Refs |
|---|---|---|
| Angiopoietin-2 (Ang2) | Recruit innate immune cells | [96] |
| Basic fibroblast growth factor (bFGF) | Organogenesis and tumorigenesis | [89,97] |
| Biglycan | Stimulate tumor cell intravasation | [79] |
| Bone morphogenetic protein-2, 4 (BMP2, 4) | Tumorigenesis | [98] |
| Calcineurin | Vascular stabilization and promote metastatic outgrowth | [99] |
| C-X-C motif chemokine 12 (CXCL12) | Tumorigenesis and tumor progression | [100,101] |
| Endothelin-1 | Promote tumor growth | [102] |
| Granulocyte colony stimulating factor (G-CSF) | Promote leukemic cell proliferation | [103] |
| Granulocyte macrophage colony stimulating factor (GM-CSF) | Angiogenesis | [104] |
| Insulin growth factor binding protein-7 (IGFBP7) | Tumor-suppressive checkpoint | [105] |
| Insulin growth factor-1 (IGF1) | Stimulate chemoresistance and angiogenesis | [105,106] |
| Interleukin-3 (IL-3) | Promote leukemic cell proliferation | [103] |
| Interleukin-6 (IL-6) | Macrophage activation and tumor progression | [107] |
| Interleukin-8 (IL-8) | Angiogenesis and tumor progression | [108] |
| Jagged-1 (Jag1) | Promote tumor invasiveness and chemoresistance | [90] |
| laminin α4 (LAMA4) | Tumorigenesis | [109] |
| Lysyl oxidase (Lox) | Angiogenesis and stimulate tumor cell intravasation | [30] |
| Nitric oxide (NO) | Tumorigenesis | [110] |
| Platelet-derived growth factor (PDGF) | Angiogenesis and tumorigenesis | [94] |
| Placental growth factor (PGF) | Angiogenesis and tumorigenesis | [111] |
| Pentraxin 3 (PTX3) | Stimulate TEC proliferation | [31] |
| Slit2 | Tumor suppression | [95] |
| Suprabasin | Angiogenesis | [112] |
| Transforming growth factor beta (TGF-β) | Tumorigenesis and tumor progression | [113] |
| Vascular endothelial growth factor-A (VEGFA) | Angiogenesis and autocrine loop | [114] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers 2019, 11, 1511. https://doi.org/10.3390/cancers11101511
Maishi N, Annan DA, Kikuchi H, Hida Y, Hida K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers. 2019; 11(10):1511. https://doi.org/10.3390/cancers11101511
Chicago/Turabian StyleMaishi, Nako, Dorcas A. Annan, Hiroshi Kikuchi, Yasuhiro Hida, and Kyoko Hida. 2019. "Tumor Endothelial Heterogeneity in Cancer Progression" Cancers 11, no. 10: 1511. https://doi.org/10.3390/cancers11101511
APA StyleMaishi, N., Annan, D. A., Kikuchi, H., Hida, Y., & Hida, K. (2019). Tumor Endothelial Heterogeneity in Cancer Progression. Cancers, 11(10), 1511. https://doi.org/10.3390/cancers11101511