HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Responses of Leukemic Cells to LBH589
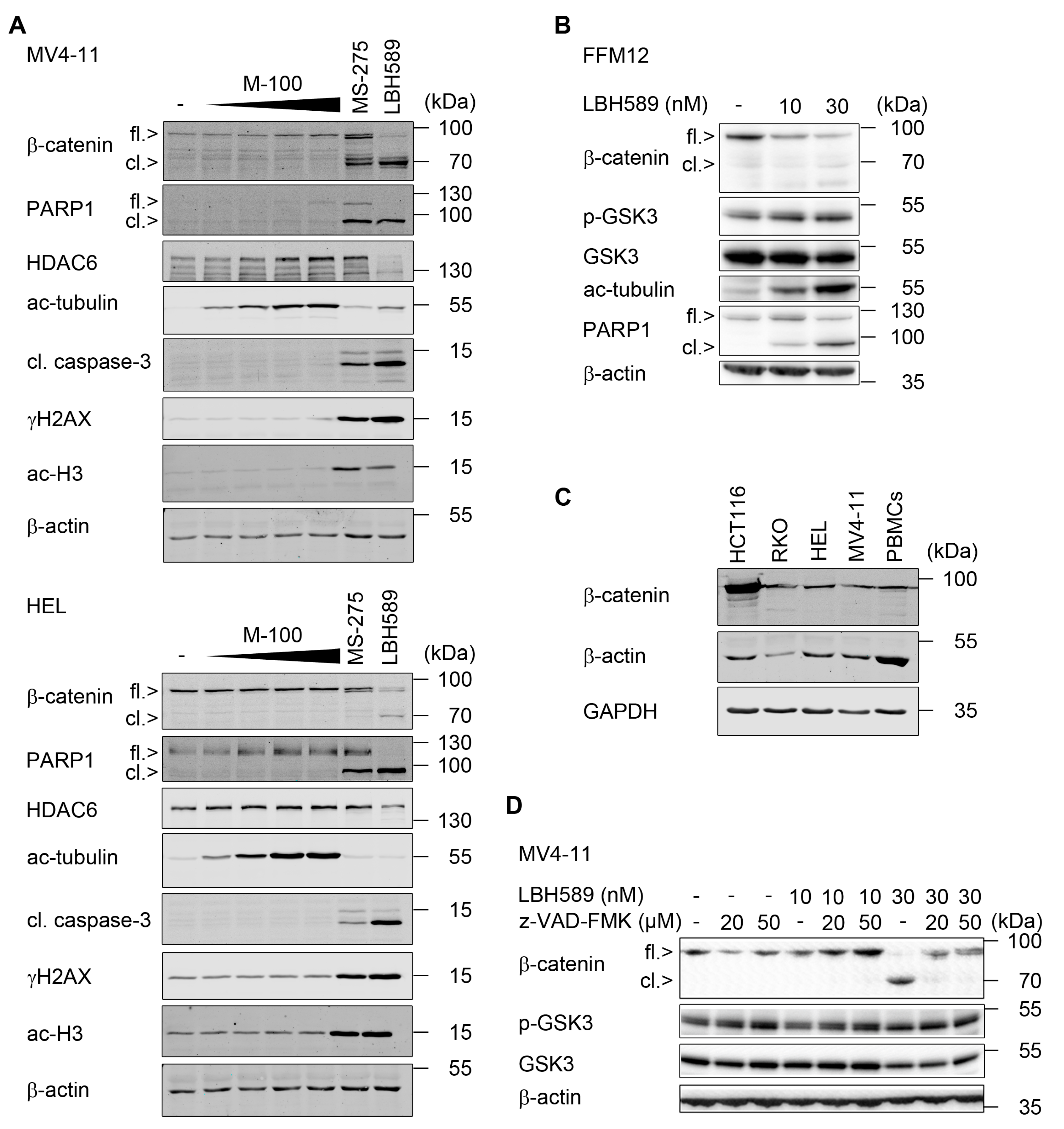
2.2. Reduction of β-Catenin upon Class I HDAC Inhibition
2.3. Indomethacin Can Accentuate Anti-Proliferative Effects of LBH589
2.4. HDAC3 Activity Is Required to Maintain β-Catenin, MYC, and WT1
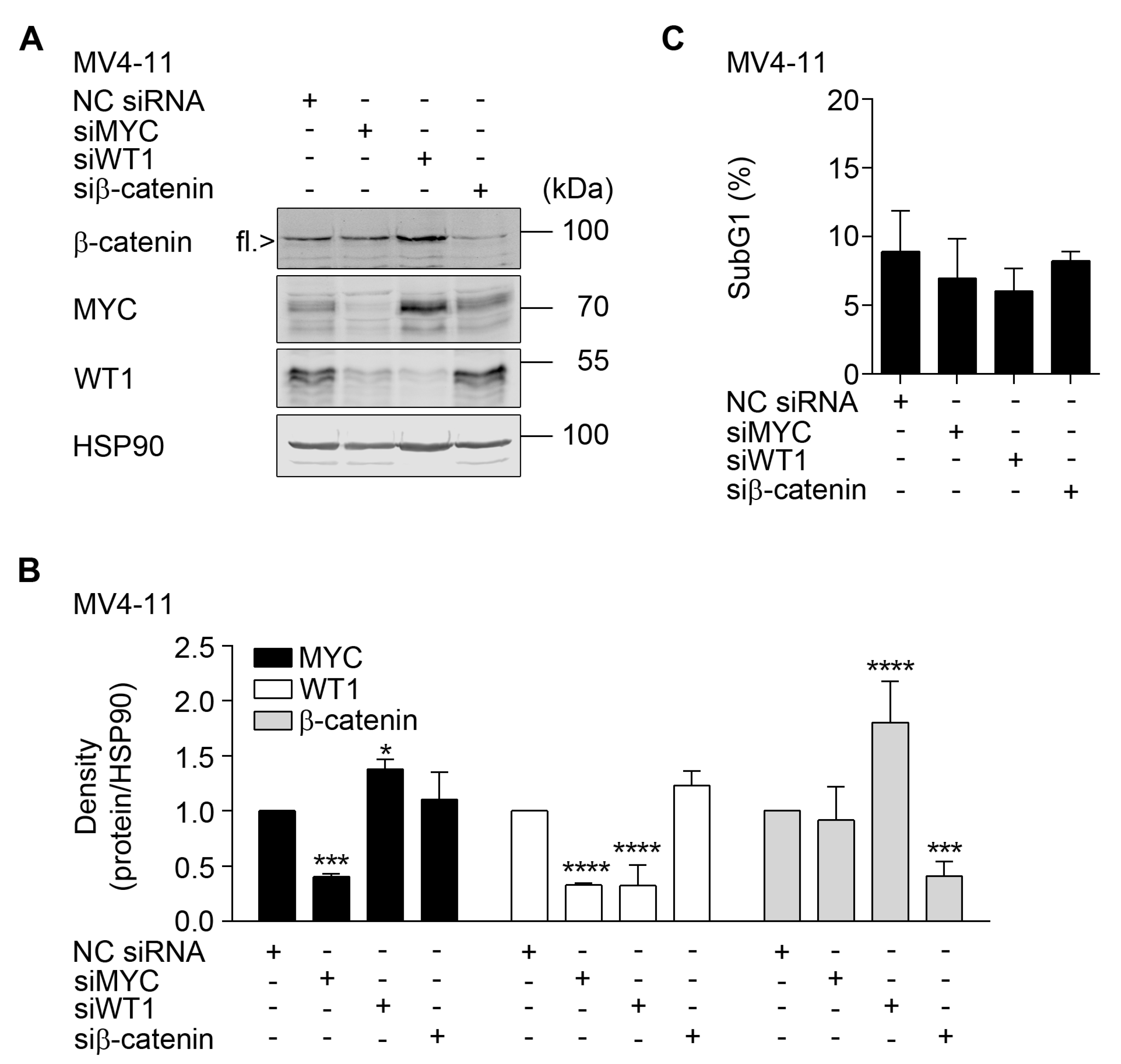
2.5. Interplay between β-Catenin, MYC, and WT1
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Apoptosis and Proliferation Assays
4.3. Western Blotting and Immunofluorescence
4.4. Real-Time PCR-TaqMan (qRT-PCR)
4.5. RNAi
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval and Consent to Participate
Consent to Publish
Data Availability
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Golding, M.C.; Srivastava, P.; Povinelli, B.J.; James, S.R.; Ford, L.A.; Wetzler, M.; Wang, E.S.; Nemeth, M.J. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica 2015, 100, e49–e52. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Gau, J.P.; You, J.Y.; Lee, K.D.; Yu, Y.B.; Lu, C.H.; Lin, J.T.; Lan, C.; Lo, W.H.; Liu, J.M.; et al. Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am. J. Hematol. 2009, 84, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Serinsoz, E.; Neusch, M.; Busche, G.; Wasielewski, R.; Kreipe, H.; Bock, O. Aberrant expression of beta-catenin discriminates acute myeloid leukaemia from acute lymphoblastic leukaemia. Br. J. Haematol. 2004, 126, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Ysebaert, L.; Chicanne, G.; Demur, C.; De Toni, F.; Prade-Houdellier, N.; Ruidavets, J.B.; Mansat-De Mas, V.; Rigal-Huguet, F.; Laurent, G.; Payrastre, B.; et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006, 20, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef]
- Morgan, R.G.; Ridsdale, J.; Payne, M.; Heesom, K.J.; Wilson, M.C.; Davidson, A.; Greenhough, A.; Davies, S.; Williams, A.C.; Blair, A.; et al. LEF-1 drives aberrant beta-catenin nuclear localization in myeloid leukemia cells. Haematologica 2019. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Gandillet, A.; Park, S.; Lassailly, F.; Griessinger, E.; Vargaftig, J.; Filby, A.; Lister, T.A.; Bonnet, D. Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia 2011, 25, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Famili, F.; Garcia Perez, L.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.A.; Jackson, L.; Helgason, V.; Holyoake, T.L. Stem Cell Guardians—Old and New Perspectives in LSC Biology. Curr. Drug Targets 2017, 18, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Wang, Y.J.; Lo Celso, C.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Knauer, S.K.; Mahendrarajah, N.; Roos, W.P.; Krämer, O.H. The inducible E3 ubiquitin ligases SIAH1 and SIAH2 perform critical roles in breast and prostate cancers. Cytokine Growth Factor Rev. 2015, 26, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Steinhusen, U.; Badock, V.; Bauer, A.; Behrens, J.; Wittman-Liebold, B.; Dorken, B.; Bommert, K. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J. Biol. Chem. 2000, 275, 16345–16353. [Google Scholar] [CrossRef] [PubMed]
- Van de Craen, M.; Berx, G.; Van den Brande, I.; Fiers, W.; Declercq, W.; Vandenabeele, P. Proteolytic cleavage of beta-catenin by caspases: An in vitro analysis. FEBS Lett. 1999, 458, 167–170. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef]
- Nikolova, T.; Kiweler, N.; Krämer, O.H. Interstrand Crosslink Repair as a Target for HDAC Inhibition. Trends Pharmacol. Sci. 2017, 38, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Mak, R.; Tanguturi, S.K.; Mazitschek, R.; Haggarty, S.J.; Ross, K.; Chang, C.Y.; Bosco, J.; West, N.; Morse, E.; et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl. Acad. Sci. USA 2010, 107, 12617–12622. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Schofield, A.V.; Gamell, C.; Bernard, O. Tubulin polymerization promoting protein 1 (TPPP1) increases beta-catenin expression through inhibition of HDAC6 activity in U2OS osteosarcoma cells. Biochem. Biophys. Res. Commun. 2013, 436, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.B.; Nixon, A.M.; Kittanakom, S.; Stewart, J.M.; Chen, G.I.; Curak, J.; Gingras, A.C.; Mazitschek, R.; Neel, B.G.; Stagljar, I.; et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012, 2, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Sellmer, A.; Stangl, H.; Beyer, M.; Grünstein, E.; Leonhardt, M.; Pongratz, H.; Eichhorn, E.; Elz, S.; Striegl, B.; Jenei-Lanzl, Z.; et al. Marbostat-100 Defines a New Class of Potent and Selective Antiinflammatory and Antirheumatic Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2018, 61, 3454–3477. [Google Scholar] [CrossRef] [PubMed]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 2019, 145, 735–747. [Google Scholar] [CrossRef]
- Noack, K.; Mahendrarajah, N.; Hennig, D.; Schmidt, L.; Grebien, F.; Hildebrand, D.; Christmann, M.; Kaina, B.; Sellmer, A.; Mahboobi, S.; et al. Analysis of the interplay between all-trans retinoic acid and histone deacetylase inhibitors in leukemic cells. Arch. Toxicol. 2017, 91, 2191–2208. [Google Scholar] [CrossRef]
- Pons, M.; Nagel, G.; Zeyn, Y.; Beyer, M.; Laguna, T.; Brachetti, T.; Sellmer, A.; Mahboobi, S.; Conradi, R.; Butter, F.; et al. Human platelet lysate as validated replacement for animal serum to assess chemosensitivity. ALTEX Altern. Anim. Exp. 2018. [Google Scholar] [CrossRef]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef]
- Gardner, S.H.; Hawcroft, G.; Hull, M.A. Effect of nonsteroidal anti-inflammatory drugs on beta-catenin protein levels and catenin-related transcription in human colorectal cancer cells. Br. J. Cancer 2004, 91, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Steinert, G.; Oancea, C.; Roos, J.; Hagemeyer, H.; Maier, T.; Ruthardt, M.; Puccetti, E. Sulindac sulfide reverses aberrant self-renewal of progenitor cells induced by the AML-associated fusion proteins PML/RARalpha and PLZF/RARalpha. PLoS ONE 2011, 6, e22540. [Google Scholar] [CrossRef]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Raji, I.; Yadudu, F.; Janeira, E.; Fathi, S.; Szymczak, L.; Kornacki, J.R.; Komatsu, K.; Li, J.D.; Mrksich, M.; Oyelere, A.K. Bifunctional conjugates with potent inhibitory activity towards cyclooxygenase and histone deacetylase. Bioorg. Med. Chem. 2017, 25, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, G.; Wang, A.; Zhang, Z.; Merchan, J.R.; Halmos, B. Combined histone deacetylase and cyclooxygenase inhibition achieves enhanced antiangiogenic effects in lung cancer cells. Mol. Carcinog. 2013, 52, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2018. [Google Scholar] [CrossRef]
- Minke, K.S.; Staib, P.; Puetter, A.; Gehrke, I.; Gandhirajan, R.K.; Schlosser, A.; Schmitt, E.K.; Hallek, M.; Kreuzer, K.A. Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur. J. Haematol. 2009, 82, 165–175. [Google Scholar] [CrossRef]
- Romanski, A.; Schwarz, K.; Keller, M.; Wietbrauk, S.; Vogel, A.; Roos, J.; Oancea, C.; Brill, B.; Krämer, O.H.; Serve, H.; et al. Deacetylase inhibitors modulate proliferation and self-renewal properties of leukemic stem and progenitor cells. Cell Cycle 2012, 11, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Labisso, W.L.; Wirth, M.; Stojanovic, N.; Stauber, R.H.; Schnieke, A.; Schmid, R.M.; Krämer, O.H.; Saur, D.; Schneider, G. MYC directs transcription of MCL1 and eIF4E genes to control sensitivity of gastric cancer cells toward HDAC inhibitors. Cell Cycle 2012, 11, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Heinzel, T.; Englert, C. TSA downregulates Wilms tumor gene 1 (Wt1) expression at multiple levels. Nucleic Acids Res. 2008, 36, 4067–4078. [Google Scholar] [CrossRef]
- Green, L.M.; Wagner, K.J.; Campbell, H.A.; Addison, K.; Roberts, S.G. Dynamic interaction between WT1 and BASP1 in transcriptional regulation during differentiation. Nucleic Acids Res. 2009, 37, 431–440. [Google Scholar] [CrossRef]
- Hewitt, S.M.; Hamada, S.; McDonnell, T.J.; Rauscher, F.J., 3rd; Saunders, G.F. Regulation of the proto-oncogenes bcl-2 and c-myc by the Wilms’ tumor suppressor gene WT1. Cancer Res. 1995, 55, 5386–5389. [Google Scholar]
- Lee, K.Y.; Jeon, Y.J.; Kim, H.G.; Ryu, J.; Lim, D.Y.; Jung, S.K.; Yu, D.H.; Chen, H.; Bode, A.M.; Dong, Z. The CUG-translated WT1, not AUG-WT1, is an oncogene. Carcinogenesis 2017, 38, 1228–1240. [Google Scholar] [CrossRef]
- Chang, H.; Gao, F.; Guillou, F.; Taketo, M.M.; Huff, V.; Behringer, R.R. Wt1 negatively regulates beta-catenin signaling during testis development. Development 2008, 135, 1875–1885. [Google Scholar] [CrossRef]
- Zhang, T.F.; Yu, S.Q.; Guan, L.S.; Wang, Z.Y. Inhibition of breast cancer cell growth by the Wilms’ tumor suppressor WT1 is associated with a destabilization of beta-catenin. Anticancer Res. 2003, 23, 3575–3584. [Google Scholar] [PubMed]
- Zhou, J.; Yuan, W.; Zhuge, Y. Down-regulation of Wt1 activates Wnt/beta-catenin signaling through modulating endocytic route of LRP6 in podocyte dysfunction in vitro. Cell. Signal. 2015, 27, 1772–1780. [Google Scholar]
- Kim, M.S.; Yoon, S.K.; Bollig, F.; Kitagaki, J.; Hur, W.; Whye, N.J.; Wu, Y.P.; Rivera, M.N.; Park, J.Y.; Kim, H.S.; et al. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J. Biol. Chem. 2010, 285, 14585–14593. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Li, X.; Jia, Y.; Huai, L.; He, K.; Yu, P.; Wang, M.; Xing, H.; Rao, Q.; et al. Role of the Wilms’ tumor 1 gene in the aberrant biological behavior of leukemic cells and the related mechanisms. Oncol. Rep. 2014, 32, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Reichardt, C.M.; Hennig, D.; Nathan, A.; Kiweler, N.; Stocking, C.; Wichmann, C.; Christmann, M.; Butter, F.; Reichardt, S.; et al. Loss of Wilms tumor 1 protein is a marker for apoptosis in response to replicative stress in leukemic cells. Arch. Toxicol. 2018, 92, 2119–2135. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Shibata, T.; Sakamoto, M.; Hirohashi, S. Target disruption of the mutant beta-catenin gene in colon cancer cell line HCT116: Preservation of its malignant phenotype. Oncogene 2002, 21, 5906–5911. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, X.; Halicka, H.D.; Traganos, F.; Tanaka, T.; Kurose, A.; Darzynkiewicz, Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005, 38, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Summers, A.R.; Fischer, M.A.; Stengel, K.R.; Zhao, Y.; Kaiser, J.F.; Wells, C.E.; Hunt, A.; Bhaskara, S.; Luzwick, J.W.; Sampathi, S.; et al. HDAC3 is essential for DNA replication in hematopoietic progenitor cells. J. Clin. Investig. 2013, 123, 3112–3123. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Kim, W.; Bariteau, M.; Davis, T.; Vempati, S.; Minehart, J.; Witkin, M.; Qi, J.; Krivtsov, A.V.; Bradner, J.E.; et al. Selective Inhibition of HDAC1 and HDAC2 as a Potential Therapeutic Option for B-ALL. Clin. Cancer Res. 2015, 21, 2348–2358. [Google Scholar] [CrossRef]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K.; et al. The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef]
- Rainsford, K.D. Anti-inflammatory drugs in the 21st century. Sub-Cell. Biochem. 2007, 42, 3–27. [Google Scholar] [PubMed]
- Wells, C.E.; Bhaskara, S.; Stengel, K.R.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS ONE 2013, 8, e68915. [Google Scholar] [CrossRef] [PubMed]
- Conti, C.; Leo, E.; Eichler, G.S.; Sordet, O.; Martin, M.M.; Fan, A.; Aladjem, M.I.; Pommier, Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010, 70, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, J.E.; Grenon, M.; Lowndes, N.F. 53BP1: Function and mechanisms of focal recruitment. Biochem. Soc. Trans. 2009, 37, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Masamoto, Y.; Kurokawa, M. Targeting chronic myeloid leukemia stem cells: Can transcriptional program be a druggable target for cancers? Stem Cell Investig. 2018, 5, 10. [Google Scholar] [CrossRef][Green Version]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef]
- Göder, A.; Emmerich, C.; Nikolova, T.; Kiweler, N.; Schreiber, M.; Kühl, T.; Imhof, D.; Christmann, M.; Heinzel, T.; Schneider, G.; et al. HDAC1 and HDAC2 integrate checkpoint kinase phosphorylation and cell fate through the phosphatase-2A subunit PR130. Nat. Commun. 2018, 9, 764. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Guitian, R.; Moreno, J.; de Toro, F.J.; Galdo, F. Effect of antiinflammatory drugs on COX-1 and COX-2 activity in human articular chondrocytes. J. Rheumatol. 1999, 26, 1366–1373. [Google Scholar] [PubMed]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H.; et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/beta-catenin signaling. Mol Cancer Ther. 2013, 12, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Pfeilschifter, J.; Geisslinger, G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 Inhibition Specifically Targets Inv (16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; San-Marina, S.; Liu, J.; Minden, M.D. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene 2004, 23, 6933–6941. [Google Scholar] [CrossRef] [PubMed]
- Englert, C.; Maheswaran, S.; Garvin, A.J.; Kreidberg, J.; Haber, D.A. Induction of p21 by the Wilms’ tumor suppressor gene WT1. Cancer Res. 1997, 57, 1429–1434. [Google Scholar] [PubMed]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.M.; Schlamp, C.L.; Nickells, R.W. Targeting HDAC3 Activity with RGFP966 Protects Against Retinal Ganglion Cell Nuclear Atrophy and Apoptosis After Optic Nerve Injury. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2018, 34, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schafer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Gao, S.; Yang, J.; Li, H.; Huang, H.; Zheng, X. WT1 protein is cleaved by caspase-3 in apoptotic leukemic cells. Leuk. Lymphoma 2018, 59, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Bug, G. Establishment and Characterization of Long-Term Cultures Derived from Primary Acute Myeloid Leukemia Cells for HDAC Inhibitor Research. Methods Mol. Biol. 2017, 1510, 127–148. [Google Scholar] [PubMed]
- Beyer, M.; Kiweler, N.; Mahboobi, S.; Krämer, O.H. How to Distinguish Between the Activity of HDAC1-3 and HDAC6 with Western Blot. Methods Mol. Biol. 2017, 1510, 355–364. [Google Scholar] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyer, M.; Romanski, A.; Mustafa, A.-H.M.; Pons, M.; Büchler, I.; Vogel, A.; Pautz, A.; Sellmer, A.; Schneider, G.; Bug, G.; et al. HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1. Cancers 2019, 11, 1436. https://doi.org/10.3390/cancers11101436
Beyer M, Romanski A, Mustafa A-HM, Pons M, Büchler I, Vogel A, Pautz A, Sellmer A, Schneider G, Bug G, et al. HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1. Cancers. 2019; 11(10):1436. https://doi.org/10.3390/cancers11101436
Chicago/Turabian StyleBeyer, Mandy, Annette Romanski, Al-Hassan M. Mustafa, Miriam Pons, Iris Büchler, Anja Vogel, Andrea Pautz, Andreas Sellmer, Günter Schneider, Gesine Bug, and et al. 2019. "HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1" Cancers 11, no. 10: 1436. https://doi.org/10.3390/cancers11101436
APA StyleBeyer, M., Romanski, A., Mustafa, A.-H. M., Pons, M., Büchler, I., Vogel, A., Pautz, A., Sellmer, A., Schneider, G., Bug, G., & Krämer, O. H. (2019). HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of β-catenin, MYC, and WT1. Cancers, 11(10), 1436. https://doi.org/10.3390/cancers11101436