Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer

 ,
, 
 , , , ,
, , , ,  and
and 
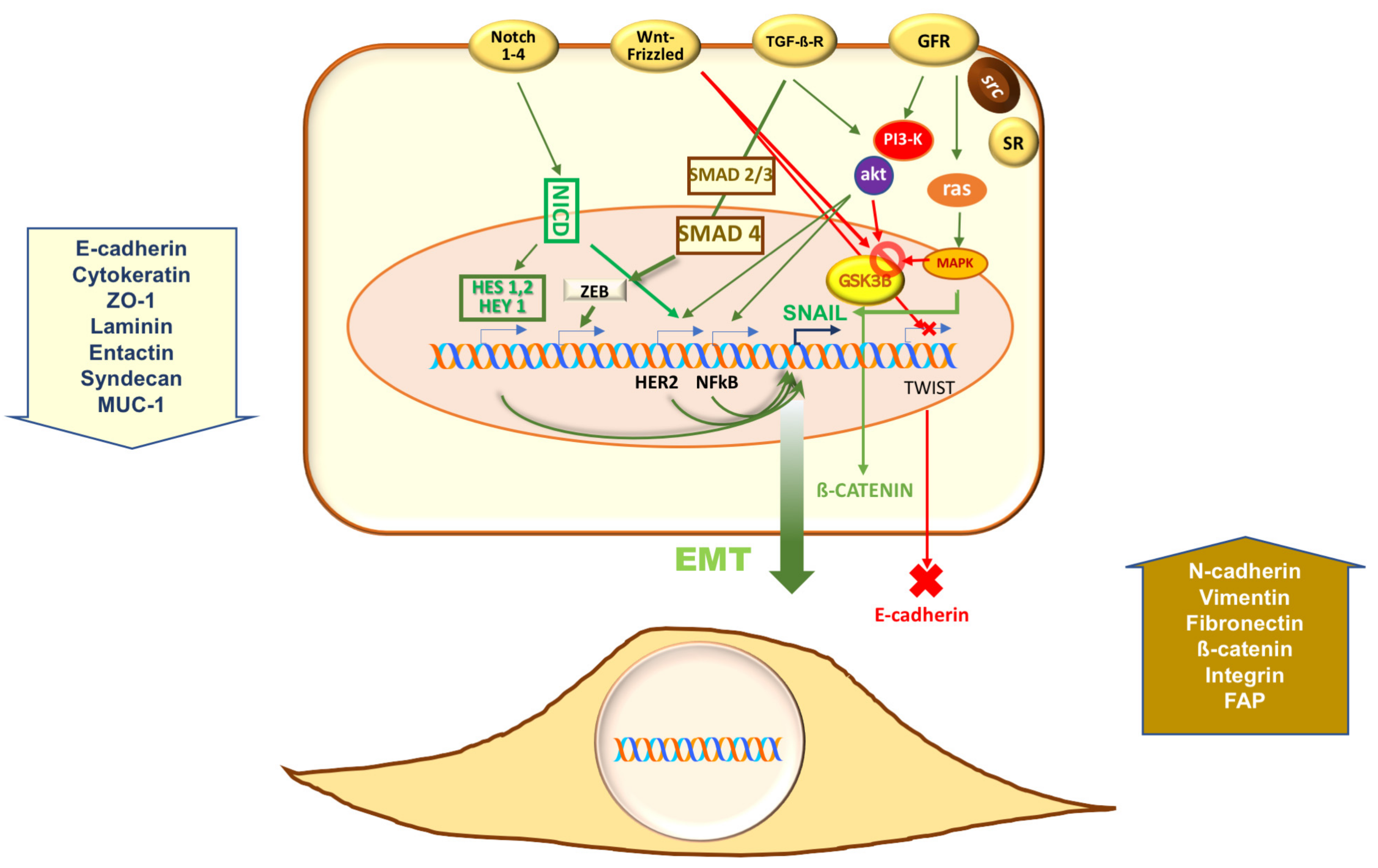{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epithelial-Mesenchymal Transition (EMT) in Prostate Cancer (PC)
3. Estrogen Receptors (ERs) in Prostate Cancer (PC)
4. Estrogen Receptor (ER) α in Epithelial Mesenchymal Transition (EMT) of Benign Prostatic Hyperplasia (BPH)
5. Estrogen Receptor (ER) α in Epithelial Mesenchymal Transition (EMT) of Prostate Cancer (PC)
6. Estrogen Receptor (ER) β in Epithelial Mesenchymal Transition (EMT) of Prostate Cancer (PC)
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ADT | androgen deprivation therapy |
| AF-2 | activation function 2 |
| AR | androgen receptor |
| BC | breast cancer |
| BPH | benign prostatic hyperplasia |
| CAFs | cancer-associated fibroblasts |
| CRPC | castration-resistant prostate cancer |
| CTCs | circulating tumor cells |
| 17β-diol | 5α-androstane-3β |
| DPN | 2,3-bis(4-Hydroxyphenyl)-propionitrile |
| EGF | epidermal growth factor |
| EGF-R | epidermal growth factor recpetor |
| EMT | epithelial-mesenchymal transition |
| ER | estrogen receptor |
| ERR | estrogen related receptor |
| FGF | fibroblast growth factor |
| GFR | Growth factor receptors |
| GSK3β | Glycogen synthase kinase- 3β |
| HES | hairy and enhancer of split |
| HEY | HES 1 related |
| HGF | hepatocyte-derived growth factor |
| HIF | hypoxia-induced factor |
| IGF-1 | insulin-like growth factor 1 |
| IGF-1R | insulin-like growth factor 1 receptor |
| IHC | immunohistochemistry |
| LBD | ligand binding domain |
| Lnc RNA | long non-coding RNA |
| MAPK | mitogen-activated protein kinase |
| MET | mesenchymal-epithelial transition |
| NEAT1 | nuclear enriched abundant transcript 1 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NICD | notch intracellular domain |
| eNOS | endothelial nitric oxide synthase |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| PDGF | platelet-derived growth factor |
| PD-1 | programmed cell death 1 |
| PC | prostate cancer |
| PHD2 | propyl hydroxylase domain 2 |
| PI-3K | Phosphoinositide 3- kinase |
| PPT | 4,4′,4″-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol |
| RUNX2 | regulator Runt-related transcription factor 2 |
| SERM | selective estrogen receptor modulator |
| sFRP | secreted frizzled-related proteins |
| SMAD | small mother against decapentaplegic |
| SNAI | Snail Family Transcriptional Repressor 2 |
| SOX4 | sex determining region Y-box |
| SRs | steroid receptors |
| TERT | telomerase Reverse Transcriptase |
| TGF-β | transforming growth factor β |
| TGF-βR | transforming growth factor β receptor |
| TWIST1 | Twist-related protein 1 |
| VEGF-A | vascular endothelial growth factor A |
| Wnt | Wingless-INT |
| ZEB | Zinc finger E-box-binding homeobox |
References
- Bishop, J.L.; Davies, A.; Ketola, K.; Zoubeidi, A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr Relat Cancer 2015, 22, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA 2018, 68, 7–30. [Google Scholar] [PubMed]
- Cuccurullo, V.; Di Stasio, G.D.; Evangelista, L.; Castoria, G.; Mansi, L. Biochemical and Pathophysiological Premises to Positron Emission Tomography With Choline Radiotracers. J Cell. Physiol. 2017, 232, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Calabria, F.; Chiaravalloti, A.; Cicciò, C.; Gangemi, V.; Gullà, D.; Rocca, F.; Gallo, G.; Cascini, G.L.; Schillaci, O. PET/CT with 18F-Choline: Physiological whole bio-Distribution in male and female subjects and diagnostic pitfalls on 1000 prostate cancer patients: 18F-choline PET/CT bio-Distribution and pitfalls. A southern Italian experience. Nucl. Med. Biol. 2017, 51, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Fanti, S.; Minozzi, S.; Antoch, G.; Banks, I.; Briganti, A.; Carrio, I.; Chiti, A.; Clarke, N.; Eiber, M.; De Bono, J.; et al. Consensus on molecular imaging and theranostics in prostate cancer. Lancet Oncol. 2018, 19, 696–708. [Google Scholar] [CrossRef]
- Attard, G.; Antonarakis, E.S. Prostate cancer: AR aberrations and resistance to abiraterone or enzalutamide. Nat. Rev. Urol. 2016, 13, 697–698. [Google Scholar] [CrossRef]
- Baciarello, G.; Gizzi, M.; Fizazi, K. Advancing therapies in metastatic castration-Resistant prostate cancer. Expert Opin. Pharmacother. 2018, 19, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-Resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-Mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-Mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-Like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Umbas, R.; Isaacs, W.B.; Bringuier, P.P.; Schaafsma, H.E.; Karthaus, H.F.; Oosterhof, G.O.; Debruyne, F.M.; Schalken, J.A. Decreased E-Cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994, 54, 3929–3933. [Google Scholar] [PubMed]
- Richmond, P.J.; Karayiannakis, A.J.; Nagafuchi, A.; Kaisary, A.V.; Pignatelli, M. Aberrant E-Cadherin and alpha-Catenin expression in prostate cancer: Correlation with patient survival. Cancer Res. 1997, 57, 3189–3193. [Google Scholar] [PubMed]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A switch from E-Cadherin to N-Cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; Nagle, R.B.; Cress, A.E.; Heimark, R.L. N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-Mesenchymal transformation mediating adhesion withStromal cells. Am. J. Pathol. 1999, 155, 787–798. [Google Scholar] [CrossRef]
- Bussemakers, M.J.; Van Bokhoven, A.; Tomita, K.; Jansen, C.F.; Schalken, J.A. Complex cadherin expression in human prostate cancer cells. Int. J. Cancer 2000, 85, 446–450. [Google Scholar] [CrossRef]
- Jennbacken, K.; Gustavsson, H.; Welén, K.; Vallbo, C.; Damber, J.E. Prostate cancer progression into androgen independency is associated with alterations in cell adhesion and invasivity. Prostate 2006, 66, 1631–1640. [Google Scholar] [CrossRef]
- Cavallaro, U. N-Cadherin as an invasion promoter: A novel target for antitumor therapy? Curr. Opin. Investig. Drugs 2004, 5, 1274–1278. [Google Scholar] [PubMed]
- Mariotti, A.; Perotti, A.; Sessa, C.; Rüegg, C. N-Cadherin as a therapeutic target in cancer. Expert Opin. Investig. Drugs 2007, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of N-Cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen deprivation causes epithelial-Mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Aytes, A.; Mitrofanova, A.; Kinkade, C.W.; Lefebvre, C.; Lei, M.; Phelan, V.; LeKaye, H.C.; Koutcher, J.A.; Cardiff, R.D.; Califano, A.; et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 3506–3515. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; De Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-Induced androgen receptor-Oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-Kinase in concert with Src promotes the S-Phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Castoria, G.; Auricchio, F. Src-Dependent signalling pathway regulation by sex-steroid hormones: Therapeutic implications. Int. J. Biochem. Cell Biol. 2007, 39, 1343–1348. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-Associated fibroblasts stimulates epithelial-Mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA methylation variations are required for epithelial-to-Mesenchymal transition induced by cancer-Associated fibroblasts in prostate cancer cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef] [PubMed]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-Mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-Mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, J.T.; Henry, M.D. Epithelial-to-Mesenchymal transition in prostate cancer: Paradigm or puzzle? Nat. Rev. Urol. 2011, 8, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.N.; McDermott, N.; Meunier, A.; Marignol, L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2014, 11, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.A. Human estrogen receptor beta-Gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef]
- Bonkhoff, H. Estrogen receptor signaling in prostate cancer: Implications for carcinogenesis and tumor progression. Prostate 2018, 78, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.W.; Tilley, W.D.; Neal, D.E.; Carroll, J.S. Estrogen receptor beta in prostate cancer: Friend or foe? Endocr.-Relat. Cancer 2014, 21, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; De Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: Steroid antagonist action. Cancer Res. 2005, 65, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Pisolato, R.; Lombardi, A.P.; Vicente, C.M.; Lucas, T.F.; Lazari, M.F.; Porto, C.S. Expression and regulation of the estrogen receptors in PC-3 human prostate cancer cells. Steroids 2016, 107, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Di Zazzo, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize With the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Lam, H.M.; Wu, S.; Song, D.; Levin, L.; Cheng, L.; Wu, C.L.; Ho, S.M. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr.-Relat. Cancer 2010, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.W.; Groen, A.J.; Miller, J.L.; Warren, A.Y.; Holmes, K.A.; Tarulli, G.A.; Tilley, W.D.; Katzenellenbogen, B.S.; Hawse, J.R.; Gnanapragasam, V.J.; et al. Comprehensive assessment of estrogen receptor beta antibodies in cancer cell line models and tissue reveals critical limitations in reagent specificity. Mol. Cell. Endocrinol. 2017, 440, 138–150. [Google Scholar] [CrossRef]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramström, M.; Söderberg, O.; et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef]
- Gustafsson, J.A.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. 2019, 191, 105312. [Google Scholar] [CrossRef]
- Wu, D.; Cheung, A.; Wang, Y.; Yu, S.; Chan, F.L. The emerging roles of orphan nuclear receptors in prostate cancer. Biochim. Biophys. Acta 2016, 1866, 23–36. [Google Scholar] [CrossRef]
- Rossi, V.; Staibano, S.; Abbondanza, C.; Pasquali, D.; De Rosa, C.; Mascolo, M.; Bellastella, G.; Visconti, D.; De Bellis, A.; Moncharmont, B.; et al. Expression of RIZ1 protein (Retinoblastoma-Interacting zinc-Finger protein 1) in prostate cancerepithelial cells changes with cancer grade progression and is modulated in vitro by DHT and E2. J. Cell. Physiol. 2009, 221, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Madak-Erdogan, Z.; Charn, T.H.; Jiang, Y.; Liu, E.T.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Integrative genomics of gene and metabolic regulation by estrogenreceptors α and β, and their coregulators. Mol. Syst. Biol. 2013, 9, 676. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.S.; Lombardi, A.P.G.; Vicente, C.M.; Lucas, T.F.G.; Erustes, A.G.; Pereira, G.J.S.; Porto, C.S. Estrogen receptors localization and signaling pathways in DU-145 human prostate cancer cells. Mol. Cell. Endocrinol. 2019, 483, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Grubisha, M.J.; DeFranco, D.B. Local endocrine, paracrine and redox signaling networks impact estrogen and androgen crosstalk in the prostate cancer microenvironment. Steroids 2013, 78, 538–541. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karamouzis, M.V.; Papavassiliou, K.A.; Adamopoulos, C.; Papavassiliou, A.G. Targeting Androgen/Estrogen Receptors Crosstalk in Cancer. Trends Cancer 2016, 2, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.; Huang, B.; Gustafsson, J.A. Estrogen Receptor β as a Pharmaceutical Target. Trends Pharmacol. Sci. 2017, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-Mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef]
- Kozák, I.; Bartsch, W.; Krieg, M.; Voigt, K.D. Nuclei of stroma: Site of highest estrogen concentration in human benign prostatic hyperplasia. Prostate 1982, 3, 433–438. [Google Scholar] [CrossRef]
- Schulze, H.; Claus, S. Histological localization of estrogen receptors in normal and diseased human prostates by immunocytochemistry. Prostate 1990, 16, 331–343. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Brössner, C.; Reiner, A.; Cheng, G.; Sugiyama, N.; Warner, M.; Gustafsson, J.A. A role for epithelial-Mesenchymal transition in the etiology of benign prostatic hyperplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 2859–2863. [Google Scholar] [CrossRef]
- Shao, R.; Shi, J.; Liu, H.; Shi, X.; Du, X.; Klocker, H.; Lee, C.; Zhu, Y.; Zhang, J. Epithelial-to-Mesenchymal transition and estrogen receptor α mediated epithelial dedifferentiation mark the development of benign prostatic hyperplasia. Prostate 2014, 74, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Peng, Y.; Du, X.; Liu, H.; Klocker, H.; Lin, Q.; Shi, J.; Zhang, J. Estradiol promotes epithelial-to-Mesenchymal transition in human benign prostatic epithelial cells. Prostate 2017, 77, 1424–1437. [Google Scholar] [CrossRef]
- Dhasarathy, A.; Kajita, M.; Wade, P.A. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef]
- Huang, J.W.; Guan, B.Z.; Yin, L.H.; Liu, F.N.; Hu, B.; Zheng, Q.Y.; Li, F.L.; Zhong, Y.X.; Chen, Y. Effects of estrogen-Related receptor alpha (ERRα) on proliferation and metastasis of human lung cancer A549 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 875–881. [Google Scholar] [CrossRef]
- Yoriki, K.; Mori, T.; Kokabu, T.; Matsushima, H.; Umemura, S.; Tarumi, Y.; Kitawaki, J. Estrogen-Related receptor alpha induces epithelial-mesenchymal transition through cancer-Stromal interactions in endometrial cancer. Sci. Rep. 2019, 9, 6697. [Google Scholar] [CrossRef]
- Mishra, S.; Tai, Q.; Gu, X.; Schmitz, J.; Poullard, A.; Fajardo, R.J.; Mahalingam, D.; Chen, X.; Zhu, X.; Sun, L.Z. Estrogen and estrogen receptor alpha promotes malignancy and osteoblastic tumorigenesis in prostate cancer. Oncotarget 2015, 6, 44388–44402. [Google Scholar] [CrossRef]
- Chakravarty, D.; Sboner, A.; Nair, S.S.; Giannopoulou, E.; Li, R.; Hennig, S.; Mosquera, J.M.1; Pauwels, J.; Park, K.; Kossai, M.; et al. The oestrogen receptor alpha-Regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 2014, 5, 5383. [Google Scholar] [CrossRef]
- Shen, Y.; Cao, J.; Liang, Z.; Lin, Q.; Wang, J.; Yang, X.; Zhang, R.; Zong, J.; Du, X.; Peng, Y.; et al. Estrogen receptor α-NOTCH1 axis enhances basal stem-like cells and epithelial-Mesenchymal transition phenotypes in prostate cancer. Cell Commun Signal. 2019, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.R.; Da, J.; Song, W.; Fazili, A.; Yeh, S. Estrogen receptors in prostate development and cancer. Am. J. Clin. Exp. Urol. 2014, 2, 161–168. [Google Scholar] [PubMed]
- Christoforou, P.; Christopoulos, P.F.; Koutsilieris, M. The role of estrogen receptor β in prostate cancer. Mol. Med. 2014, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Jędroszka, D.; Orzechowska, M.; Hamouz, R.; Górniak, K.; Bednarek, A.K. Markers of epithelial-to-Mesenchymal transition reflect tumor biology according to patient age and Gleason score in prostate cancer. PLoS ONE 2017, 12, e0188842. [Google Scholar] [CrossRef] [PubMed]
- Bergan, R.C.; Reed, E.; Myers, C.E.; Headlee, D.; Brawley, O.; Cho, H.K.; Figg, W.D.; Tompkins, A.; Linehan, W.M.; Kohler, D.; et al. A Phase II study of high-dose tamoxifen in patients with hormone-Refractory prostate cancer. Clin. Cancer Res. 1999, 5, 2366–2373. [Google Scholar] [PubMed]
- Stein, S.; Zoltick, B.; Peacock, T.; Holroyde, C.; Haller, D.; Armstead, B.; Malkowicz, S.B.; Vaughn, D.J. Phase II trial of toremifene in androgen-Independent prostate cancer: A Penn cancer clinical trials group trial. Am. J. Clin. Oncol. 2001, 24, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Price, D.; Stein, B.; Sieber, P.; Tutrone, R.; Bailen, J.; Goluboff, E.; Burzon, D.; Bostwick, D.; Steiner, M. Toremifene for the prevention of prostate cancer in men with high grade prostatic intraepithelial neoplasia: Results of a double-Blind, placebo controlled, phase IIB clinical trial. J. Urol. 2006, 176, 965–970. [Google Scholar] [CrossRef]
- Steiner, M.S.; Raghow, S. Antiestrogens and selective estrogen receptor modulators reduce prostate cancer risk. World J. Urol. 2003, 21, 31–36. [Google Scholar]
- Ho, S.M. Estrogens and anti-Estrogens: Key mediators of prostatecarcinogenesis and new therapeutic candidates. J. Cell. Biochem. 2004, 91, 491–503. [Google Scholar] [CrossRef]
- Saltzstein, D.; Sieber, P.; Morris, T.; Gallo, J. Prevention and management of bicalutamide-Induced gynecomastia and breast pain: Randomized endocrinologic and clinical studies with tamoxifen and anastrozole. Prostate Cancer Prostatic Dis. 2005, 8, 75–83. [Google Scholar] [CrossRef]
- Kunath, F.; Keck, B.; Antes, G.; Wullich, B.; Meerpohl, J.J. Tamoxifen for the management of breast events induced by non-Steroidal antiandrogens in patients with prostate cancer: A systematic review. BMC Med. 2012, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Morton, R.A.; Barnette, K.G.; Sieber, P.R.; Malkowicz, S.B.; Rodriguez, D.; Hancock, M.L.; Steiner, M.S. Toremifene to reduce fracture risk in men receiving androgen deprivation therapy for prostate cancer. J. Urol. 2013, 189, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Takahashi, S.; Kume, H.; Urano, T.; Takayama, K.; Yamada, Y.; Suzuki, M.; Fukuhara, H.; Nakagawa, T.; Inoue, S.; et al. Toremifene, a selective estrogen receptor modulator, significantly improved biochemical recurrence in bone metastatic prostate cancer: A randomized controlled phase II a trial. BMC Cancer 2015, 15, 836. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Takayama, K.; Takahashi, S.; Inoue, S. Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine. Cancers 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Lau, K.M.; Adams, J.Y.; McNeal, J.E.; Taplin, M.E.; Wang, J.; Singh, H.; Ho, S.M. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am. J. Pathol. 2001, 159, 79–92. [Google Scholar] [CrossRef]
- Hurtado, A.; Pinós, T.; Barbosa-Desongles, A.; López-Avilés, S.; Barquinero, J.; Petriz, J.; Santamaria-Martínez, A.; Morote, J.; De Torres, I.; Bellmunt, J.; et al. Estrogen receptor beta displays cell cycle-Dependent expression and regulates the G1 phase through a non-Genomic mechanism in prostate carcinoma cells. Anal. Cell. Pathol. 2008, 30, 349–365. [Google Scholar]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-Beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef] [PubMed]
- Latil, A.; Bièche, I.; Vidaud, D.; Lidereau, R.; Berthon, P.; Cussenot, O.; Vidaud, M. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by real-Time quantitative reverse transcription-Polymerase chain reaction assays. Cancer Res. 2001, 61, 1919–1926. [Google Scholar] [PubMed]
- Zhu, X.; Leav, I.; Leung, Y.K.; Wu, M.; Liu, Q.; Gao, Y.; McNeal, J.E.; Ho, S.M. Dynamic regulation of estrogen receptor-Beta expression by DNA methylation during prostate cancer development and metastasis. Am. J. Pathol. 2004, 164, 2003–2012. [Google Scholar] [CrossRef]
- Slusarz, A.; Jackson, G.A.; Day, J.K.; Shenouda, N.S.; Bogener, J.L.; Browning, J.D.; Fritsche, K.L.; MacDonald, R.S.; Besch-Williford, C.L.; Lubahn, D.B. Aggressive prostate cancer is prevented in ERαKO mice and stimulated in ERβKO TRAMP mice. Endocrinology 2012, 153, 4160–4170. [Google Scholar] [CrossRef]
- Fixemer, T.; Remberger, K.; Bonkhoff, H. Differential expression of the estrogen receptor beta (ERbeta) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. Prostate 2003, 54, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Guerini, V.; Sau, D.; Scaccianoce, E.; Rusmini, P.; Ciana, P.; Maggi, A.; Martini, P.G.; Katzenellenbogen, B.S.; Martini, L.; Motta, M.; et al. The androgen derivative 5alpha-Androstane-3beta,17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res. 2005, 65, 5445–5453. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Dondi, D.; Piccolella, M.; Biserni, A.; Della Torre, S.; Ramachandran, B.; Locatelli, A.; Rusmini, P.; Sau, D.; Caruso, D.; Maggi, A.; et al. Estrogen receptor beta and the progression of prostate cancer: Role of 5alpha-androstane-3beta,17beta-Diol. Endocr.-Relat. Cancer. 2010, 17, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Piccolella, M.; Crippa, V.; Messi, E.; Tetel, M.J.; Poletti, A. Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacol. Res. 2014, 79, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Ganju, A.; Sikander, M.; Kashyap, V.K.; Hafeez, Z.B.; Chauhan, N.; Malik, S.; Massey, A.E.; Tripathi, M.K.; Halaweish, F.T.; et al. Ormeloxifene Suppresses Prostate Tumor Growth and Metastatic Phenotypes via Inhibition of Oncogenic β-Catenin Signaling and EMT Progression. Mol. Cancer Ther. 2017, 16, 2267–2280. [Google Scholar] [CrossRef]
- Brown, J.M. The hypoxic cell: A target for selective cancer therapy—Eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar]
- Mak, P.; Chang, C.; Pursell, B.; Mercurio, A.M. Estrogen receptor β sustains epithelial differentiation by regulating prolyl hydroxylase 2 transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 4708–4713. [Google Scholar] [CrossRef]
- Horvath, L.G.; Henshall, S.M.; Lee, C.S.; Head, D.R.; Quinn, D.I.; Makela, S.; Delprado, W.; Golovsky, D.; Brenner, P.C.; O’Neill, G.; et al. Frequent loss of estrogen receptor-Beta expression in prostate cancer. Cancer Res. 2001, 61, 5331–5335. [Google Scholar]
- Zellweger, T.; Stürm, S.; Rey, S.; Zlobec, I.; Gsponer, J.R.; Rentsch, C.A.; Terracciano, L.M.; Bachmann, A.; Bubendorf, L.; Ruiz, C. Estrogen receptor β expression and androgen receptor phosphorylation correlate with a poor clinical outcome in hormone-Naive prostate cancer and are elevated in castration-Resistant disease. Endocr.-Relat. Cancer 2013, 20, 403–413. [Google Scholar] [CrossRef]
- .Nanni, S.; Benvenuti, V.; Grasselli, A.; Priolo, C.; Aiello, A.; Mattiussi, S.; Colussi, C.; Lirangi, V.; Illi, B.; D’Eletto, M.; et al. Endothelial NOS, estrogen receptor beta, and HIFs cooperate in the activation of a prognostic transcriptional pattern in aggressive human prostate cancer. J. Clin. Investig. 2009, 119, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, J.; Wang, L.; Shen, C.; Su, B.; Qi, M.; Hu, J.; Gao, W.; Tan, W.; Han, B. Estrogen induces androgen-Repressed SOX4 expression to promote progression of prostate cancer cells. Prostate 2015, 75, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Yang, X.; Chang, Y.W.; Qi, M.; Zhou, Z.; Zhang, J.; Han, B. SOX4 is associated with poor prognosis in prostate cancer and promotes epithelial-Mesenchymal transition in vitro. Prostate Cancer Prostatic Dis. 2013, 16, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Lu, B.; Leygue, E.; Murphy, L.C. Putative functional characteristics of human estrogen receptor-beta isoforms. J. Mol. Endocrinol. 2003, 30, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Velazquez-Villegas, L.A.; Faria, M.; Turner, A.; Jonsson, P.; Webb, P.; Williams, C.; Gustafsson, J.Å.; Ström, A.M. Estrogen Receptor β2 Induces Hypoxia Signature of Gene Expression by Stabilizing HIF-1α in Prostate Cancer. PLoS ONE 2015, 10, e0128239. [Google Scholar] [CrossRef]
- Dey, P.; Jonsson, P.; Hartman, J.; Williams, C.; Ström, A.; Gustafsson, J.Å. Estrogen receptors β1 and β2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol. Endocrinol. 2012, 26, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Dago, D.N.; Scafoglio, C.; Rinaldi, A.; Memoli, D.; Giurato, G.; Nassa, G.; Ravo, M.; Rizzo, F.; Tarallo, R.; Weisz, A. Estrogen receptor beta impacts hormone-Induced alternative mRNA splicing in breast cancer cells. BMC Genom. 2015, 16, 367. [Google Scholar] [CrossRef]
- Genua, M.; Pandini, G.; Sisci, D.; Castoria, G.; Maggiolini, M.; Vigneri, R.; Belfiore, A. Role of cyclic AMP response element-Binding protein in insulin-Like growth factor-I receptor up-Regulation by sex steroids in prostate cancer cells. Cancer Res. 2009, 69, 7270–7277. [Google Scholar] [CrossRef]
- Mottet, N.; De Santis, M.; Briers, E.; Bourke, L.; Gillessen, S.; Grummet, J.P.; Lam, T.B.; van der Poel, H.G.; Rouvière, O.; van den Bergh, R.C.; et al. Updated Guidelines for Metastatic Hormone-Sensitive Prostate Cancer: Abiraterone Acetate Combined with Castration Is Another Standard. Eur. Urol. 2017, 73, 316–321. [Google Scholar] [CrossRef]
- Voon, D.C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers 2019, 11, 1418. https://doi.org/10.3390/cancers11101418
Di Zazzo E, Galasso G, Giovannelli P, Di Donato M, Bilancio A, Perillo B, Sinisi AA, Migliaccio A, Castoria G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers. 2019; 11(10):1418. https://doi.org/10.3390/cancers11101418
Chicago/Turabian StyleDi Zazzo, Erika, Giovanni Galasso, Pia Giovannelli, Marzia Di Donato, Antonio Bilancio, Bruno Perillo, Antonio A. Sinisi, Antimo Migliaccio, and Gabriella Castoria. 2019. "Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer" Cancers 11, no. 10: 1418. https://doi.org/10.3390/cancers11101418
APA StyleDi Zazzo, E., Galasso, G., Giovannelli, P., Di Donato, M., Bilancio, A., Perillo, B., Sinisi, A. A., Migliaccio, A., & Castoria, G. (2019). Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers, 11(10), 1418. https://doi.org/10.3390/cancers11101418