MET Inhibitors in Small Cell Lung Cancer: From the Bench to the Bedside

 , , ,
, , ,
Abstract
1. Introduction
2. Description of the MET Molecule and the Downstream Pathway
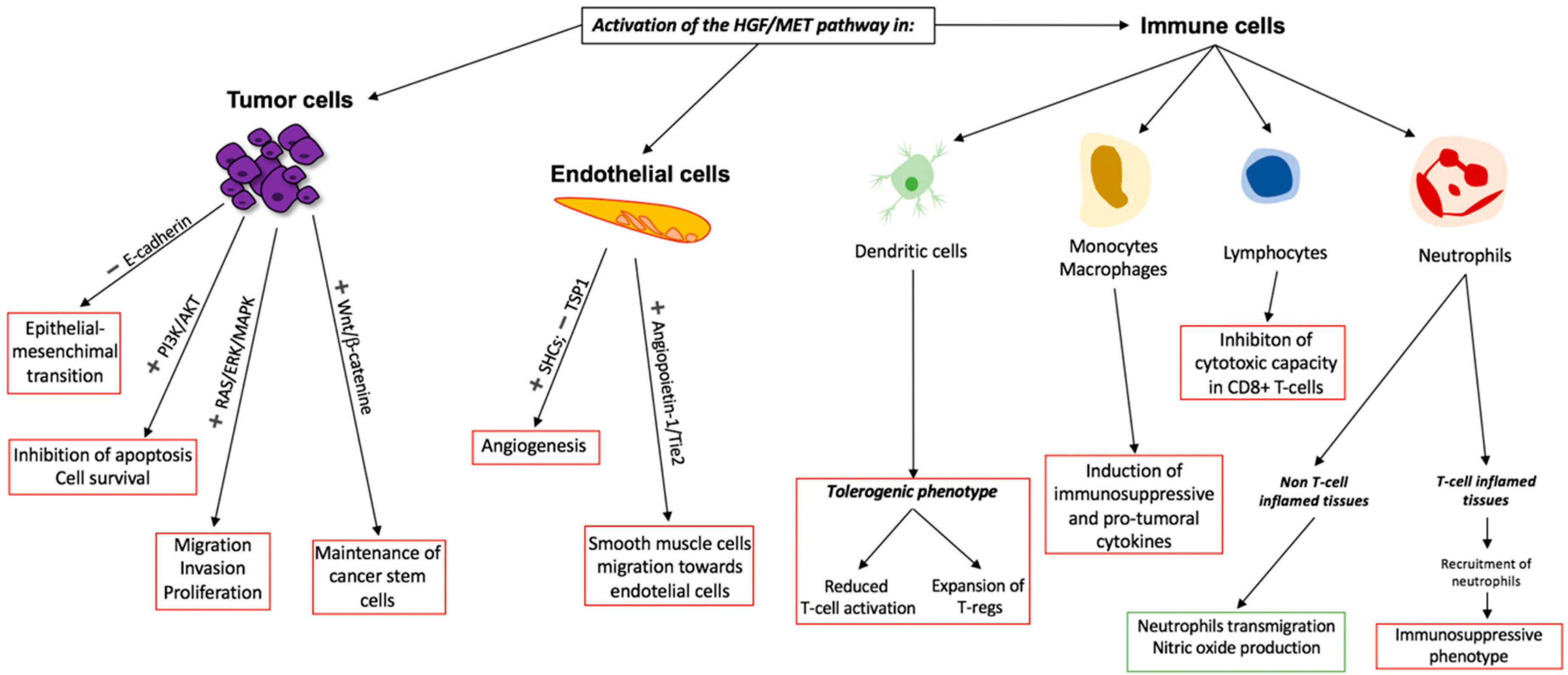
3. Role of the HGF/MET Axis in Cancer
4. MET in SCLC
4.1. Emergence of MET in the SCLC Biomarker Spectrum
4.2. Small MET Inhibitor Molecules
4.3. Combination of MET Inhibitors with Chemotherapy
4.4. Clinical Trials
4.5. Prevalence of MET Alterations in SCLC
5. Rationale for MET Inhibition in Combination with Immunotherapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. Cancer Statistics Review, 1975–2014; National Cancer Institute: Bethesda, MD, USA, 2017. [Google Scholar]
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: Analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006, 24, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.A. Effect of cigarette smoking on major histological types of lung cancer: A meta-analysis. Lung Cancer 2000, 31, 139–148. [Google Scholar] [CrossRef]
- Ou, S.H.I.; Ziogas, A.; Zell, J.A. Prognostic factors for survival in extensive stage small cell lung cancer (ED-SCLC): The importance of smoking history, socioeconomic and marital statuses, and ethnicity. J. Thorac. Oncol. 2009, 4, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, M.; Dolly, S.; Faria, A.; Myerson, J.S.; Popat, S.; O’Brien, M.E.R. Treatment options for small cell lung cancer—Do we have more choice? Br. J. Cancer 2010, 102, 629–638. [Google Scholar] [CrossRef]
- Nicholson, A.G.; Chansky, K.; Crowley, J.; Beyruti, R.; Kubota, K.; Turrisi, A.; Eberhardt, W.E.E.; Van Meerbeeck, J.; Rami-Porta, R. The international association for the study of lung cancer lung cancer staging project: Proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the tnm classification for lung cancer. J. Thorac. Oncol. 2016, 11, 300–311. [Google Scholar] [CrossRef]
- Oze, I.; Hotta, K.; Kiura, K.; Ochi, N.; Takigawa, N.; Fujiwara, Y.; Tabata, M.; Tanimoto, M. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: Disappointing results. PLoS ONE 2009, 4, e7835. [Google Scholar] [CrossRef]
- Sabari, J.K.; Lok, B.H.; Laird, J.H.; Poirier, J.T.; Rudin, C.M. Unravelling the biology of SCLC: Implications for therapy. Nat. Rev. Clin. Oncol. 2017, 14, 549. [Google Scholar] [CrossRef]
- Wang, S.; Tang, J.; Sun, T.; Zheng, X.; Li, J.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; Cheng, Z.; et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci. Rep. 2017, 7, 1339. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Hardy-Werbin, M.; Rocha, P.; Arpi, O.; Taus, Á.; Nonell, L.; Durán, X.; Villanueva, X.; Joseph-Pietras, D.; Nolan, L.; Danson, S.; et al. Serum cytokine levels as predictive biomarkers of benefit from ipilimumab in small cell lung cancer. Oncoimmunology 2019, 8, e1593810. [Google Scholar] [CrossRef] [PubMed]
- Hardy-Werbin, M.; Arpí, O.; Taus, A.; Rocha, P.; Joseph-Pietras, D.; Nolan, L.; Danson, S.; Griffiths, R.; Lopez-Botet, M.; Rovira, A.; et al. Assessment of neuronal autoantibodies in patients with small cell lung cancer treated with chemotherapy with or without ipilimumab. Oncoimmunology 2018, 7, e1395125. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretia, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Ready, N.E.; Pang, H.H.; Gu, L.; Otterson, G.A.; Thomas, S.P.; Miller, A.A.; Baggstrom, M.; Masters, G.A.; Graziano, S.L.; Crawford, J.; et al. Chemotherapy with or without Maintenance Sunitinib for Untreated Extensive-Stage Small-Cell Lung Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase II Study—CALGB 30504 (Alliance). J. Clin. Oncol. 2015, 33, 1660–1665. [Google Scholar] [CrossRef]
- Arnold, A.M.; Seymour, L.; Smylie, M.; Ding, K.; Ung, Y.; Findlay, B.; Lee, C.W.; Djurfeldt, M.; Whitehead, M.; Ellis, P.; et al. Phase II study of vandetanib or placebo in small-cell lung cancer patients after complete or partial response to induction chemotherapy with or without radiation therapy: National Cancer Institute of Canada Clinical Trials Group Study BR.20. J. Clin. Oncol. 2007, 25, 4278–4284. [Google Scholar] [CrossRef]
- Cañadas, I.; Rojo, F.; Taus, Á.; Arpí, O.; Arumí-Uría, M.; Pijuan, L.; Menéndez, S.; Zazo, S.; Dómine, M.; Salido, M.; et al. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin. Cancer Res. 2014, 20, 938–950. [Google Scholar] [CrossRef]
- Arriola, E.; Cañadas, I.; Arumí-Uría, M.; Dómine, M.; Lopez-Vilarĩo, J.A.; Arpí, O.; Salido, M.; Menéndez, S.; Grande, E.; Hirsch, F.R.; et al. MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines. Br. J. Cancer 2011, 105, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Park, M.; Le Beau, M.M.; Robins, T.S.; Diaz, M.O.; Rowley, J.D.; Blair, D.G.; Vande Woude, G.F. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 1985, 318, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; Di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904. [Google Scholar] [CrossRef]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Gherardi, E.; Chirgadze, D.Y.; Hepple, J.P.; Zhou, H.; Byrd, R.A.; Blundell, T.L. Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nat. Struct. Biol. 1999, 6, 72–79. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Ma, P.C.; Tretiakova, M.S.; Nallasura, V.; Jagadeeswaran, R.; Husain, A.N.; Salgia, R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: Implications for tumour invasion. Br. J. Cancer 2007, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Vande Woude, G.F. HGF/SF-Met signaling in the control of branching morphogenesis and invasion. J. Cell. Biochem. 2003, 88, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; Di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef]
- Chmielowiec, J.; Borowiak, M.; Morkel, M.; Stradal, T.; Munz, B.; Werner, S.; Wehland, J.; Birchmeier, C.; Birchmeier, W. C-Met is essential for wound healing in the skin. J. Cell Biol. 2007, 177, 151–162. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. Escape from Cbl-mediated downregulation: A recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 2003, 3, 519–523. [Google Scholar] [CrossRef]
- Hammond, D.E.; Urbé, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef]
- Palka, H.L.; Park, M.; Tonks, N.K. Hepatocyte growth factor receptor tyrosine kinase Met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J. Biol. Chem. 2003, 278, 5728–5735. [Google Scholar] [CrossRef]
- Rong, S.; Segal, S.; Anver, M.; Resau, J.H.; Vande Woude, G.F. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc. Natl. Acad. Sci. USA 2006, 91, 4731–4735. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Bodescot, M.; Blair, D.; Dunn, J.; Nakamura, T.; Mizuno, K.; Park, M.; Chan, A.; Aaronson, S.; Vande Woude, G.F. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol. Cell. Biol. 2015, 12, 5152–5158. [Google Scholar] [CrossRef] [PubMed]
- Abounader, R.; Lal, B.; Luddy, C.; Koe, G.; Davidson, B.; Rosen, E.M.; Laterra, J. In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J. 2002, 16, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.; Warn, A.; Newman, P.; Perry, L.J.; Ball, R.Y.; Warn, R.M. Immunoreactivity for hepatocyte growth factor/scatter factor and its receptor, met, in human lung carcinomas and malignant mesotheliomas. J. Pathol. 1996, 180, 389–394. [Google Scholar] [CrossRef]
- Oh, R.R.; Park, J.Y.; Lee, J.H.; Shin, M.S.; Kim, H.S.; Lee, S.K.; Kim, Y.S.; Lee, S.H.; Lee, S.N.; Yang, Y.M.; et al. Expression of HGF/SF and Met protein is associated with genetic alterations of VHL gene in primary renal cell carcinomas. APMIS 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.-M.; Schmitz, F.; Kiehne, K.; Stechele, H.U.; Banasiewicz, T.; Krokowicz, P.; Nakamura, T.; Fölsch, U.R.; Herzig, K.-H. Functional Expression of HGF and Its Receptor in Human Colorectal Cancer. Digestion 2000, 61, 237–246. [Google Scholar] [CrossRef]
- Edakuni, G.; Sasatomi, E.; Satoh, T.; Tokunaga, O.; Miyazaki, K. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol. Int. 2001, 51, 172–178. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Trink, B.; Tzai, T.-S.; Liu, H.-S.; Chan, S.-H.; Ho, C.-L.; Sidransky, D.; Chow, N.-H. Overexpression of c-met as a Prognostic Indicator for Transitional Cell Carcinoma of the Urinary Bladder: A Comparison with p53 Nuclear Accumulation. J. Clin. Oncol. 2002, 20, 1544–1550. [Google Scholar] [CrossRef]
- Joseph, A.; Weiss, G.H.; Jin, L.; Fuchs, A.; Chowdhury, S.; O’Shaugnessy, P.; Goldberg, I.D.; Rosen, E.M. Expression of Scatter Factor in Human Bladder Carcinoma. JNCI J. Natl. Cancer Inst. 1995, 87, 372–377. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar] [PubMed]
- de Aguirre, I.; Salvatierra, A.; Font, A.; Mate, J.L.; Perez, M.; Botia, M.; Taron, M.; Rosell, R. c-Met Mutational Analysis in the Sema and Juxtamembrane Domains in Small-Cell-Lung-Cancer. Transl. Oncogenom. 2006, 1, 11–18. [Google Scholar]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.; Tory, K.; Rigó, J.; Nagy, B.; Papp, Z. Evaluation of the tyrosine kinase domain of the Met proto-oncogene in sporadic ovarian carcinomas. Pathol. Oncol. Res. 1999, 5, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Schmidt, L.; Nakaigawa, N.; Webb, C.P.; Weirich, G.; Kishida, T.; Zbar, B.; Vande Woude, G.F. Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; De Stefani, A.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Han, S.-U.; Cho, H.; Jennings, B.; Gerrard, B.; Dean, M.; Schmidt, L.; Zbar, B.; Vande Woude, G.F. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene 2000, 19, 4947–4953. [Google Scholar] [CrossRef]
- Craene, B. De Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Grotegut, S.; Von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Liu, C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin. Exp. Metastasis 2011, 28, 721–731. [Google Scholar] [CrossRef]
- Yano, S.; Takeuchi, S.; Nakagawa, T.; Yamada, T. Ligand-triggered resistance to molecular targeted drugs in lung cancer: Roles of hepatocyte growth factor and epidermal growth factor receptor ligands. Cancer Sci. 2012, 103, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte Growth Factor Induces Gefitinib Resistance of Lung Adenocarcinoma with Epidermal Growth Factor Receptor-Activating Mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Finisguerra, V.; Prenen, H.; Mazzone, M. Preclinical and clinical evaluation of MET functions in cancer cells and in the tumor stroma. Oncogene 2016, 35, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Woude, G. Vande Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Su, Y.; Volpert, O.V.; Woude, G.F.V. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.; Ma, P.C.; Maulik, G.; Singh, R.; Khan, E.; Skarin, A.T.; Salgia, R. Haptoglobin α-Subunit and Hepatocyte Growth Factor can Potentially Serve as Serum Tumor Biomarkers in Small Cell Lung Cancer. Anticancer Res. 2004, 24, 1031–1038. [Google Scholar] [PubMed]
- Masuya, D.; Huang, C.; Liu, D.; Nakashima, T.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br. J. Cancer 2004, 90, 1555–1562. [Google Scholar] [CrossRef]
- Ghoussoub, R.A.D.; Dillon, D.A.; D’Aquila, T.; Rimm, E.B.; Fearon, E.R.; Rimm, D.L. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer 1998, 82, 1513–1520. [Google Scholar] [CrossRef]
- Cañadas, I.; Taus, A.; González, I.; Villanueva, X.; Gimeno, J.; Pijuan, L.; Dómine, M.; Sánchez-Font, A.; Vollmer, I.; Menéndez, S.; et al. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget 2014, 5, 5246–5256. [Google Scholar] [CrossRef]
- Rygaard, K.; Nakamura, T.; Spang-Thomsen, M. Expression of the proto-oncogenes c-met and c-kit and their ligands, hepatocyte growth factor/scatter factor and stem cell factor, in sclc cell lines and xenografts. Br. J. Cancer 1993, 67, 37–46. [Google Scholar] [CrossRef]
- Maulik, G.; Kijima, T.; Ma, P.C.; Ghosh, S.K.; Lin, J.; Shapiro, G.I.; Schaefer, E.; Tibaldi, E.; Johnson, B.E.; Salgia, R. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin. Cancer Res. 2002, 8, 620–627. [Google Scholar] [PubMed]
- Webb, C.P.; Hose, C.D.; Koochekpour, S.; Jeffers, M.; Oskarsson, M.; Sausville, E.; Monks, A.; Vande Woude, G.F. The geldanamycins are potent inhibitors of the hepatocyte growth factor/scatter factor-Met-urokinase plasminogen activator-plasmin proteolytic network. Cancer Res. 2000, 60, 342–349. [Google Scholar] [PubMed]
- Cho, S.H.; Kim, J.I.; Kim, H.S.; Park, S.D.; Jang, K.W. The antitumor effect of c-terminus of hsp70-interacting protein via degradation of c-met in small cell lung cancer. Korean J. Thorac. Cardiovasc. Surg. 2017, 50, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Maulik, G.; Madhiwala, P.; Brooks, S.; Ma, P.C.; Kijima, T.; Tibaldi, E.V.; Schaefer, E.; Parmar, K.; Salgia, R. Activated c-Met signals through PI3K with dramatic effects on cytoskeletal functions in small cell lung cancer. J. Cell. Mol. Med. 2002, 6, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 3657–3660. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Pride, Y.B.; Ma, P.; Gramlich, J.L.; Chu, S.C.; Quinnan, L.A.; Shirazian, S.; Liang, C.; Podar, K.; Christensen, J.G.; et al. A novel small molecule Met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003, 63, 5462–5469. [Google Scholar]
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A.; et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005, 65, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Schaefer, E.; Christensen, J.G.; Salgia, R. A selective small molecule c-MET inhibitor, PHA665752, cooperates with rapamycin. Clin. Cancer Res. 2005, 11, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Khramtsov, A.; Ahmed, S.; Nallasura, V.; Hetzel, J.T.; Jagadeeswaran, R.; Karczmar, G.; Salgia, R. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007, 67, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Rolle, C.E.; Kanteti, R.; Surati, M.; Nandi, S.; Dhanasingh, I.; Yala, S.; Tretiakova, M.; Arif, Q.; Hembrough, T.; Brand, T.M.; et al. Combined MET Inhibition and Topoisomerase I Inhibition Block Cell Growth of Small Cell Lung Cancer. Mol. Cancer Ther. 2013, 13, 576–584. [Google Scholar] [CrossRef]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar] [PubMed]
- Ettinger, D.S. New drugs for treating small cell lung cancer. Lung Cancer 1995, 12, S53–S61. [Google Scholar] [CrossRef]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, H.; Oguri, T.; Maeno, K.; Takakuwa, O.; Kunii, E.; Yagi, Y.; Uemura, T.; Kasai, D.; Miyazaki, M.; Niimi, A. Significance of c-MET overexpression in cytotoxic anticancer drug-resistant small-cell lung cancer cells. Cancer Sci. 2014, 105, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Inoue, H.; Ohba, S.; Kohda, Y.; Usami, I.; Masuda, T.; Kawada, M.; Nomoto, A. New metastatic model of human small-cell lung cancer by orthotopic transplantation in mice. Cancer Sci. 2015, 106, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Takeuchi, S.; Arai, S.; Fukuda, K.; Sakamoto, S.; Kawada, M.; Yamaguchi, H.; Mukae, H.; Yano, S. Impact of MET inhibition on small-cell lung cancer cells showing aberrant activation of the hepatocyte growth factor/MET pathway. Cancer Sci. 2017, 108, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Pennacchietti, S.; Vigna, E.; Chiriaco, C.; Arena, S.; Bardelli, A.; Valdembri, D.; Serini, G.; Michieli, P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin. Cancer Res. 2013, 19, 2381–2392. [Google Scholar] [CrossRef] [PubMed]
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Jänne, P.A. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 2015, 9, 260–269. [Google Scholar] [CrossRef]
- Liu, S.V.; Groshen, S.G.; Kelly, K.; Reckamp, K.L.; Belani, C.; Synold, T.W.; Goldkorn, A.; Gitlitz, B.J.; Cristea, M.C.; Gong, I.Y.; et al. A phase I trial of topotecan plus tivantinib in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 82, 723–732. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT02608411 (accessed on 11 May 2019).
- Glisson, B.; Besse, B.; Dols, M.C.; Dubey, S.; Schupp, M.; Jain, R.; Jiang, Y.; Menon, H.; Nackaerts, K.; Orlov, S.; et al. A Randomized, Placebo-Controlled, Phase 1b/2 Study of Rilotumumab or Ganitumab in Combination With Platinum-Based Chemotherapy as First-Line Treatment for Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 615–625.e8. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Horn, L.; Ghandi, J.; Kloecker, G.; Owonikoko, T.; Waqar, S.N.; Krzakowski, M.; Cardnell, R.J.; Fujimoto, J.; Taverna, P.; et al. A phase 2, open-label, multi-center study of amuvatinib in combination with platinum etoposide chemotherapy in platinum-refractory small cell lung cancer patients. Oncotarget 2017, 8, 81441–81454. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Bordi, P.; Tiseo, M.; Barbieri, F.; Bavieri, M.; Sartori, G.; Marchetti, A.; Buttitta, F.; Bortesi, B.; Ambrosini-Spaltro, A.; Gnetti, L.; et al. Gene mutations in small-cell lung cancer (SCLC): Results of a panel of 6 genes in a cohort of Italian patients. Lung Cancer 2014, 86, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Voortman, J.; Harada, T.; Chang, R.P.; Killian, J.K.; Suuriniemi, M.; Smith, W.I.; Meltzer, P.S.; Lucchi, M.; Wang, Y.; Giaccone, G. Detection and Therapeutic Implications of c-Met Mutations in Small Cell Lung Cancer and Neuroendocrine Tumors. Curr. Pharm. Des. 2013, 19, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Passioura, T.; Sato, H.; Ito, K.; Furuhashi, H.; Umitsu, M.; Takagi, J.; Kato, Y.; Mukai, H.; Warashina, S.; et al. Macrocyclic peptide-based inhibition and imaging of hepatocyte growth factor. Nat. Chem. Biol. 2019, 15, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Schag, K.; Schmidt, S.M.; Müller, M.R.; Weinschenk, T.; Appel, S.; Weck, M.M.; Grünebach, F.; Stevanovic, S.; Rammensee, H.G.; Brossart, P. Identification of C-Met oncogene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin. Cancer Res. 2004, 10, 3658–3666. [Google Scholar] [CrossRef]
- Liu, Y.; Wilkinson, F.L.; Kirton, J.P.; Jeziorska, M.; Iizasa, H.; Sai, Y.; Nakashima, E.; Heagerty, A.M.; Canfield, A.E.; Alexander, M.Y. Hepatocyte growth factor and c-Met expression in pericytes: Implications for atherosclerotic plaque development. J. Pathol. 2007, 212, 12–19. [Google Scholar] [CrossRef]
- Chen, Q.; De, F.M.; Zarnegar, R. Induction of met proto-oncogene (hepatocyte growth factor receptor) expression during human monocyte-macrophage differentiation. Cell Growth Differ 1996, 7, 821–832. [Google Scholar]
- Benkhoucha, M.; Santiago-Raber, M.-L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6424–6429. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Molnarfi, N.; Kaya, G.; Belnoue, E.; Bjarnadóttir, K.; Dietrich, P.; Walker, P.R.; Martinvalet, D.; Derouazi, M.; Lalive, P.H. Identification of a novel population of highly cytotoxic c-Met-expressing CD8 + T lymphocytes. EMBO Rep. 2017, 18, 1545–1558. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.R.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e9. [Google Scholar] [CrossRef]
- Schrock, A.B.; Li, S.D.; Frampton, G.M.; Suh, J.; Braun, E.; Mehra, R.; Buck, S.C.; Bufill, J.A.; Peled, N.; Karim, N.A.; et al. Pulmonary Sarcomatoid Carcinomas Commonly Harbor Either Potentially Targetable Genomic Alterations or High Tumor Mutational Burden as Observed by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2017, 12, 932–942. [Google Scholar] [CrossRef]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Greenall, S.A.; Adams, T.E.; Johns, T.G. Incomplete target neutralization by the anti-cancer antibody rilotumumab. MAbs 2016, 8, 246–252. [Google Scholar] [CrossRef]
- Adjei, A.A.; Schwartz, B.; Garmey, E. Early Clinical Development of ARQ 197, a Selective, Non-ATP-Competitive Inhibitor Targeting MET Tyrosine Kinase for the Treatment of Advanced Cancers. Oncologist 2011, 16, 788–799. [Google Scholar] [CrossRef]


{kind=link}
| Anti-MET Drug | Regimen | Reference | Test Setting |
|---|---|---|---|
| NSC122750 (geldanamycin) | Monotherapy | Maulik et al. (2002) [64] | In vitro |
| SU11274 | Monotherapy | Sattler et al. (2003) [69] Ma et al. (2007) [31] Ma et al. (2005) [70] | In vitro In vitro In vitro |
| SU11274 | Combination with irinotecan | Rolle et al. (2013) [74] Osaza et al. (2014) [78] | In vitro In vitro |
| PHA665752 | Monotherapy | Puri et al. (2007) [72] Arriola et al. (2011) [20] Sakamoto et al. (2015) [79] | In vivo In vitro In vitro |
| PF2341066 (criozotinib) | Combination with etoposide | Cañadas et al. (2014) [19] Taniguchi et al. (2017) [80] | In vitro/in vivo In vitro/in vivo |
| E7050 (golvatinib) | Monotherapy | Taniguchi et al. (2017) [80] | In vitro/in vivo |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardy-Werbin, M.; del Rey-Vergara, R.; Galindo-Campos, M.A.; Moliner, L.; Arriola, E. MET Inhibitors in Small Cell Lung Cancer: From the Bench to the Bedside. Cancers 2019, 11, 1404. https://doi.org/10.3390/cancers11101404
Hardy-Werbin M, del Rey-Vergara R, Galindo-Campos MA, Moliner L, Arriola E. MET Inhibitors in Small Cell Lung Cancer: From the Bench to the Bedside. Cancers. 2019; 11(10):1404. https://doi.org/10.3390/cancers11101404
Chicago/Turabian StyleHardy-Werbin, Max, Raúl del Rey-Vergara, Miguel Alejandro Galindo-Campos, Laura Moliner, and Edurne Arriola. 2019. "MET Inhibitors in Small Cell Lung Cancer: From the Bench to the Bedside" Cancers 11, no. 10: 1404. https://doi.org/10.3390/cancers11101404
APA StyleHardy-Werbin, M., del Rey-Vergara, R., Galindo-Campos, M. A., Moliner, L., & Arriola, E. (2019). MET Inhibitors in Small Cell Lung Cancer: From the Bench to the Bedside. Cancers, 11(10), 1404. https://doi.org/10.3390/cancers11101404