Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers




Abstract
1. Introduction
2. Results
2.1. KRTs Are Differentially Expressed in Colon and Breast Cancers
2.2. Knockdown of KRT19 Differentially Regulates Properties of Colon and Breast Cancers
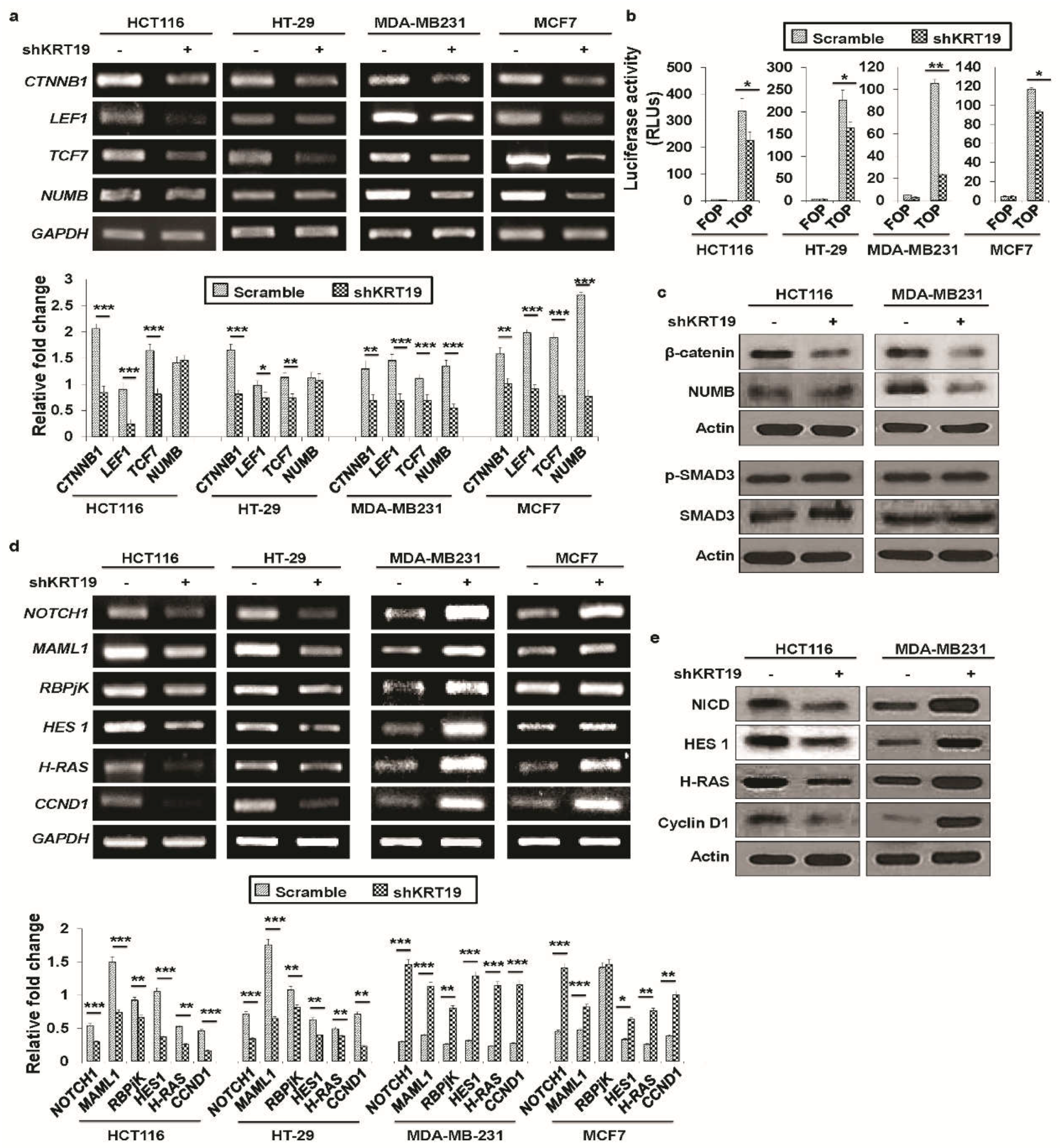
2.3. KRT19 Differentially Modulates Wnt/β-Catenin/Notch Signaling Pathways in Colon and Breast Cancers
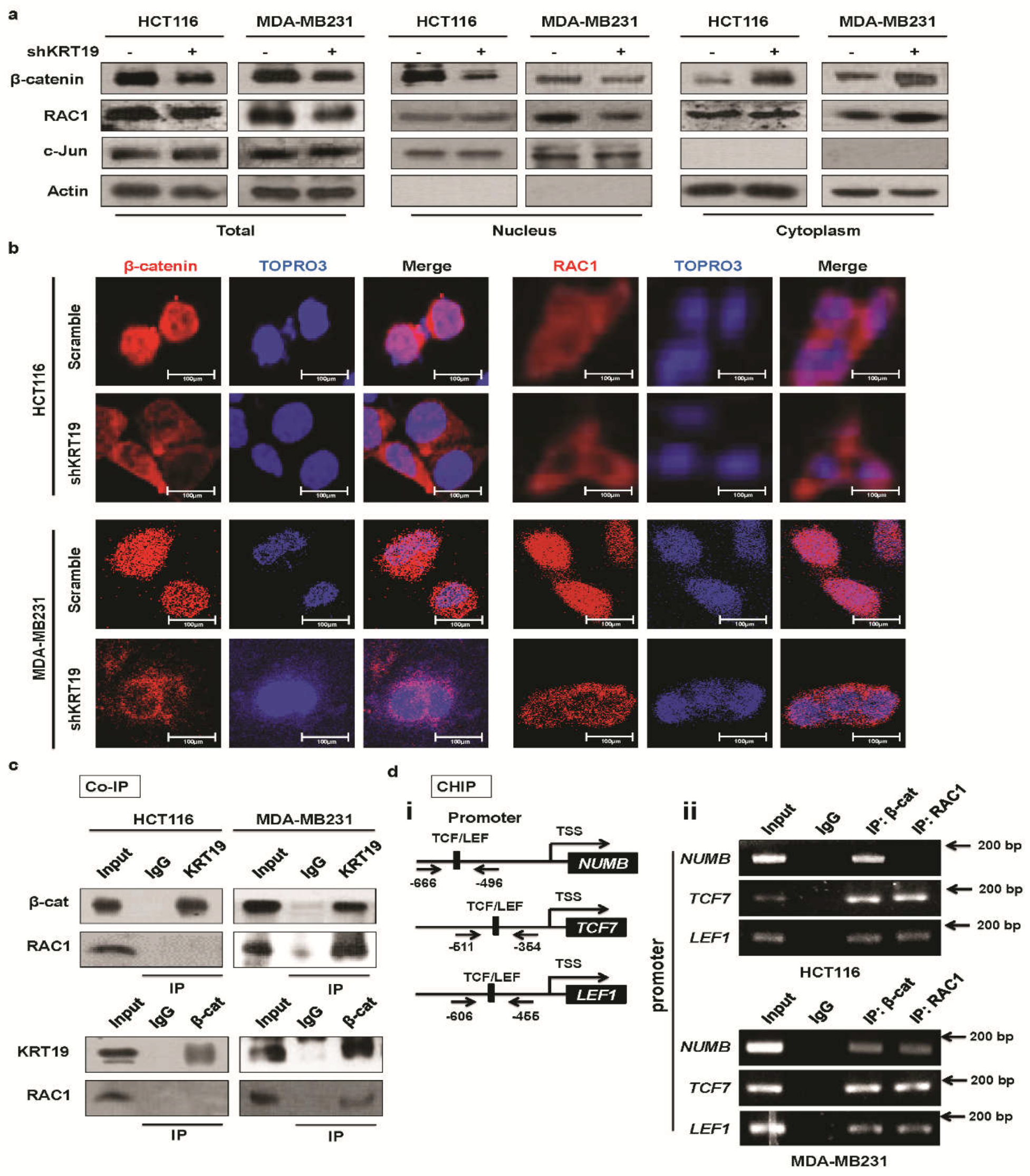
2.4. KRT19 Regulates β-Catenin Localization and Differential RAC1 Localization
2.5. RAC1 Differentially Binds to the KRT19/β-Catenin Complex
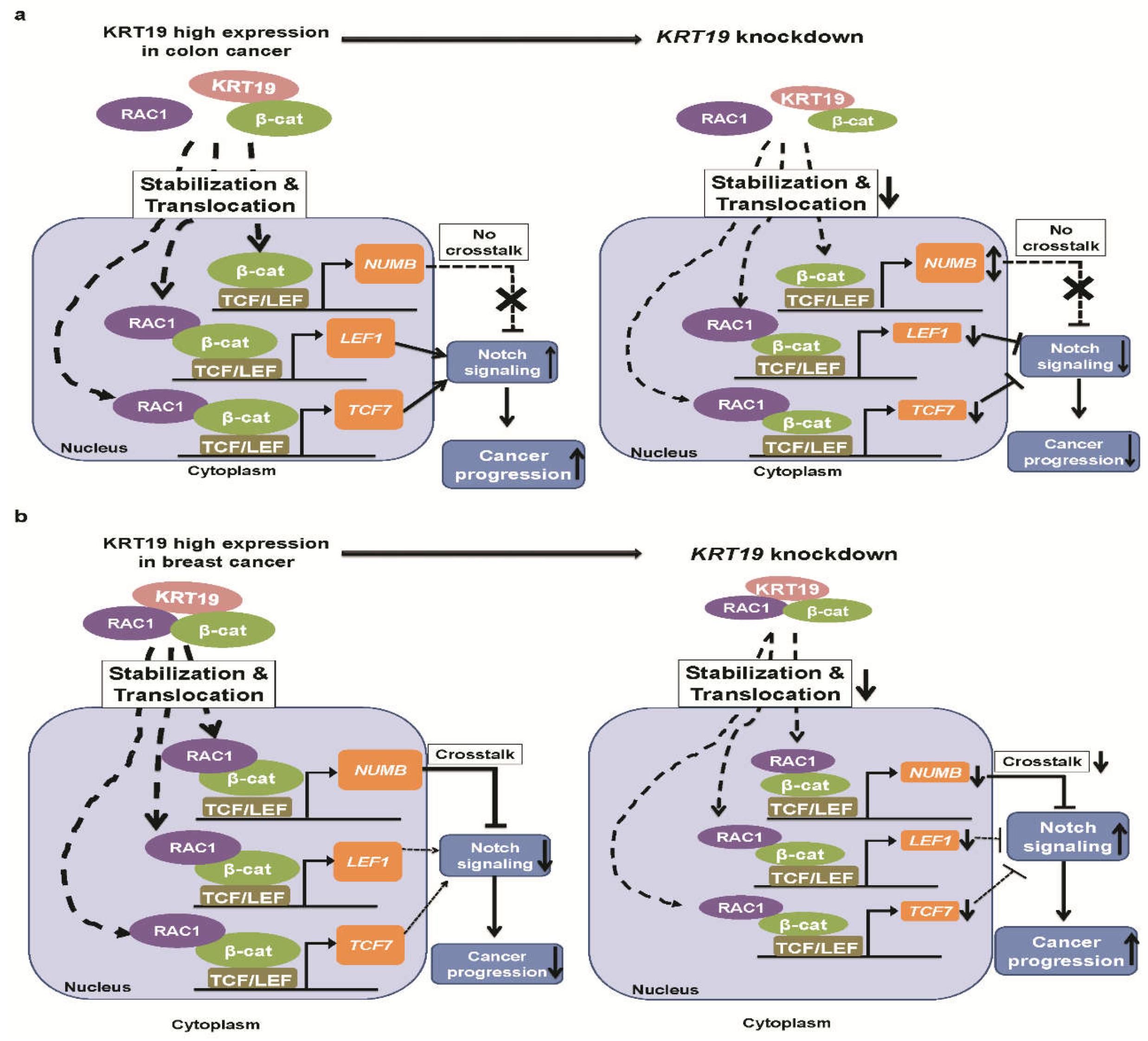
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. Cell Culture
4.3. Knockdown of KRT19 Using a Short Hairpin RNA (shRNA) Construct
4.4. Lentivirus Production and Transduction
4.5. RNA Extraction and RT-PCR
4.6. Western Blotting
4.7. Cell Proliferation and Viability Assay
4.8. Wound Healing Cell Migration Assay
4.9. Sphere Formation Assay
4.10. Luciferase Reporter Assay
4.11. Subcellular Fractionation Assay
4.12. Immunocytochemistry
4.13. Co-Immunoprecipitation (Co-IP) Assay
4.14. Chromatin Immunoprecipitation (ChIP) Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.Y.; Crespi, C.M.; Maxwell, A.E. Colorectal cancer among Koreans living in South Korea versus California: Incidence, mortality, and screening rates. Ethn. Health 2014, 19, 406–423. [Google Scholar] [CrossRef]
- Ponce, N.A.; Tsui, J.; Knight, S.J.; Afable-Munsuz, A.; Ladabaum, U.; Hiatt, R.A.; Haas, J.S. Disparities in cancer screening in individuals with a family history of breast or colorectal cancer. Cancer 2012, 118, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.; Kim, K.-Z.; Jung, K.-W.; Park, S.; Won, Y.-J.; Kim, J.; Kim, D.Y.; Oh, J.H. Increasing trend of colorectal cancer incidence in Korea, 1999–2009. Cancer Res. Treat. 2012, 44, 219. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Fan, T.; Zhao, Q.; Zeng, W.; Zaslavsky, E.; Chen, J.J.; Frohman, M.A.; Golightly, M.G.; Madajewicz, S.; Chen, W.T. Isolation of circulating epithelial and tumor progenitor cells with an invasive phenotype from breast cancer patients. Int. J. Cancer 2010, 126, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 Loss in Epithelial Cancer Cells Increases Collective Cell Migration and Cisplatin Sensitivity through Claudin1 Up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Escobar-Hoyos, L.F.; Shah, R.; Roa-Pena, L.; Vanner, E.A.; Najafian, N.; Banach, A.; Nielsen, E.; Al-Khalil, R.; Akalin, A.; Talmage, D.; et al. Keratin-17 Promotes p27KIP1 Nuclear Export and Degradation and Offers Potential Prognostic Utility. Cancer Res. 2015, 75, 3650–3662. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Rheinwald, J.G. A new small (40 kd) keratin filament protein made by some cultured human squamous cell carcinomas. Cell 1981, 25, 627–635. [Google Scholar] [CrossRef]
- Stasiak, P.C.; Purkis, P.E.; Leigh, I.M.; Lane, E.B. Keratin 19: Predicted amino acid sequence and broad tissue distribution suggest it evolved from keratinocyte keratins. J. Investig. Dermatol. 1989, 92, 707–716. [Google Scholar] [CrossRef]
- Bartek, J.; Taylor-Papadimitriou, J.; Miller, N.; Millis, R. Patterns of expression of keratin 19 as detected with monoclonal antibodies in human breast tissues and tumours. Int. J. Cancer 1985, 36, 299. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Morgan, P.R.; Thomas, J.A.; Lane, E.B. Expression of keratin 14 and 19 mRNA and protein in normal oral epithelia, hairy leukoplakia, tongue biting and white sponge nevus. J. Oral Pathol. Med. 1993, 22, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Choi, H.; Kim, B.; Dayem, A.; Yang, G.; Kim, K.; Yin, Y.; Cho, S. KRT19 directly interacts with β-catenin/RAC1 complex to regulate NUMB-dependent NOTCH signaling pathway and breast cancer properties. Oncogene 2017, 36, 332. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-H.; Yang, W.; Lee, K.; Oh, S.; Nam, K.; Shim, S.; Shin, S.Y.; Gye, M.C.; Chu, I.-S.; Shin, I. Regulation of cell proliferation and migration by keratin19-induced nuclear import of early growth response-1 in breast cancer cells. Clin. Cancer Res. 2013, 19, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.H.; Oh, S.; Lee, K.M.; Yang, W.; Nam, K.S.; Moon, H.G.; Noh, D.Y.; Kim, C.G.; Park, G.; Park, J.B.; et al. Cytokeratin19 induced by HER2/ERK binds and stabilizes HER2 on cell membranes. Cell Death Differ. 2015, 22, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Sakaguchi, M.; Yamamoto, H.; Tomida, S.; Takata, K.; Shien, K.; Hashida, S.; Miyata-Takata, T.; Watanabe, M.; Suzawa, K. Interaction of cytokeratin 19 head domain and HER2 in the cytoplasm leads to activation of HER2-Erk pathway. Sci. Rep. 2016, 6, 39557. [Google Scholar] [CrossRef]
- Govaere, O.; Komuta, M.; Berkers, J.; Spee, B.; Janssen, C.; de Luca, F.; Katoonizadeh, A.; Wouters, J.; van Kempen, L.C.; Durnez, A. Keratin 19: A key role player in the invasion of human hepatocellular carcinomas. Gut 2013, 63, 674–685. [Google Scholar] [CrossRef]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; Yasuda, K.; Fukumitsu, K.; Mizumoto, M.; et al. Keratin 19, a Cancer Stem Cell Marker in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2015, 21, 3081–3091. [Google Scholar] [CrossRef]
- Tang, J.; Zhuo, H.; Zhang, X.; Jiang, R.; Ji, J.; Deng, L.; Qian, X.; Zhang, F.; Sun, B. A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis. 2014, 5, e1549. [Google Scholar] [CrossRef]
- Govaere, O.; Petz, M.; Wouters, J.; Vandewynckel, Y.-P.; Scott, E.J.; Topal, B.; Nevens, F.; Verslype, C.; Anstee, Q.M.; Van Vlierberghe, H. The PDGFRα-laminin B1-keratin 19 cascade drives tumor progression at the invasive front of human hepatocellular carcinoma. Oncogene 2017, 36, 6605–6616. [Google Scholar] [CrossRef]
- Kim, H.; Choi, G.H.; Na, D.C.; Ahn, E.Y.; Kim, G.I.; Lee, J.E.; Cho, J.Y.; Yoo, J.E.; Choi, J.S.; Park, Y.N. Human hepatocellular carcinomas with “Stemness”-related marker expression: Keratin 19 expression and a poor prognosis. Hepatology 2011, 54, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; Von Burstin, J.; Mastracci, T.L.; Worthley, D.L. Krt19+/Lgr5− cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Kim, K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. Cytokeratin 19 (KRT19) has a Role in the Reprogramming of Cancer Stem Cell-Like Cells to Less Aggressive and More Drug-Sensitive Cells. Int. J. Mol. Sci. 2018, 19, 1423. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Ross, J.S.; Royce, M.; McKenna, E.F.; Perou, C.M.; Avisar, E.; Wu, L. TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine +/- trastuzumab. Breast Cancer Res. Treat. 2012, 132, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ki, D.H.; Jeung, H.C.; Park, C.H.; Kang, S.H.; Lee, G.Y.; Lee, W.S.; Kim, N.K.; Chung, H.C.; Rha, S.Y. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int. J. Cancer 2007, 121, 2005–2012. [Google Scholar] [CrossRef]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]
- Goswami, C.P.; Nakshatri, H. PROGgeneV2: Enhancements on the existing database. BMC Cancer 2014, 14, 970. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, M.N.; Tan, X.H.; Li, T.F.; Zhang, Y.E.; Chen, D. Smad3 Prevents beta-Catenin Degradation and Facilitates beta-Catenin Nuclear Translocation in Chondrocytes. J. Biol. Chem. 2010, 285, 8703–8710. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, J.; Tan, T.K.; Lyons, J.G.; Zhao, H.; Niu, B.; Lee, S.R.; Tsatralis, T.; Zhao, Y.; Wang, Y. Association of β-catenin with P-Smad3 but not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-β1. J. Cell Sci. 2013, 126, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N. Macrophage derived Wnt signalling opposes Notch signalling in a Numb mediated manner to specify HPC fate in chronic liver disease in human and mouse. Nat. Med. 2012, 18, 572. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. NUMB is a break of WNT-Notch signaling cycle. Int. J. Mol. Med. 2006, 18, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Huber, T.L.; Chen, V.C.; Gadue, P.; Keller, G.M. Numb mediates the interaction between Wnt and Notch to modulate primitive erythropoietic specification from the hemangioblast. Development 2008, 135, 3447–3458. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Fabris, L. The balance between Notch/Wnt signaling regulates progenitor cells’ commitment during liver repair: Mystery solved? J. Hepatol. 2013, 58, 181–183. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.A.; McGlade, C.J. Mammalian numb proteins promote notch1 receptor ubiquitination and degradation of the notch1 intracellular domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef] [PubMed]
- Centelles, J.J. General aspects of colorectal cancer. ISRN Oncol. 2012, 2012, 139268. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Waterman, M.L. Nuclear localization and formation of β-catenin–lymphoid enhancer factor 1 complexes are not sufficient for activation of gene expression. Mol. Cell. Biol. 1999, 19, 4503–4515. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tu, X.; Joeng, K.S.; Hilton, M.J.; Williams, D.A.; Long, F. Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell 2008, 133, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Pethe, V.V.; Charames, G.S.; Bapat, B. Rac1b recruits Dishevelled and β-catenin to Wnt target gene promoters independent of Wnt3A stimulation. Int. J. Oncol. 2011, 39, 805–810. [Google Scholar] [PubMed]
- Jamieson, C.; Lui, C.; Brocardo, M.G.; Martino-Echarri, E.; Henderson, B.R. Rac1 augments Wnt signaling by stimulating β-catenin-LEF-1 complex assembly independent of β-catenin nuclear import. J. Cell Sci. 2015, 128, 3933–3946. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Esufali, S.; Bapat, B. Cross-talk between Rac1 GTPase and dysregulated Wnt signaling pathway leads to cellular redistribution of β-catenin and TCF/LEF-mediated transcriptional activation. Oncogene 2004, 23, 8260–8271. [Google Scholar] [CrossRef] [PubMed]
- Gartner, J.J.; Parker, S.C.J.; Prickett, T.D.; Dutton-Regester, K.; Stitzel, M.L.; Lin, J.C.; Davis, S.; Simhadri, V.L.; Jha, S.; Katagiri, N.; et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 13481–13486. [Google Scholar] [CrossRef]
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous Mutations Frequently Act as Driver Mutations in Human Cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-H.; Wu, Y.; Yao, S.; Levine, A.C.; Kirschenbaum, A.; Collier, L.; Bauman, W.A.; Cardozo, C.P. Androgens upregulate transcription of the Notch inhibitor Numb in C2C12 myoblasts via Wnt/beta-catenin signaling to Tcf elements in the numb promoter. J. Biol. Chem. 2013, 288, 17990–17998. [Google Scholar] [CrossRef]
- Yang, X.R.; Xu, Y.; Shi, G.M.; Fan, J.; Zhou, J.; Ji, Y.; Sun, H.C.; Qiu, S.J.; Yu, B.; Gao, Q.; et al. Cytokeratin 10 and cytokeratin 19: Predictive markers for poor prognosis in hepatocellular carcinoma patients after curative resection. Clin. Cancer Res. 2008, 14, 3850–3859. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.F.; Jiang, G.L.; Fu, X.L.; Wang, L.J.; Qian, H.; Wu, K.L.; Zhao, S. CK19 mRNA expression measured by reverse-transcription polymerase chain reaction (RT-PCR) in the peripheral blood of patients with non-small cell lung cancer treated by chemo-radiation: An independent prognostic factor. Lung Cancer 2007, 56, 105–114. [Google Scholar] [CrossRef]
- Stathopoulos, E.N.; Sanidas, E.; Kafousi, M.; Mavroudis, D.; Askoxylakis, J.; Bozionelou, V.; Perraki, M.; Tsiftsis, D.; Georgoulias, V. Detection of CK-19 mRNA-positive cells in the peripheral blood of breast cancer patients with histologically and immunohistochemically negative axillary lymph nodes. Ann. Oncol. 2005, 16, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Husic, N.; Lin, Y.; Snider, B.J. Production of Lentiviral Vectors for Transducing Cells from the Central Nervous System. JoVE-J. Vis. Exp. 2012, 63, e4031. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Xenidis, N.; Perraki, M.; Apostolaki, S.; Politaki, E.; Kafousi, M.; Stathopoulos, E.N.; Stathopoulou, A.; Lianidou, E.; Chlouverakis, G.; et al. Different prognostic value of cytokeratin-19 mRNA positive circulating tumor cells according to estrogen receptor and HER2 status in early-stage breast cancer. J. Clin. Oncol. 2007, 25, 5194–5202. [Google Scholar] [CrossRef] [PubMed]
- Bozionellou, V.; Mavroudis, D.; Perraki, M.; Papadopoulos, S.; Apostolaki, S.; Stathopoulos, E.; Stathopoulou, A.; Lianidou, E.; Georgoulias, V. Trastuzumab Administration Can Effectively Target Chemotherapy-Resistant Cytokeratin-19 Messenger RNA–Positive Tumor Cells in the Peripheral Blood and Bone Marrow of Patients With Breast Cancer. Clin. Cancer Res. 2004, 10, 8185–8194. [Google Scholar] [CrossRef]
- Zhang, J.C.; Shao, X.M.; Sun, H.Y.; Liu, K.; Ding, Z.H.; Chen, J.T.; Fang, L.J.; Su, W.; Hong, Y.; Li, H.S.; et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016, 7, 61036–61053. [Google Scholar] [CrossRef] [PubMed]
- Al Thawadi, H.; Abu-Kaoud, N.; Al Farsi, H.; Hoarau-Véchot, J.; Rafii, S.; Rafii, A.; Pasquier, J. VE-cadherin cleavage by ovarian cancer microparticles induces β-catenin phosphorylation in endothelial cells. Oncotarget 2016, 7, 5289–5305. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Kueanjinda, P.; Roytrakul, S.; Palaga, T. A Novel Role of Numb as A Regulator of Pro-inflammatory Cytokine Production in Macrophages in Response to Toll-like Receptor 4. Sci. Rep. 2015, 5, 12784. [Google Scholar] [CrossRef] [PubMed]
- Abrusán, G.; Marsh, J.A. Alpha Helices Are More Robust to Mutations than Beta Strands. PLOS Comput. Biol. 2016, 12, e1005242. [Google Scholar] [CrossRef]
- Buongiorno, P.; Pethe, V.V.; Charames, G.S.; Esufali, S.; Bapat, B. Rac1 GTPase and the Rac1 exchange factor Tiam1 associate with Wnt-responsive promoters to enhance beta-catenin/TCF-dependent transcription in colorectal cancer cells. Mol. Cancer 2008, 7, 73. [Google Scholar] [CrossRef]
- Salmon, P.; Trono, D. Production and Titration of Lentiviral Vectors. Curr. Protoc. Hum. Genet. 2007, 54. [Google Scholar] [CrossRef]
- Nasri, M.; Karimi, A.; Allahbakhshian Farsani, M. Production, purification and titration of a lentivirus-based vector for gene delivery purposes. Cytotechnology 2014, 66, 1031–1038. [Google Scholar] [CrossRef]
- Saito, Y.; Nakahata, S.; Yamakawa, N.; Kaneda, K.; Ichihara, E.; Suekane, A.; Morishita, K. CD52 as a molecular target for immunotherapy to treat acute myeloid leukemia with high EVI1 expression. Leukemia 2011, 25, 921–931. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession no. | Gene Name | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|---|
| NM_002272.3 | KRT4 | AGATACCTTGGGCAATGACA | CTTGTTCAGGTAGGCAGCAT |
| NM_001256282 | KRT8 | CCTCATCAAGAAGGATGTGGA | CACCACAGATGTGTCCGAGA |
| NM_002275 | KRT15 | AGGGTCAGGAGGAGGATATG | TTTTCTCATTGCCAGAGAGG |
| NM_002276.4 | KRT19 | TCGAGCATGAGGTATCCAGT | GTAGCGGTTCTTCGTGTCTT |
| NM_019010 | KRT20 | AACGCCAGAACAACGAATAC | CTTCCAGGGTGCTTAACTGA |
| NM_001005743.1 | NUMB | TCCCACCTCTCCTACTTCTG | TGCCTCCCCTTCTACTTCTG |
| NM_001904.3 | CTNNB1 | AAAATGGCAGTGCGTTTAG | TTTGAAGGCAGTCTGTCGTA |
| NM_003202.3 | TCF7 | GACATCAGCCAGAAGCAAG | CACCAGAACCTAGCATCAAG |
| NM_016269.4 | LEF1 | CCTGGTCCCCACACAACTG | GGCTCCTGCTCCTTTCTCTG |
| NM_017617.3 | NOTCH1 | GGGTACAAGTGCGACTGTGA | CGGCAACGTCGTCAATACAC |
| NM_014757.4 | MAML1 | CACCAGCCACCGAGTAACTT | AACAGGGAGTTCTGCTCGTG |
| NM_005349.3 | RBPjK | GAACAAATGGAACGCGATGG | GATGACTTTTATCCGCTTGCTG |
| NM_005524.3 | HES1 | GGCTAAGGTGTTTGGAGGCT | GGTGGGTTGGGGAGTTTAGG |
| NM_001130442.1 | H-RAS | TTCTACACGTTGGTGCGTGA | CACAAGGGAGGCTGCTGAC |
| NM_053056.2 | CCND1 | CACACGGACTACAGGGGAGT | ATGGTTTCCACTTCGCAGCA |
| NM_002046.5 | GAPDH | AATCCCATCACCATCTTCCAG | CACGATACCAAAGTTGTCATGG |
| EMT markers | |||
| NM_001205255 | OCLN | CTTCAGGCAGCCTCGTTACA | TCCTCCTCCAGCTCATCACA |
| NM_000474 | TWIST1 | CTCAGCTACGCCTTCTCG | ACTGTCCATTTTCTCCTTCTCTG |
| NM_001792 | N-CADHRIN | GACAATGCCCCTCAAGTGTT | GACAATGCCCCTCAAGTGTT |
| Stemness markers | |||
| NM_001285986.1 | OCT4 | GTCCCAGGACATCAAAGCTC | CTCCAGGTTGCCTCTCACTC |
| NM_003106.3 | SOX2 | ACACCAATCCCATCCACACT | GCAAGAAGCCTCTCCTTGAA |
| NM_024865.3 | NANOG | ATACCTCAGCCTCCAGCAGA | GCAGGACTGCAGAGATTCCT |
| NM_002467.4 | c-MYC | CTCGGATTCTCTGCTCTC | TCGCCTCTTGACATTCTC |
| Antibody | Catalog No. | Conc. Ratio (WB) | Conc. Ratio (ICC) | Conc. Ratio (Co-IP) | Conc. Ratio (CHIP) |
|---|---|---|---|---|---|
| Primary Ab. | |||||
| Anti-KRT19 | SC-53258 | 1:1000 | - | - | - |
| Anti-Actin | SC-1616 | 1:1000 | - | - | - |
| Anti-β-catenin | SC-7199 | 1:2000 | 1:200 | 1:50 | 1:50 |
| Anti-RAC1 | SC-217 | 1:500 | 1:200 | 1:50 | 1:50 |
| Anti-c-Jun | SC-1694 | 1:1000 | - | - | - |
| Anti-p-SMAD3 | 9520S | 1:1000 | - | - | - |
| Anti-SMAD3 | 9513S | 1:1000 | - | - | - |
| Normal mouse IgG | SC-2025 | - | - | 1:50 | - |
| Normal rabbit IgG | SC-2027 | - | - | 1:50 | 1:50 |
| Second Ab. | |||||
| Anti-goat | SC-2020 | 1:1000 | - | - | - |
| Anti-rabbit | SC-2004 | 1:1000 | - | - | - |
| Anti-mouse | SC-2005 | 1:1000 | - | - | - |
| Alexa Fluor® 546 (goat anti-rabbit) | A11010 | - | 1:1000 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.K.; Yin, Y.; Chae, H.S.; Cho, S.-G. Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers. Cancers 2019, 11, 99. https://doi.org/10.3390/cancers11010099
Saha SK, Yin Y, Chae HS, Cho S-G. Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers. Cancers. 2019; 11(1):99. https://doi.org/10.3390/cancers11010099
Chicago/Turabian StyleSaha, Subbroto Kumar, Yingfu Yin, Hee Sung Chae, and Ssang-Goo Cho. 2019. "Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers" Cancers 11, no. 1: 99. https://doi.org/10.3390/cancers11010099
APA StyleSaha, S. K., Yin, Y., Chae, H. S., & Cho, S.-G. (2019). Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers. Cancers, 11(1), 99. https://doi.org/10.3390/cancers11010099