Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib

 , , ,
, , , 

Abstract
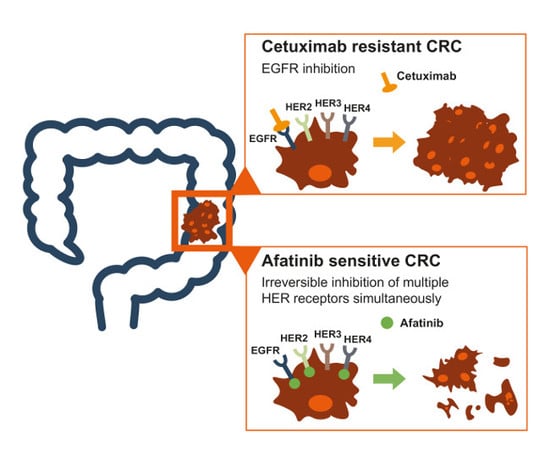
1. Introduction
2. Results
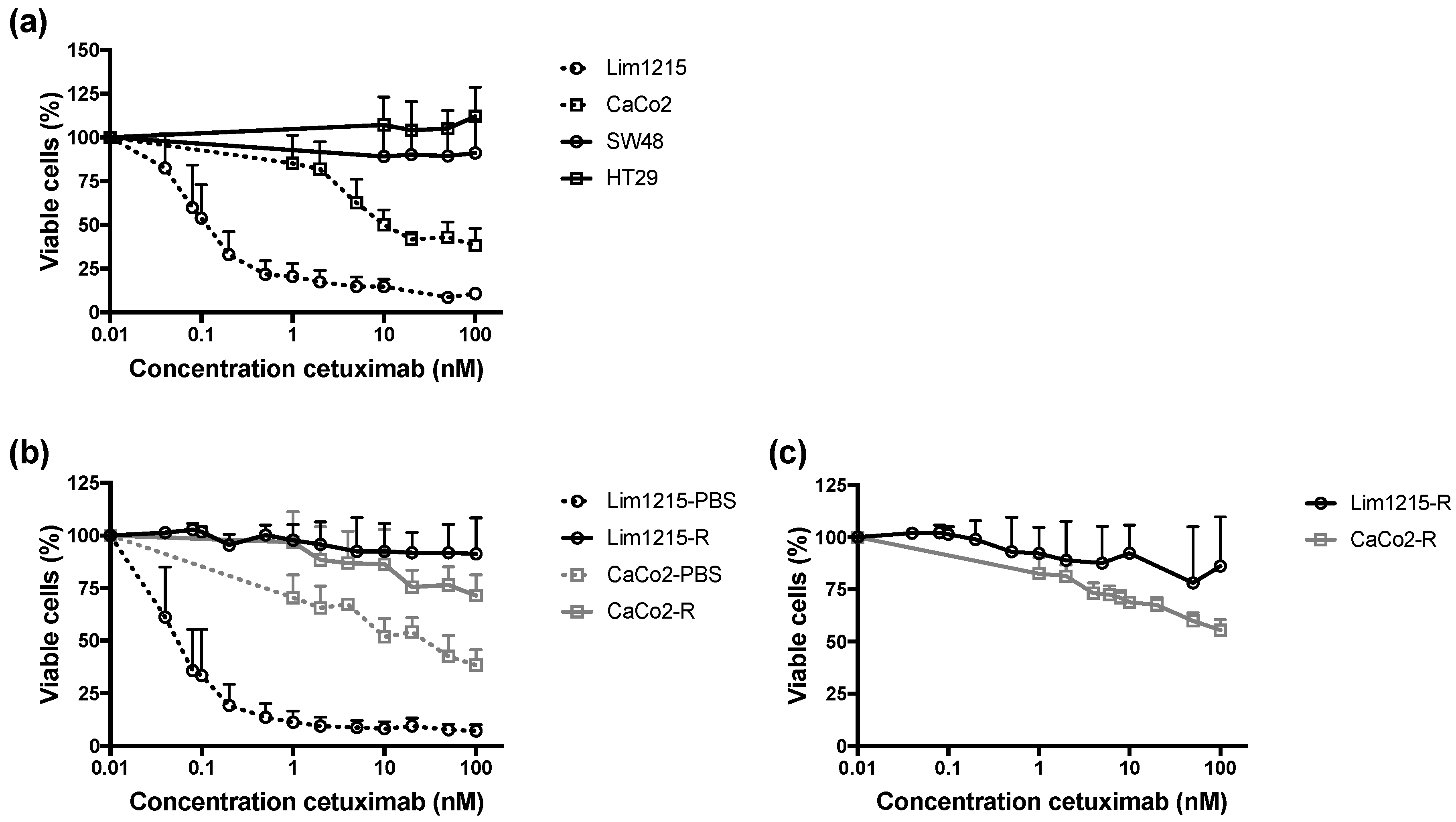
2.1. Identification of Intrinsically Cetuximab-Resistant CRC Cell Lines and Generation of Acquired Cetuximab-Resistant Cell Lines
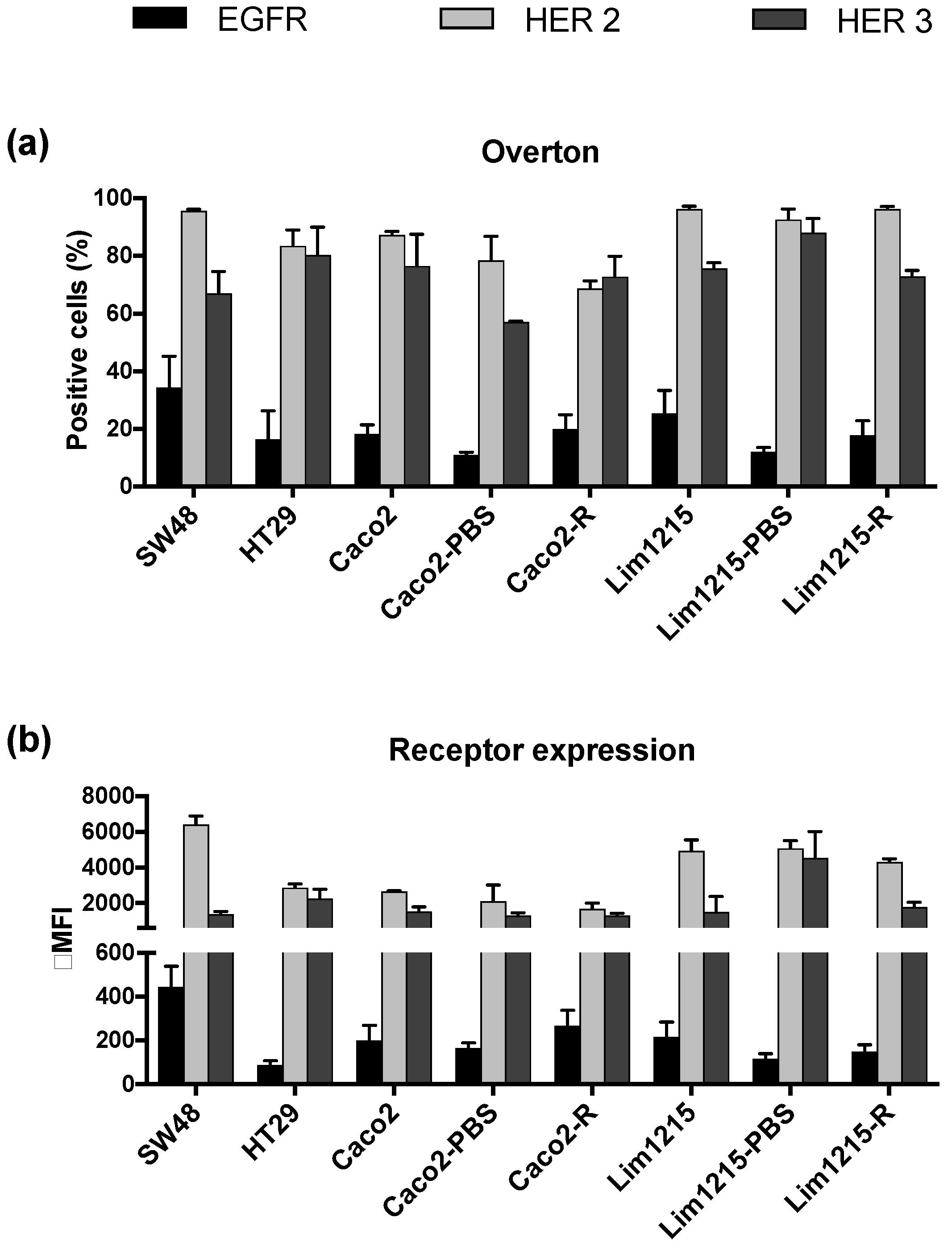
2.2. CRC Cell Lines and Patients Show Higher Expression of HER2 and HER3 than EGFR
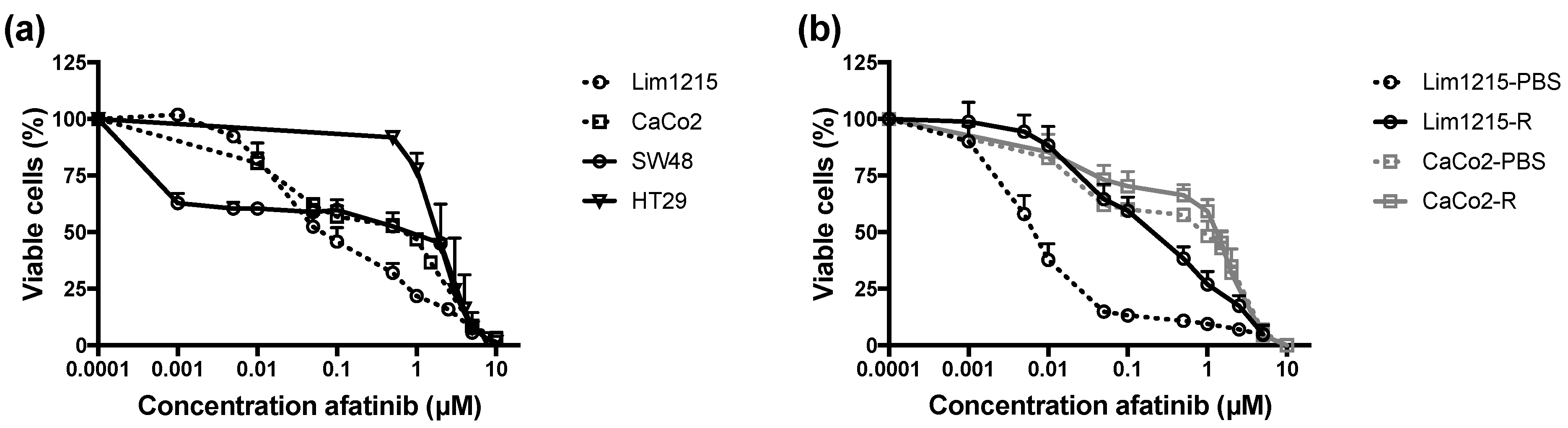
2.3. Intrinsic and Acquired Cetuximab-Resistant CRC Cell Lines Are Sensitive to Afatinib Treatment
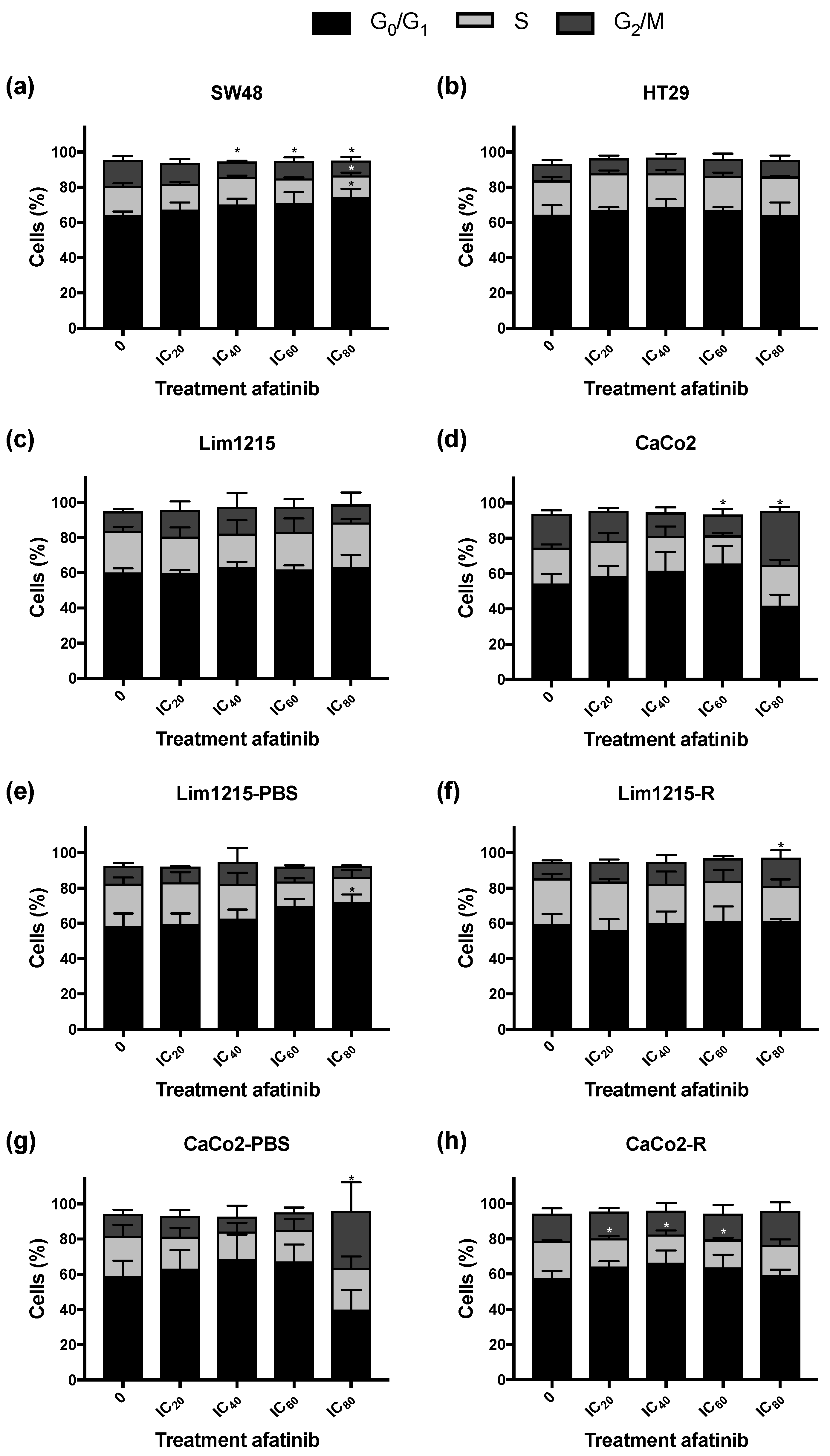
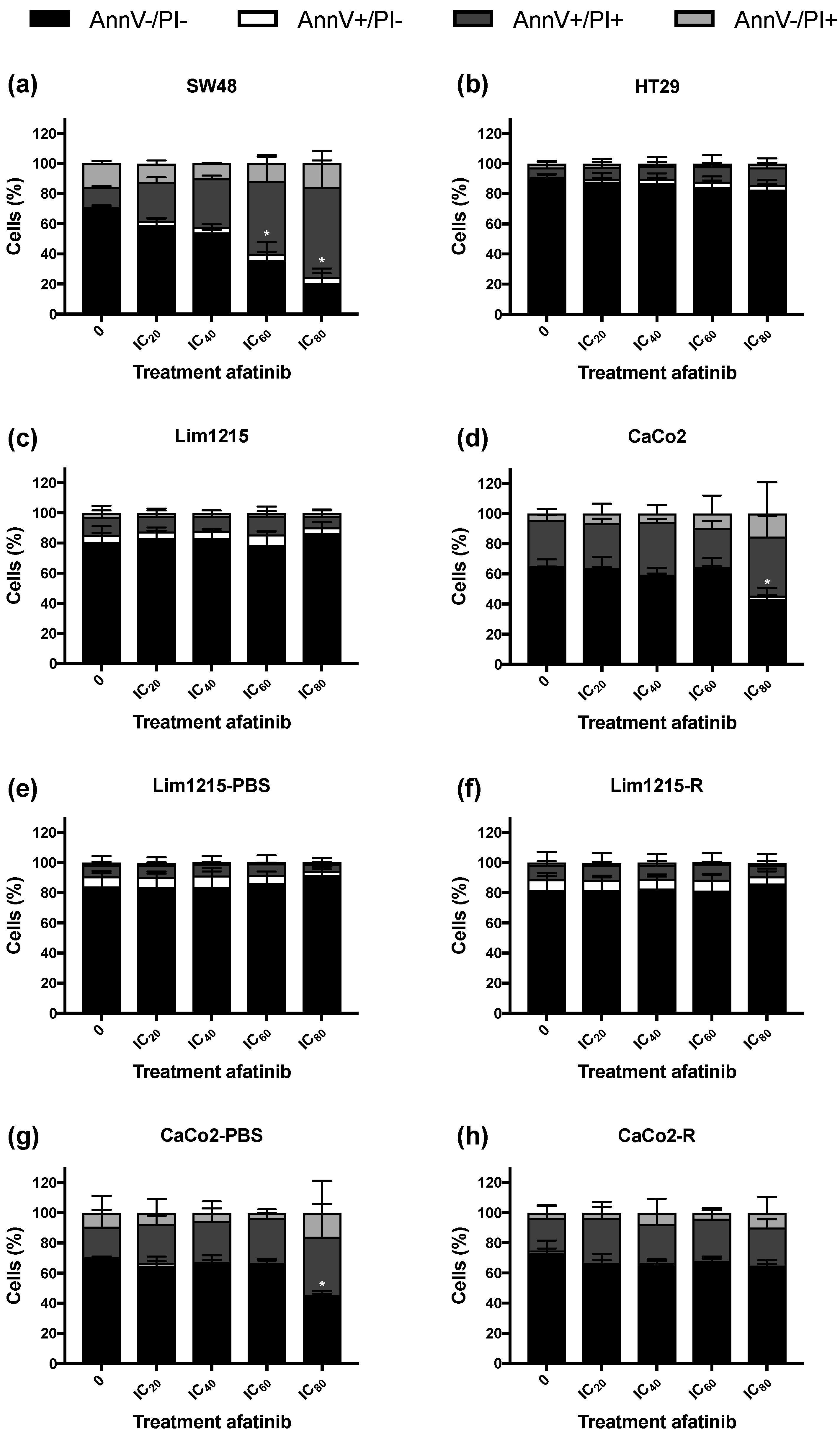
2.4. Treatment of CRC Cell Lines with Afatinib Has Little Influence on the Cell Cycle Distribution and the Induction of Apoptotic Cell Death
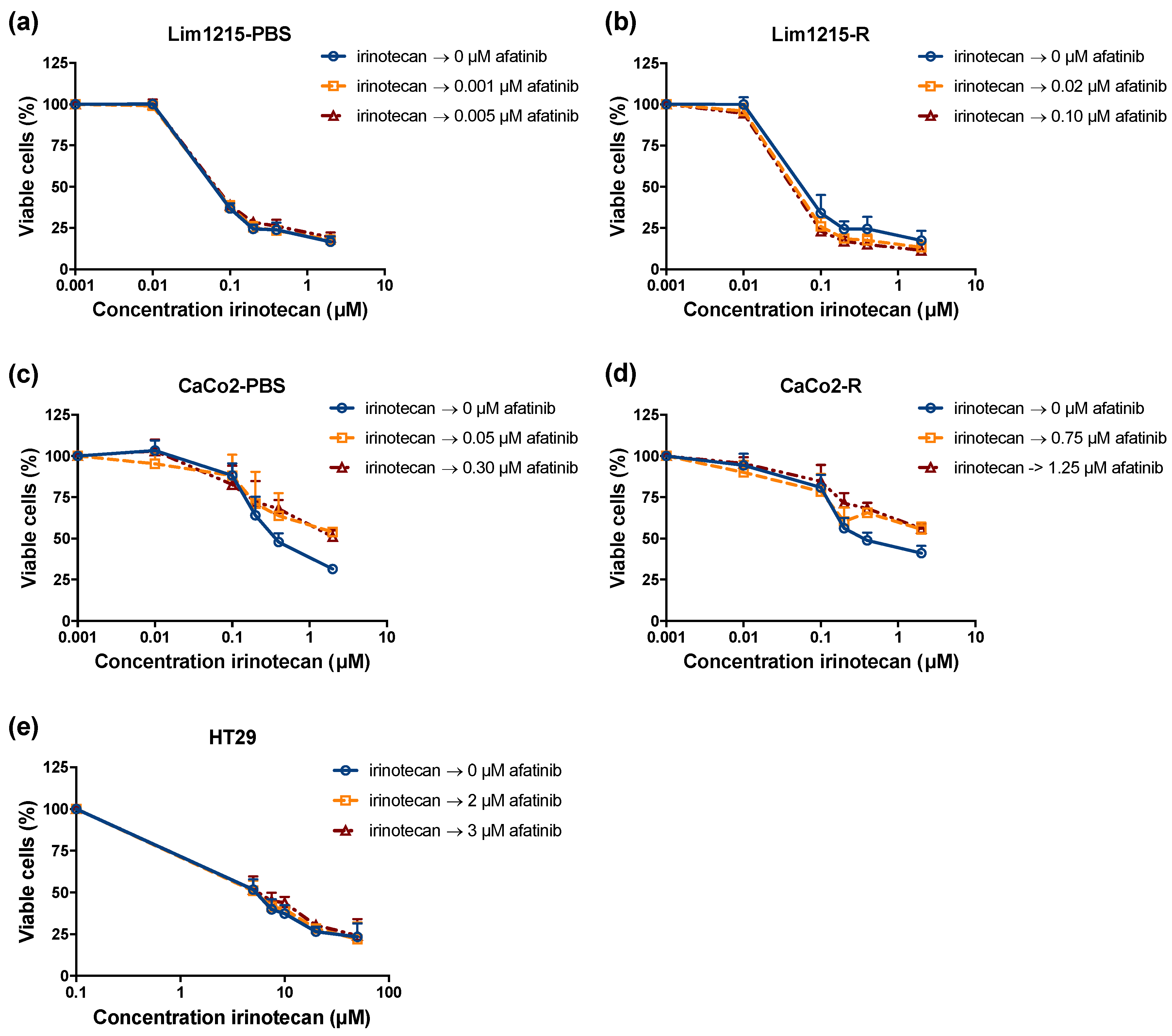
2.5. The Combination Treatment of Afatinib with Irinotecan Leads to Additive Effects in CRC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cytotoxicity Assays
- Afatinib for 72 h immediately followed by irinotecan for 24 h;
- Irinotecan for 24 h immediately followed by afatinib for 72 h.
4.3. Oxygen Conditions
4.4. Generation of Resistant Cell Clones
4.5. Expression Analysis of HER Family Members
4.6. Assays for Apoptosis and Cell Cycle Distribution
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Van Cutsem, E.; Rougier, P.; Ciardiello, F.; Heeger, S.; Schlichting, M.; Celik, I.; Kohne, C.H. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur. J. Cancer 2012, 48, 1466–1475. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Lang, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef]
- Peeters, M.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; Punt, C.J.; et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 4706–4713. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Fakih, M.; Wong, R. Efficacy of the monoclonal antibody EGFR inhibitors for the treatment of metastatic colorectal cancer. Curr. Oncol. 2010, 17 (Suppl. 1), S3–S17. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Dienstmann, R.; Tabernero, J. Development of molecular biomarkers in individualized treatment of colorectal cancer. Clin. Color. Cancer 2011, 10, 279–289. [Google Scholar] [CrossRef]
- Peeters, M.; Oliner, K.S.; Parker, A.; Siena, S.; Van Cutsem, E.; Huang, J.; Humblet, Y.; Van Laethem, J.L.; Andre, T.; Wiezorek, J.; et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 1902–1912. [Google Scholar] [CrossRef]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar]
- Ioannou, N.; Seddon, A.M.; Dalgleish, A.; Mackintosh, D.; Modjtahedi, H. Treatment with a combination of the ErbB (HER) family blocker afatinib and the IGF-IR inhibitor, NVP-AEW541 induces synergistic growth inhibition of human pancreatic cancer cells. BMC Cancer 2013, 13, 41. [Google Scholar] [CrossRef]
- De Pauw, I.; Lardon, F.; Van den Bossche, J.; Baysal, H.; Fransen, E.; Deschoolmeester, V.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Wouters, A. Simultaneous targeting of EGFR, HER2, and HER4 by afatinib overcomes intrinsic and acquired cetuximab resistance in head and neck squamous cell carcinoma cell lines. Mol. Oncol. 2018, 12, 830–854. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, I.; Wouters, A.; Van den Bossche, J.; Peeters, M.; Pauwels, P.; Deschoolmeester, V.; Vermorken, J.B.; Lardon, F. Preclinical and clinical studies on afatinib in monotherapy and in combination regimens: Potential impact in colorectal cancer. Pharmacol. Ther. 2016, 166, 71–83. [Google Scholar] [CrossRef]
- Wouters, A.; Boeckx, C.; Vermorken, J.B.; Van den Weyngaert, D.; Peeters, M.; Lardon, F. The intriguing interplay between therapies targeting the epidermal growth factor receptor, the hypoxic microenvironment and hypoxia-inducible factors. Curr. Pharm. Des. 2013, 19, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Naito, T.; Okamura, J.; Hosokawa, S.; Mineta, H.; Kawakami, J. Simple and rapid LC-MS/MS method for the absolute determination of cetuximab in human serum using an immobilized trypsin. J. Pharm. Biomed. Anal. 2017, 146, 266–272. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Leto, S.M.; Sassi, F.; Catalano, I.; Torri, V.; Migliardi, G.; Zanella, E.R.; Throsby, M.; Bertotti, A.; Trusolino, L. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2-amplified gastrointestinal carcinomas. Clin. Cancer Res. 2015, 21, 5519–5531. [Google Scholar] [CrossRef]
- Guan, S.S.; Chang, J.; Cheng, C.C.; Luo, T.Y.; Ho, A.S.; Wang, C.C.; Wu, C.T.; Liu, S.H. Afatinib and its encapsulated polymeric micelles inhibits HER2-overexpressed colorectal tumor cell growth in vitro and in vivo. Oncotarget 2014, 5, 4868–4880. [Google Scholar] [CrossRef]
- Sun, S.J.; Lin, Q.; Sun, Q.; Li, J.; Zhang, X.Y.; Tan, Z.G.; Song, Y.; Guo, Y.T.; Li, Y. High HER-2 protein levels correlate with clinicopathological features in colorectal cancer. J. Cancer Res. Ther. 2016, 12, 323–333. [Google Scholar] [CrossRef]
- Ocana, A.; Vera-Badillo, F.; Seruga, B.; Templeton, A.; Pandiella, A.; Amir, E. HER3 overexpression and survival in solid tumors: A meta-analysis. J. Natl. Cancer Inst. 2013, 105, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Cushman, S.M.; Jiang, C.; Hatch, A.J.; Shterev, I.; Sibley, A.B.; Niedzwiecki, D.; Venook, A.P.; Owzar, K.; Hurwitz, H.I.; Nixon, A.B. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: Results from CALGB 80203 (Alliance). Clin. Cancer Res. 2015, 21, 1078–1086. [Google Scholar] [CrossRef]
- Torabizadeh, Z.; Nosrati, A.; Tahvildari, S. Human Epidermal Growth Factor Receptor Expression in Colorectal Cancer and Its Relationship with Clinicopathological Characteristics. Middle East J. Dig. Dis. 2016, 8, 24–30. [Google Scholar] [CrossRef]
- Wu, S.W.; Ma, C.C.; Li, W.H. Does overexpression of HER-2 correlate with clinicopathological characteristics and prognosis in colorectal cancer? Evidence from a meta-analysis. Diagn. Pathol. 2015, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Yu, Y.; Liu, W.; Chen, S. Significance of human epidermal growth factor receptor 2 expression in colorectal cancer. Exp. Ther. Med. 2015, 9, 17–24. [Google Scholar] [CrossRef]
- Stahler, A.; Heinemann, V.; Neumann, J.; Crispin, A.; Schalhorn, A.; Stintzing, S.; Giessen-Jung, C.; Fischer von Weikersthal, L.; Vehling-Kaiser, U.; Stauch, M.; et al. Prevalence and influence on outcome of HER2/neu, HER3 and NRG1 expression in patients with metastatic colorectal cancer. Anti-Cancer Drugs 2017, 28, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, S.; Zhang, J.; Liu, J.; Xu, L.; Liu, Y.; Qu, X. Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int. J. Mol. Sci. 2014, 15, 5838–5851. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef]
- Yan, Q.; Guo, K.; Feng, G.; Shan, F.; Sun, L.; Zhang, K.; Shen, F.; Shen, M.; Ruan, S. Association between the overexpression of Her3 and clinical pathology and prognosis of colorectal cancer: A meta-analysis. Medicine 2018, 97, e12317. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.F.; Hatch, A.J.; Richman, S.D.; Elliott, F.; Jacobs, B.; Brown, S.; Hurwitz, H.; Barrett, J.H.; Quirke, P.; Nixon, A.B.; et al. Association of Tumor HER3 Messenger RNA Expression With Panitumumab Efficacy in Advanced Colorectal Cancer. JAMA Oncol. 2018, 4, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Silvestris, N.; Castiglia, M.; Galvano, A.; Passiglia, F.; Sortino, G.; Cicero, G.; Rolfo, C.; Peeters, M.; Bazan, V.; et al. New findings on primary and acquired resistance to anti-EGFR therapy in metastatic colorectal cancer: Do all roads lead to RAS? Oncotarget 2015, 6, 24780–24796. [Google Scholar] [CrossRef]
- Camidge, D.R.; Sequist, L.V.; Janne, P.A.; Weickhardt, A.J.; Dowling, E.S.; Alicea, J.; Fan, J.; Oxnard, G.R. Phase Ib Study of High-dose Intermittent Afatinib in Patients With Advanced Solid Tumors. Clin. Lung Cancer 2018, 19, e655–e665. [Google Scholar] [CrossRef] [PubMed]
- Herr, R.; Halbach, S.; Heizmann, M.; Busch, H.; Boerries, M.; Brummer, T. BRAF inhibition upregulates a variety of receptor tyrosine kinases and their downstream effector Gab2 in colorectal cancer cell lines. Oncogene 2018, 37, 1576–1593. [Google Scholar] [CrossRef]
- Khelwatty, S.A.; Essapen, S.; Seddon, A.M.; Modjtahedi, H. Growth response of human colorectal tumour cell lines to treatment with afatinib (BIBW2992), an irreversible erbB family blocker, and its association with expression of HER family members. Int. J. Oncol. 2011, 39, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Busse, D.; Doughty, R.S.; Ramsey, T.T.; Russell, W.E.; Price, J.O.; Flanagan, W.M.; Shawver, L.K.; Arteaga, C.L. Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J. Biol. Chem. 2000, 275, 6987–6995. [Google Scholar] [CrossRef]
- Poindessous, V.; Ouaret, D.; El Ouadrani, K.; Battistella, A.; Megalophonos, V.F.; Kamsu-Kom, N.; Petitprez, A.; Escargueil, A.E.; Boudou, P.; Dumont, S.; et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin. Cancer Res. 2011, 17, 6522–6530. [Google Scholar] [CrossRef]
- Wouters, A.; Pauwels, B.; Lardon, F.; Vermorken, J.B. Review: Implications of in vitro research on the effect of radiotherapy and chemotherapy under hypoxic conditions. Oncologist 2007, 12, 690–712. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Fayette, J.; Cupissol, D.; Del Campo, J.M.; Clement, P.M.; Hitt, R.; Degardin, M.; Zhang, W.; Blackman, A.; Ehrnrooth, E.; et al. A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann. Oncol. 2014, 25, 1813–1820. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Licitra, L.F.; Burtness, B.; Fayette, J.; Gauler, T.; Clement, P.M.; Grau, J.J.; Del Campo, J.M.; Mailliez, A.; Haddad, R.I.; et al. Biomarkers predict enhanced clinical outcomes with afatinib versus methotrexate in patients with second-line recurrent and/or metastatic head and neck cancer. Ann. Oncol. 2017, 28, 2526–2532. [Google Scholar] [CrossRef] [PubMed]
- Hickish, T.; Cassidy, J.; Propper, D.; Chau, I.; Falk, S.; Ford, H.; Iveson, T.; Braun, M.; Potter, V.; Macpherson, I.R.; et al. A randomised, open-label phase II trial of afatinib versus cetuximab in patients with metastatic colorectal cancer. Eur. J. Cancer 2014, 50, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Haddad, R.I.; Fayette, J.; Licitra, L.F.; Tahara, M.; Vermorken, J.B.; Clement, P.M.; Gauler, T.; Cupissol, D.; Grau, J.J.; et al. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): An open-label, randomised phase 3 trial. Lancet Oncol. 2015, 16, 583–594. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Rottey, S.; Ehrnrooth, E.; Pelling, K.; Lahogue, A.; Wind, S.; Machiels, J.P. A phase Ib, open-label study to assess the safety of continuous oral treatment with afatinib in combination with two chemotherapy regimens: Cisplatin plus paclitaxel and cisplatin plus 5-fluorouracil, in patients with advanced solid tumors. Ann. Oncol. 2013, 24, 1392–1400. [Google Scholar] [CrossRef]
- Chung, C.H.; Rudek, M.A.; Kang, H.; Marur, S.; John, P.; Tsottles, N.; Bonerigo, S.; Veasey, A.; Kiess, A.; Quon, H.; et al. A phase I study afatinib/carboplatin/paclitaxel induction chemotherapy followed by standard chemoradiation in HPV-negative or high-risk HPV-positive locally advanced stage III/IVa/IVb head and neck squamous cell carcinoma. Oral Oncol. 2016, 53, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Bouche, O.; Maindrault-Goebel, F.; Ducreux, M.; Lledo, G.; Andre, T.; Stopfer, P.; Amellal, N.; Merger, M.; De Gramont, A. Phase II trial of weekly alternating sequential BIBF 1120 and afatinib for advanced colorectal cancer. Anticancer Res. 2011, 31, 2271–2281. [Google Scholar]
- Bahleda, R.; Hollebecque, A.; Varga, A.; Gazzah, A.; Massard, C.; Deutsch, E.; Amellal, N.; Farace, F.; Ould-Kaci, M.; Roux, F.; et al. Phase I study of afatinib combined with nintedanib in patients with advanced solid tumours. Br. J. Cancer 2015, 113, 1413–1420. [Google Scholar] [CrossRef]
- Gazzah, A.; Boni, V.; Soria, J.C.; Calles, A.; Even, C.; Doger, B.; Mahjoubi, L.; Bahleda, R.; Ould-Kaci, M.; Esler, A.; et al. A phase 1b study of afatinib in combination with standard-dose cetuximab in patients with advanced solid tumours. Eur. J. Cancer 2018, 104, 1–8. [Google Scholar] [CrossRef]
- Pauwels, B.; Korst, A.E.; de Pooter, C.M.; Pattyn, G.G.; Lambrechts, H.A.; Baay, M.F.; Lardon, F.; Vermorken, J.B. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother. Pharmacol. 2003, 51, 221–226. [Google Scholar] [CrossRef]
- Limame, R.; Wouters, A.; Pauwels, B.; Fransen, E.; Peeters, M.; Lardon, F.; De Wever, O.; Pauwels, P. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS ONE 2012, 7, e46536. [Google Scholar] [CrossRef] [PubMed]
- Valeriote, F.; Lin, H. Synergistic interaction of anticancer agents: A cellular perspective. Cancer Chemother. Rep. 1975, 59, 895–900. [Google Scholar] [PubMed]
- Jonsson, E.; Fridborg, H.; Nygren, P.; Larsson, R. Synergistic interactions of combinations of topotecan with standard drugs in primary cultures of human tumor cells from patients. Eur. J. Clin. Pharmacol. 1998, 54, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; Op de Beeck, K.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Wouters, A.; Pauwels, B.; Lambrechts, H.A.; Pattyn, G.G.; Ides, J.; Baay, M.; Meijnders, P.; Dewilde, S.; Vermorken, J.B.; Lardon, F. Chemoradiation interactions under reduced oxygen conditions: Cellular characteristics of an in vitro model. Cancer Lett. 2009, 286, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Boeckx, C.; Blockx, L.; de Beeck, K.O.; Limame, R.; Camp, G.V.; Peeters, M.; Vermorken, J.B.; Specenier, P.; Wouters, A.; Baay, M.; et al. Establishment and characterization of cetuximab resistant head and neck squamous cell carcinoma cell lines: Focus on the contribution of the AP-1 transcription factor. Am. J. Cancer Res. 2015, 5, 1921–1938. [Google Scholar] [PubMed]
- De Pauw, I.; Wouters, A.; Van den Bossche, J.; Deschoolmeester, V.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Lardon, F. Dual Targeting of Epidermal Growth Factor Receptor and HER3 by MEHD7945A as Monotherapy or in Combination with Cisplatin Partially Overcomes Cetuximab Resistance in Head and Neck Squamous Cell Carcinoma Cell Lines. Cancer Biother. Radiopharm. 2017, 32, 229–238. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
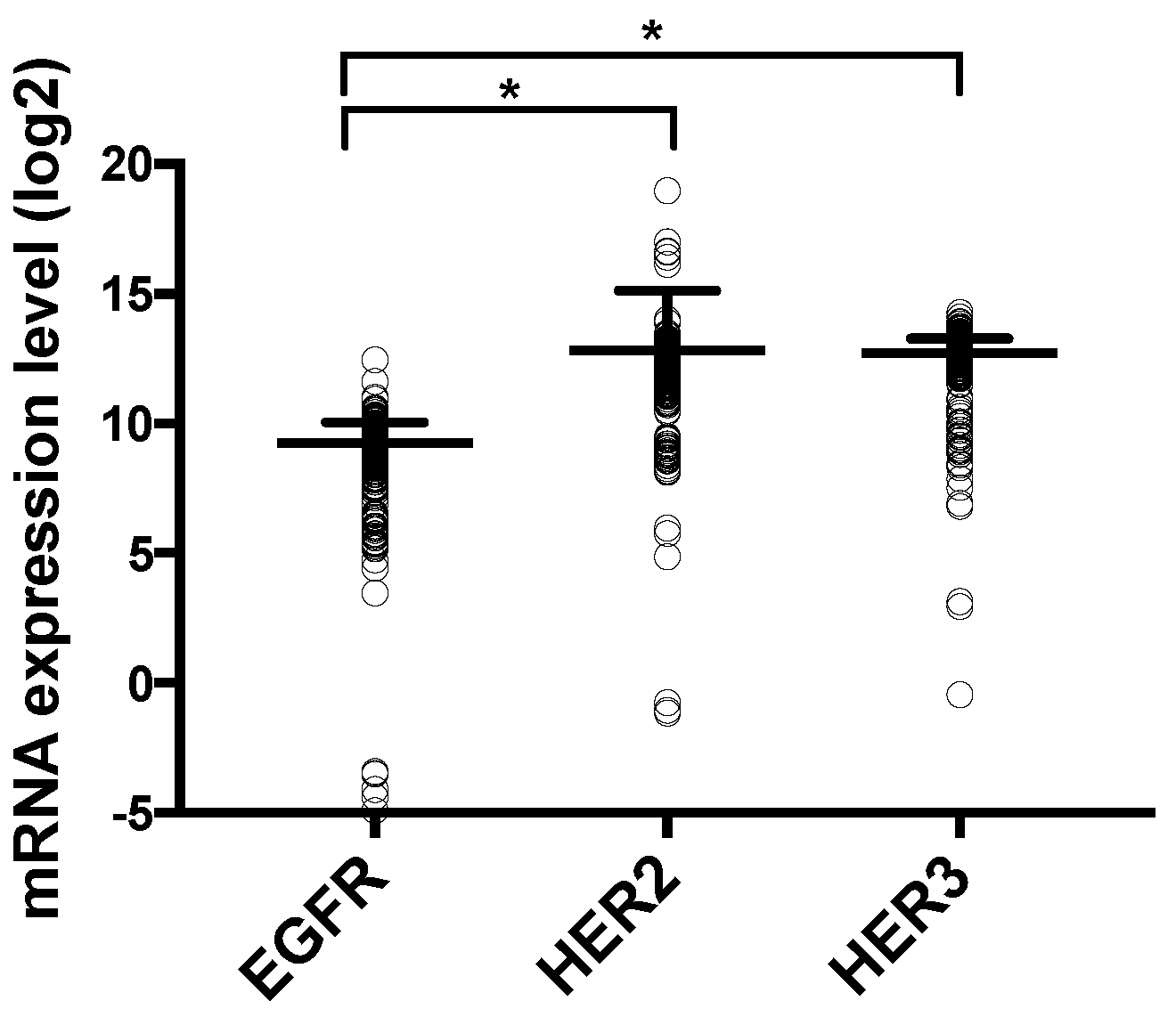{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Afatinib 72 h (μM) | |||
|---|---|---|---|
| Cell Line | Cetuximab Resistance Status | Normoxia (21% O2) | Hypoxia (1% O2) |
| Lim1215 | Sensitive | 0.081 ± 0.021 | 0.178 ± 0.055 |
| CaCo2 | Sensitive | 0.341 ± 0.199 | 0.604 ± 0.307 |
| SW48 | Intrinsically resistant | 2.379 ± 0.869 | 2.109 ± 0.691 |
| HT29 | Intrinsically resistant | 1.805 ± 0.041 | 1.816 ± 0.117 |
| Lim1215-PBS | PBS-treated control, sensitive | 0.007 ± 0.002 | 0.010 ± 0.003 |
| Lim1215-R | Acquired resistance | 0.174 ± 0.030 | 0.458 ± 0.060 |
| CaCo2-PBS | PBS-treated control, sensitive | 0.591 ± 0.384 | 0.260 ± 0.197 |
| CaCo2-R | Acquired resistance | 1.570 ± 0.264 | 1.398 ± 0.270 |
| Cell Line | Condition | IC50 (μM) | p-Value | CI |
|---|---|---|---|---|
| Lim1215-PBS | 72 h 0 μM afatinib→24 h irinotecan | 0.28 ± 0.26 | / | / |
| 72 h 0.001 μM afatinib→24 h irinotecan | 0.36 ± 0.19 | 0.757 | 0.92 ± 0.12 | |
| 72 h 0.005 μM afatinib→24 h irinotecan | 0.39 ± 0.16 | 0.681 | 0.94 ± 0.08 | |
| 24 h irinotecan→72 h 0μM afatinib | 0.11 ± 0.04 | / | / | |
| 24 h irinotecan→72 h 0.001 μM afatinib | 0.09 ± 0.04 | 0.994 | 1.02 ± 0.04 | |
| 24 h irinotecan→72 h 0.005 μM afatinib | 0.10 ± 0.04 | 0.488 | 1.10 ± 0.07 | |
| Lim1215-R | 72 h 0 μM afatinib→24 h irinotecan | 1.48 ± 0.99 | / | / |
| 72 h 0.02 μM afatinib→24 h irinotecan | 0.64 ± 0.36 | 0.458 | 0.90 ± 0.07 | |
| 72 h 0.1 μM afatinib→24 h irinotecan | 0.44 ± 0.19 | 0.417 | 0.81 ± 0.10 | |
| 24 h irinotecan→72 h 0 μM afatinib | 0.09 ± 0.02 | / | / | |
| 24 h irinotecan→72 h 0.02 μM afatinib | 0.08 ± 0.02 | 0.235 | 0.79 ± 0.10 | |
| 24 h irinotecan→72 h 0.1 μM afatinib | 0.06 ± 0.02 | 0.116 | 0.72 ± 0.13 | |
| CaCo2-PBS | 72 h 0 μM afatinib→24 h irinotecan | ND | / | / |
| 72 h 0.05 μM afatinib→24 h irinotecan | ND | / | 1.00 ± 0.07 | |
| 72 h 0.3 μM afatinib→24 h irinotecan | ND | / | 1.03 ± 0.03 | |
| 24 h irinotecan→72 h 0 μM afatinib | 0.46 ± 0.15 | / | / | |
| 24 h irinotecan→72 h 0.05 μM afatinib | ND | / | 1.22 ± 0.32 | |
| 24 h irinotecan→72 h 0.3 μM afatinib | ND | / | 1.22 ± 0.29 | |
| CaCo2-R | 72 h 0 μM afatinib→24 h irinotecan | ND | / | / |
| 72 h 0.75 μM afatinib→24 h irinotecan | ND | / | 0.99 ± 0.08 | |
| 72 h 1.25 μM afatinib→24 h irinotecan | ND | / | 1.00 ± 0.09 | |
| 24 h irinotecan→72 h 0 μM afatinib | 0.55 ± 0.28 | / | / | |
| 24 h irinotecan→72 h 0.75 μM afatinib | ND | / | 1.14 ± 0.20 | |
| 24 h irinotecan→72 h 1.25 μM afatinib | ND | / | 1.22 ± 0.18 | |
| HT29 | 72 h 0 μM afatinib→24 h irinotecan | 40.07 ± 4.59 | / | / |
| 72 h 2 μM afatinib→24 h irinotecan | 18.96 ± 3.29 | 0.051 | 1.12 ± 0.11 | |
| 72 h 3 μM afatinib→24 h irinotecan | 14.73 ± 2.77 | 0.023 | 1.10 ± 0.07 | |
| 24 h irinotecan→72 h 0 μM afatinib | 4.34 ± 1.06 | / | / | |
| 24 h irinotecan→72 h 2 μM afatinib | 4.74 ± 0.61 | 0.993 | 1.02 ± 0.06 | |
| 24 h irinotecan→72 h 3 μM afatinib | 5.52 ± 0.69 | 0.566 | 1.10 ± 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Pauw, I.; Lardon, F.; Van den Bossche, J.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Wouters, A. Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib. Cancers 2019, 11, 98. https://doi.org/10.3390/cancers11010098
De Pauw I, Lardon F, Van den Bossche J, Baysal H, Pauwels P, Peeters M, Vermorken JB, Wouters A. Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib. Cancers. 2019; 11(1):98. https://doi.org/10.3390/cancers11010098
Chicago/Turabian StyleDe Pauw, Ines, Filip Lardon, Jolien Van den Bossche, Hasan Baysal, Patrick Pauwels, Marc Peeters, Jan Baptist Vermorken, and An Wouters. 2019. "Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib" Cancers 11, no. 1: 98. https://doi.org/10.3390/cancers11010098
APA StyleDe Pauw, I., Lardon, F., Van den Bossche, J., Baysal, H., Pauwels, P., Peeters, M., Vermorken, J. B., & Wouters, A. (2019). Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib. Cancers, 11(1), 98. https://doi.org/10.3390/cancers11010098