1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and the fourth most common cause of cancer death. There are about 1,250,000 new CRC cases and more than 600,000 related deaths every year [
1]. CRC is considered a model of choice for cancer chemoprevention studies for two reasons: First, the molecular history of CRC is well established and involves early mutations in the adenomatous polyposis coli (APC) gene linked to hyperplasia followed by mutations in oncogenes and tumor repressors as in c-myc, K-ras, and p53 that lead to adenoma, adenocarcinoma, and carcinoma [
2]. Second, colonoscopy enables monitoring and removal of benign polyps, thus, allowing the monitoring of the disease and assessing the efficiency of chemoprevention treatments.
CRC chemoprevention studies using cell lines, animal models, and even clinical trials which have provided multiple candidate compounds for chemoprevention. In top of most lists of cancer chemoprevention compounds is Difluoromethylornithine (DFMO), also named eflornithine [
3,
4]. DFMO is a suicide inhibitor of ornithine decarboxylase 1 (ODC1), the limiting step in the biosynthesis of polyamines by animal cells. Therefore, an excess of polyamine biosynthesis is involved in cancer, particularly in CRC. Consistently with this view, ODC is overexpressed in most CRC and other forms of cancer [
4,
5]. ODC polymorphisms have been also reported in CRC. In addition, there is strong correlation between specific polyamines and tissue growth, and different growth stimuli induce ODC activity. Moreover, a variety of tumor promoters induces ODC1 and tumor formation [
4,
5]. Metcalf et al. reported the synthesis of DFMO, a highly targeted drug whose mechanism involved irreversible inhibition of ODC1 [
3]. DFMO inhibits colon carcinogenesis in rodent models including the
ApcMin/+ mice with increased levels of ODC and polyamines in intestinal tissues. Administration of DFMO alone is effective in suppressing carcinogenesis in the small intestines of these mice. Combinations of DFMO and non-steroidal anti-inflammatory drugs (NSAIDs) are potent inhibitors of carcinogenesis in both the large and small intestines of these mice [
5]. The grade of intestinal polyps is polyamine-dependent and the anti-carcinogenic effects can be rescued by putrescine. These findings support a role for polyamines in intestinal carcinogenesis in
ApcMin/+ mice. Moreover, in the last 10 years, several clinical trials indicate that DFMO may prevent CRC, particularly when provided in combination with NSAIDs, such as sulindac [
6,
7,
8]. In fact, there is ongoing a large clinical trial, the S0820 Preventing Adenomas of the Colon with Eflornithine and Sulindac (PACES) trial, that is presently evaluating the effectiveness of the combination of eflornithine and sulindac in preventing colon adenomas that may change CRC chemoprevention [
9]. However, in spite of the clinical relevance, the mechanisms by which polyamines affect cancer hallmarks and carcinogenesis remain to be established.
At the physiological level, polyamines have been involved in epithelial restitution, a process of transient activation of cell migration and/or proliferation after wounding for epithelial tissue repair. Recent data suggest that this process could be mediated by triggered Ca
2+ influx operated by transient receptor potential channel 1 (TRPC1) and involves changes in expression of stromal interaction molecules STIM1 and STIM2 [
10]. In addition, it has been shown that the caveolae protein caveolin1 [
11], and the small guanosine-5’-triphosphate-binding protein RhoA [
12], interact with and activates TRPC1 to stimulate rapid epithelial restitution after injury by inducing Ca
2+ signaling. TRPC1 primarily functions as a cation non-selective channel within pathways controlling Ca
2+ entry in response to cell surface receptor activation [
13,
14]. TRPC1, described for the first time in 1995 [
13], was initially reported to be the ion channel involved in store-operated Ca
2+ entry (SOCE), the most important Ca
2+ entry pathway in non-excitable cells [
15]. However, this view has been controversial since TRPC1 induces a non-selective cation current quite different from the small, Ca
2+-release activated current (CRAC) that is very selective for Ca
2+ first reported in mast cells [
16]. This was solved in 2006 after the discovery of Orai1 channels [
15]. At the molecular level, SOCE is activated after depletion of intracellular Ca
2+ stores, a process sensed by the stromal interaction protein 1 (STIM1), that oligomerizes and interacts with Orai1 channels in the plasma membrane [
17]. Now, the most extended view is that in some cells, SOCE is mediated solely by Orai1 channels while in others, TRPC1 may form ion channel complexes with Orai1 where TRPC1 tunes SOCE [
18]. Interestingly, SOCE and molecular players involved in SOCE have been recently involved in carcinogenesis of CRC and other forms of cancer [
19,
20,
21]. These data invite speculation on whether DFMO could prevent CRC acting on molecular players involved in SOCE.
We have recently reported that intracellular Ca
2+ homeostasis is remodeled in CRC [
22]. In short, CRC cells display enhanced SOCE and decreased Ca
2+ store content relative to normal colonic cells and these changes contribute to cancer hallmarks, such as increased cell proliferation, cell invasion and resistance to apoptosis [
22]. At the molecular level, enhanced SOCE is associated to increased expression of Orai1, STIM1, and TRPC1 in CRC cells, and decreased Ca
2+ store content has been associated to decreased expression of STIM2 [
22,
23] and other genes involved in intracellular Ca
2+ homeostasis [
24]. Store-operated channels (SOCs) are quite different in normal and colon cancer cells. Specifically, normal colonic cells display typical CRAC like currents driven by Orai1 channels. These are very small, Ca
2+-selective, inward rectifying currents. However, CRC cells display larger, non-selective currents with both inward and outward components that are mediated by both Orai1 and TRPC1 channels [
22]. Interestingly, aspirin, and other NSAIDs including sulindac, that have been reported to prevent CRC [
25,
26], inhibit SOCE and cell proliferation and migration in CRC cells [
27,
28] suggesting SOCE molecular players as targets for cancer chemoprevention. Inhibition of SOCE and SOCs by salicylate (aspirin) and other NSAIDs is not direct. NSAIDs act as mild mitochondrial uncouplers, thus, promoting loss of mitochondrial potential, the driving force for mitochondrial Ca
2+ uptake [
27,
28]. Accordingly, NSAIDs-induced defective mitochondrial Ca
2+ uptake promotes Ca
2+-dependent inactivation of SOCs and SOCE inhibition in CRC cells and other cell types [
29,
30]. Therefore, as SOCs are enhanced in CRC and modulated during epithelial restitution, we asked whether DFMO may influence Ca
2+ channel remodeling in CRC.
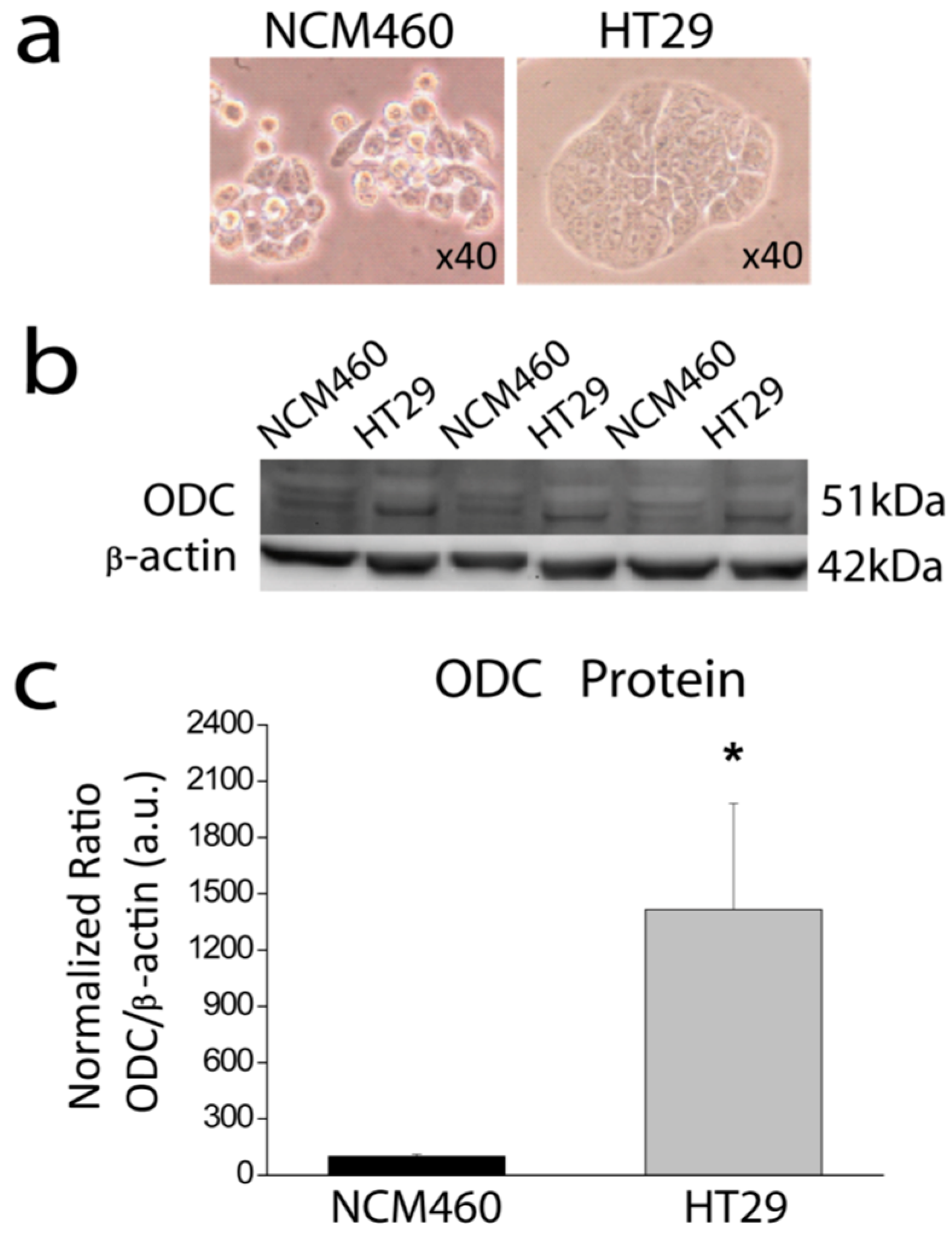
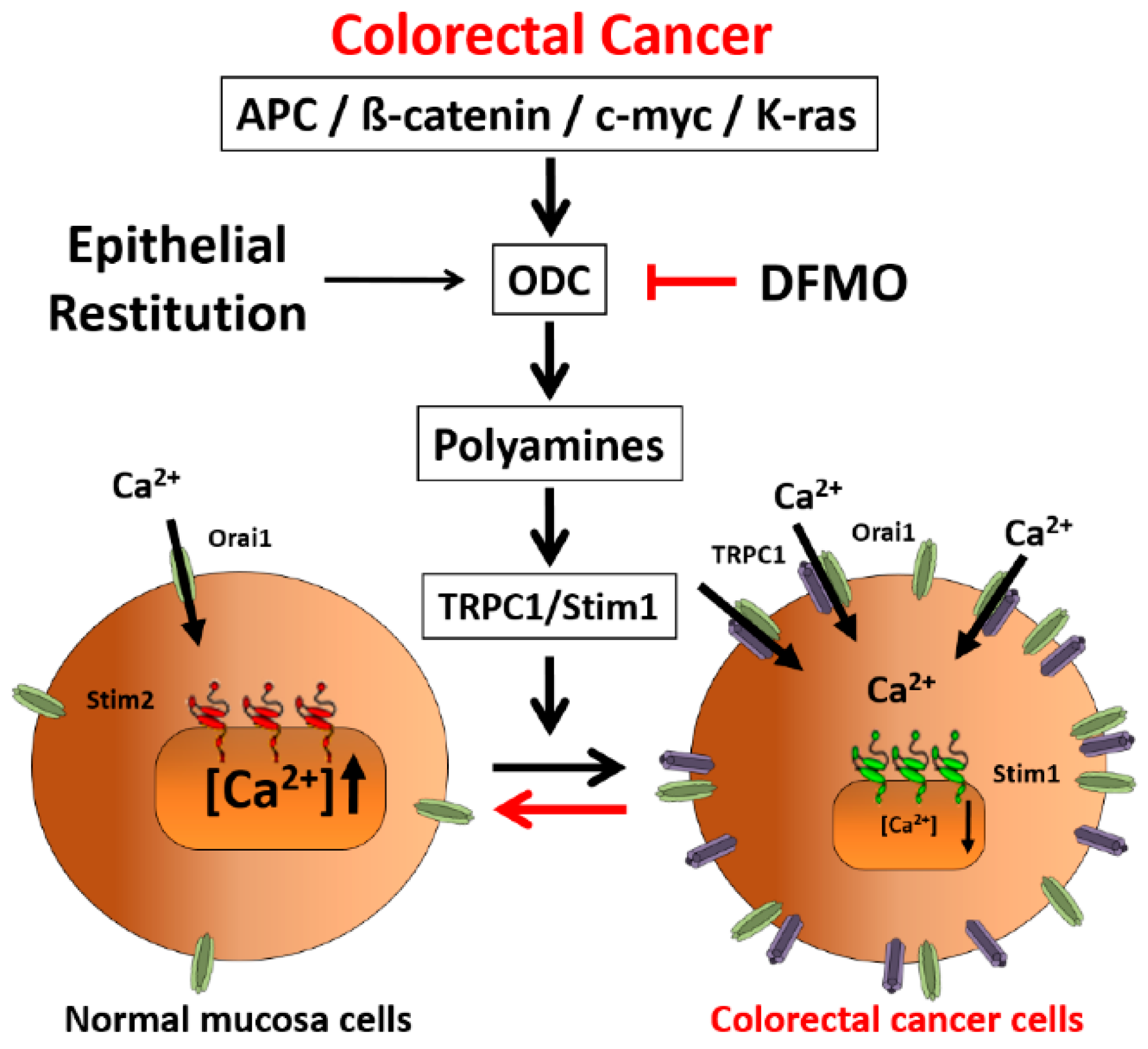
Here we have investigated the expression of ODC in CRC cells and the effects of DFMO on Ca2+ channel remodeling in CRC cells. We found that ODC is overexpressed in CRC cells. In addition, we found that ODC suicide inhibitor DFMO inhibits expression of TRPC1 and STIM1 in CRC cells, thus, reversing Ca2+ channel remodeling in CRC and providing a mechanism for cancer chemoprevention by polyamine synthesis inhibition.
3. Discussion
CRC is associated with the remodeling of intracellular Ca
2+ homeostasis [
20,
23]. Specifically, we have reported recently that colon cancer cells display decreased Ca
2+ store content and enhanced and modified SOCE [
22], the most important Ca
2+ entry pathway in non-excitable cells [
15]. In addition, we showed that these changes contributed to cancer cell hallmarks including increased apoptosis resistance and enhanced migration and proliferation of cancer cells [
22]. Here we show that DFMO, a suicide inhibitor of ODC, the limiting step in polyamine biosynthesis, previously reported to prevent CRC in vivo and in vitro, reverses this remodeling. These results highlight the role of Ca
2+ remodeling in carcinogenesis, suggest that Ca
2+ remodeling is promoted by excess polyamine biosynthesis, and finally, provide a mechanism of CRC chemoprevention by DFMO and its combination with sulindac.
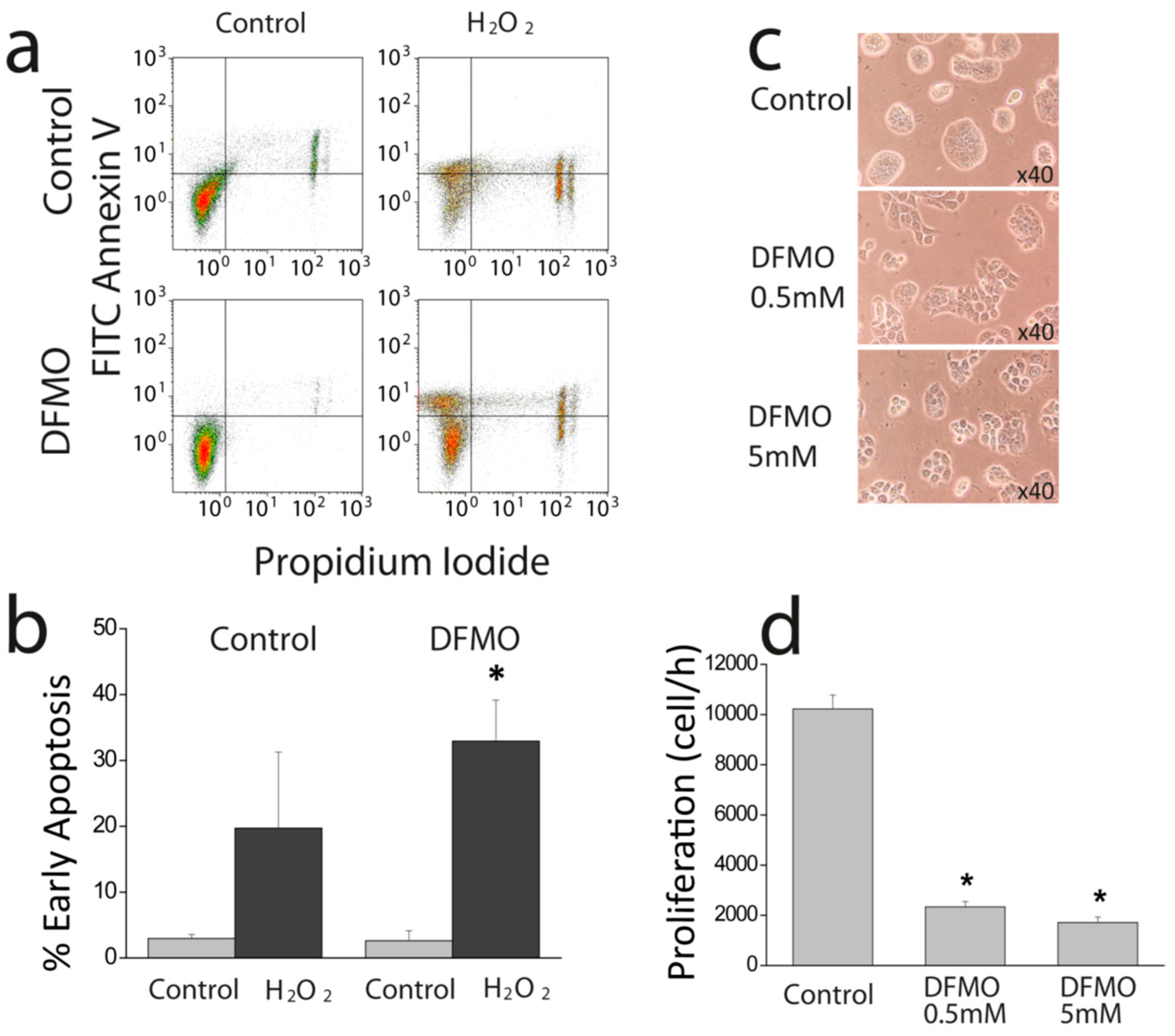
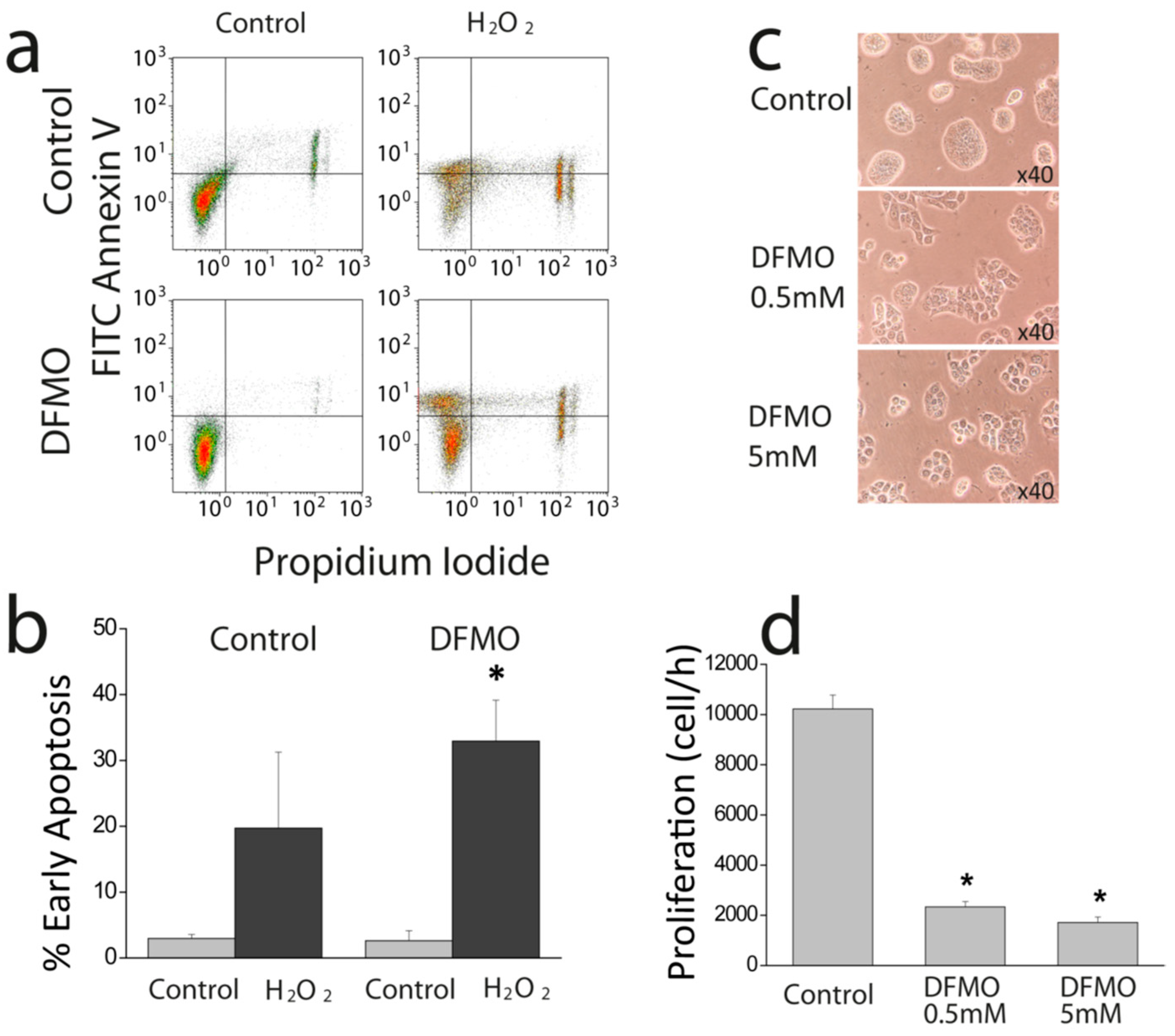
First, we show that DFMO treatment increases the rise in (Ca
2+)
cyt induced by CPA in Ca
2+ free medium and enhances early apoptosis induced by oxidative stress. This effect may be explained by enhanced Ca
2+ store content and/or defective Ca
2+ clearance in DFMO-treated cells. However, as resting levels of (Ca
2+)
cyt are not influenced by DFMO, the results suggest that DFMO modulates mainly Ca
2+ store content. Ca
2+ store content is related to the intrinsic pathway of apoptosis that involves mitochondria. In this pathway, mitochondrial Ca
2+ overload induced by Ca
2+ entry and/or release from intracellular stores favors mitochondrial permeability transition followed by cytochrome c release and activation of the apoptosome. We recently reported that colon cancer HT29 cells display decreased Ca
2+ store content relative to normal colonic NCM460 cells [
22] Consistently, HT29 cells were resistant to apoptosis induced by oxidative stress (H
2O
2) compared to normal cells [
22]. Moreover, we also showed that decreasing Ca
2+ store content in normal NCM460 colonic cells enhanced resistance to apoptosis induced by the same treatment. Therefore, it follows logically that the increased Ca
2+ store content induced by DFMO may reverse this feature of the tumor cell phenotype, thus, decreasing resistance to apoptosis induced by H
2O
2. In addition, the low Ca
2+ store content of tumor cells also favors activation of SOCE by physiological agonists that release only of a fraction the stored Ca
2+ and promote a partial depletion of intracellular stores. Accordingly, the rise in Ca
2+ store content induced by DFMO treatment may also constrain SOCE activation in tumor cells in physiological conditions.
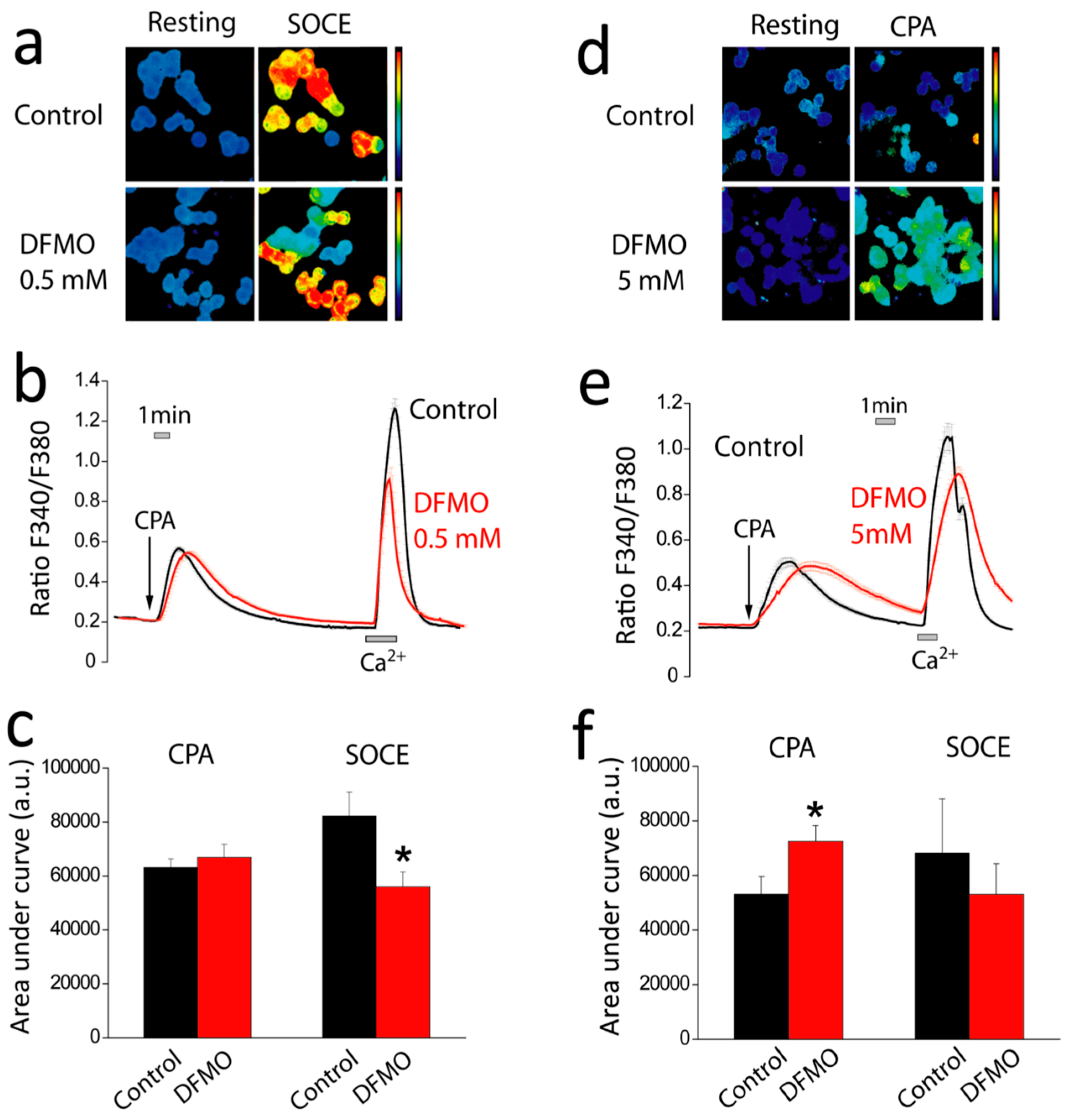
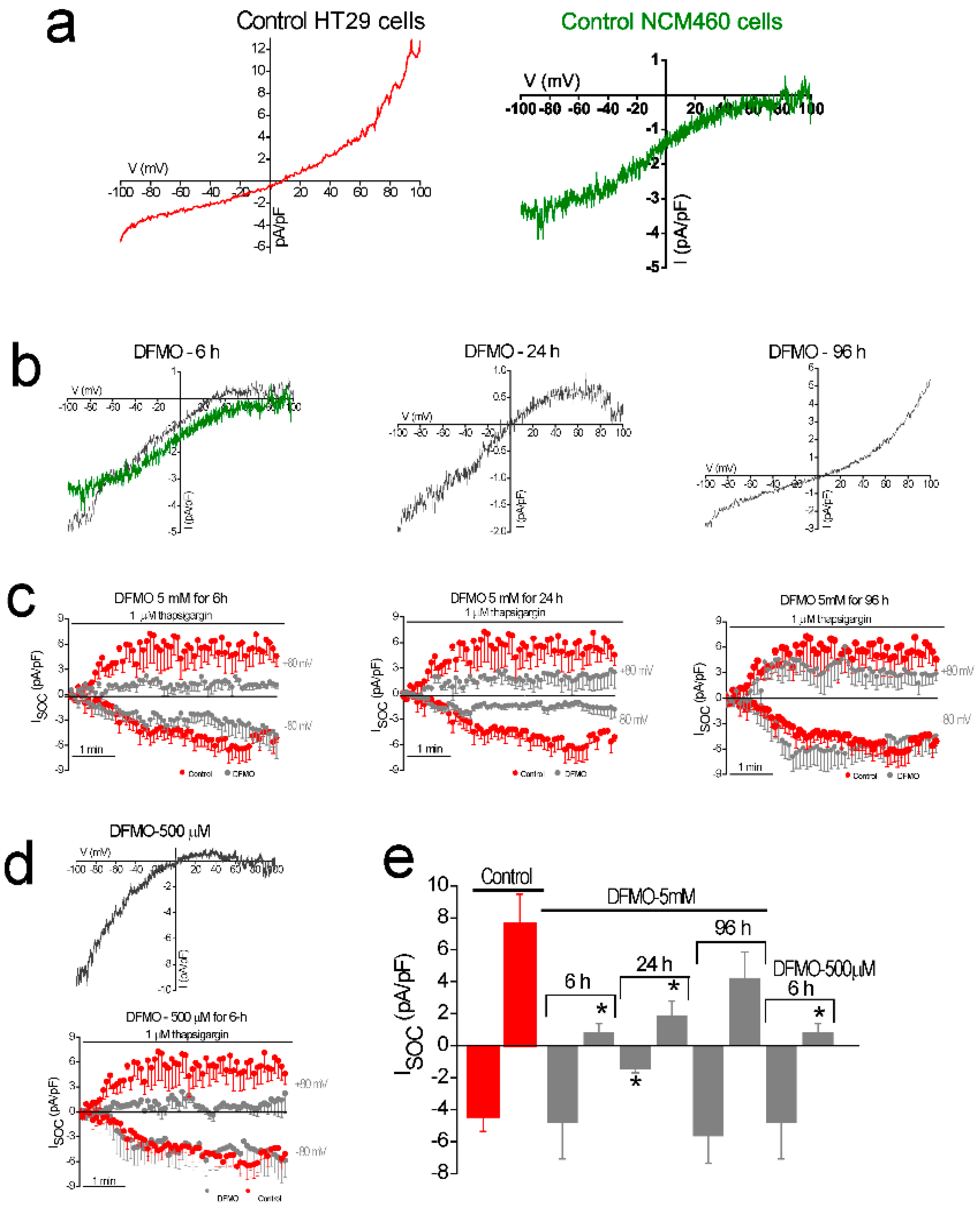
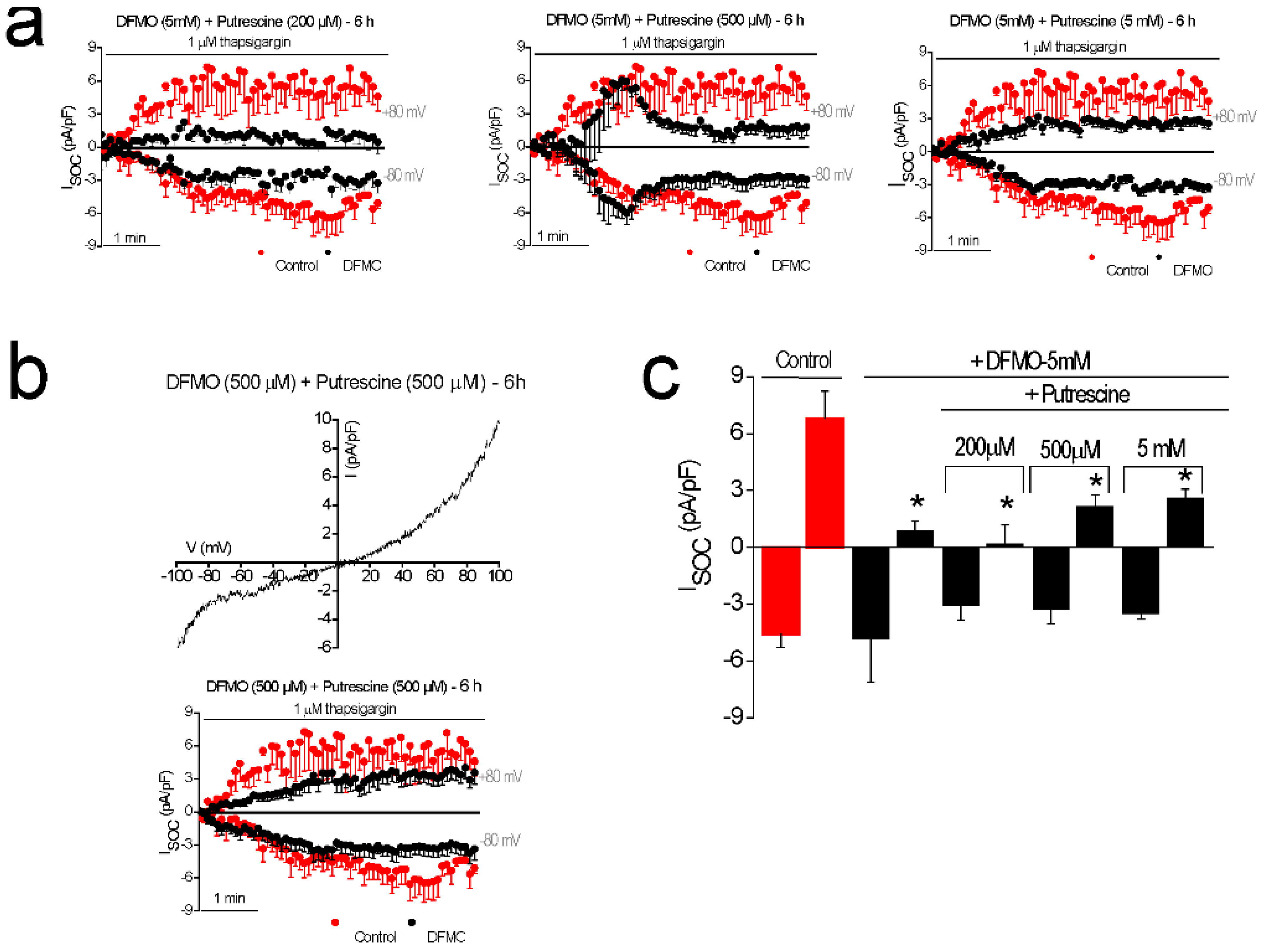
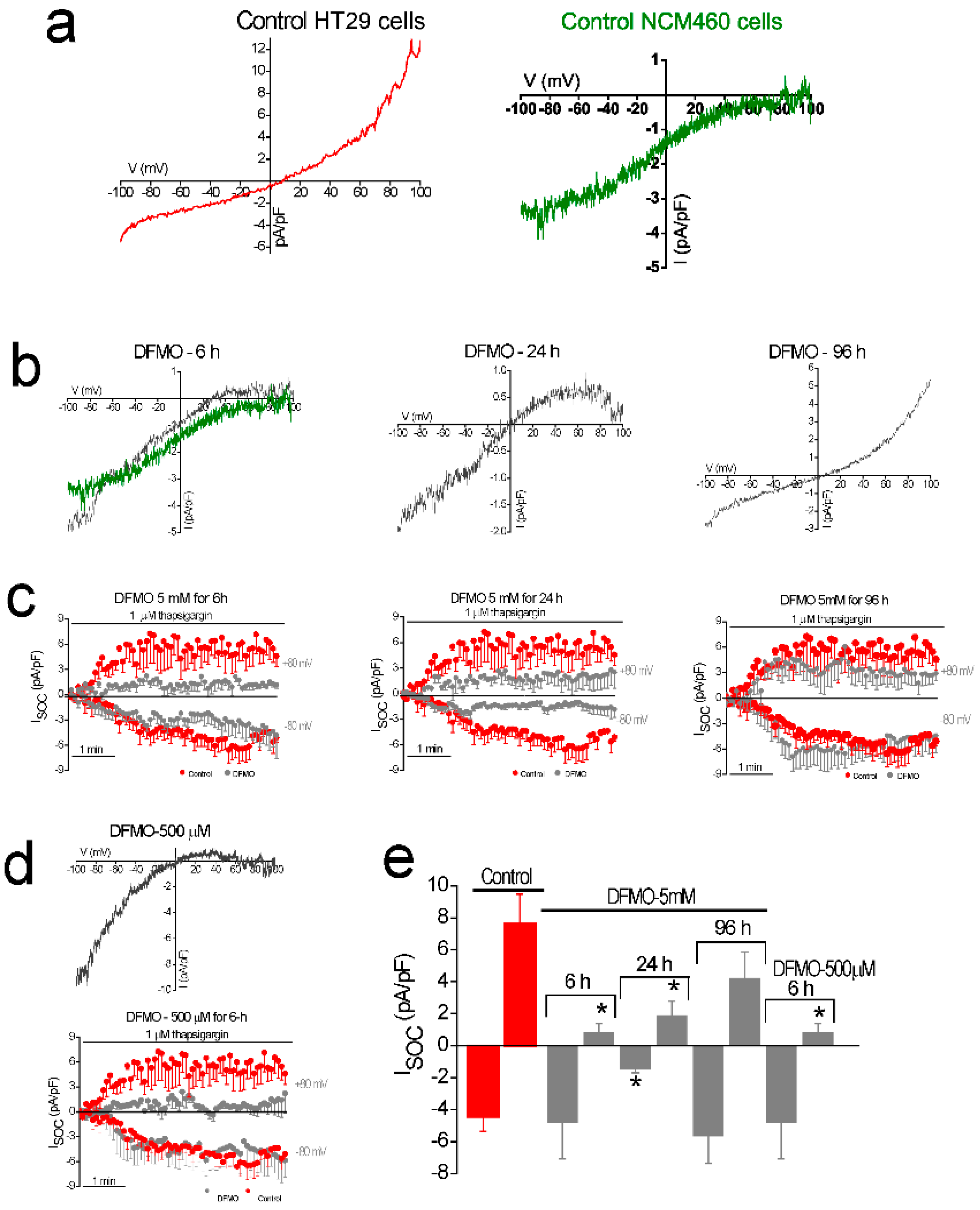
In addition, DFMO treatment removes the non-selective component of SOCs that is solely present in CRC cells but not in normal colonic cells. As stated above, CRC cells display enhanced SOCE, and this increase correlates with enhanced cell proliferation and migration [
22]. Consistently, DFMO treatment decreases both SOCE and cell proliferation in CRC cells. We reported previously that SOCs in normal colonic cells display only a Ca
2+-selective inward current driven by Orai1 channels [
22]. In contrast, CRC cells display larger currents with two components, a Ca
2+ selective inward component and a non-selective, inward and outward component [
22]. In contrast to normal cells, currents in CRC cells are driven by both Orai1 and TRPC1 channels, probably forming ion channel complexes specific of CRC cells. The I
SOC of HT29 cancer cells is significantly larger compared with the I
SOC from NCM460 normal colon cells. In addition, the I
SOC of HT29 does not display evident desensitization [
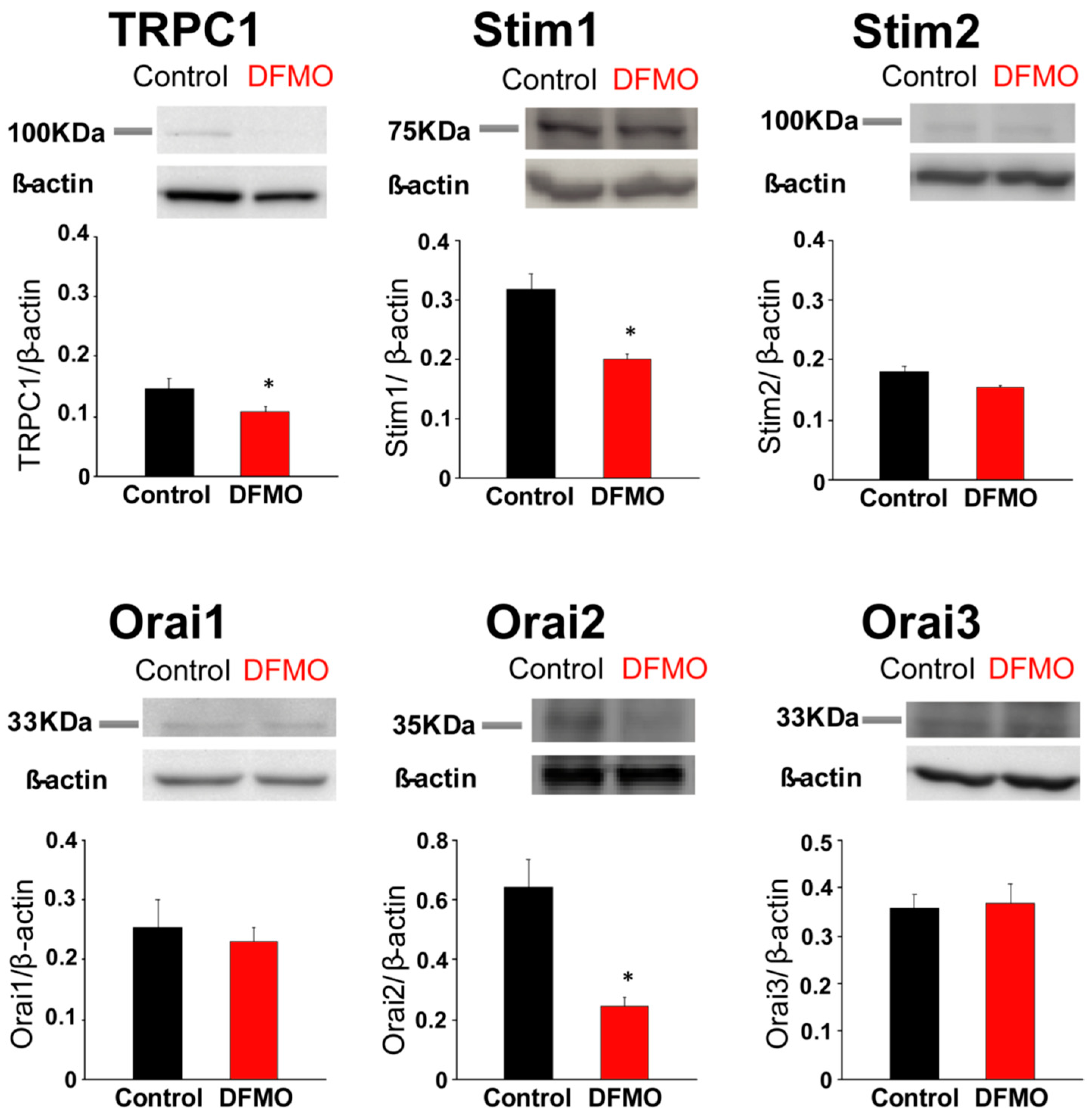
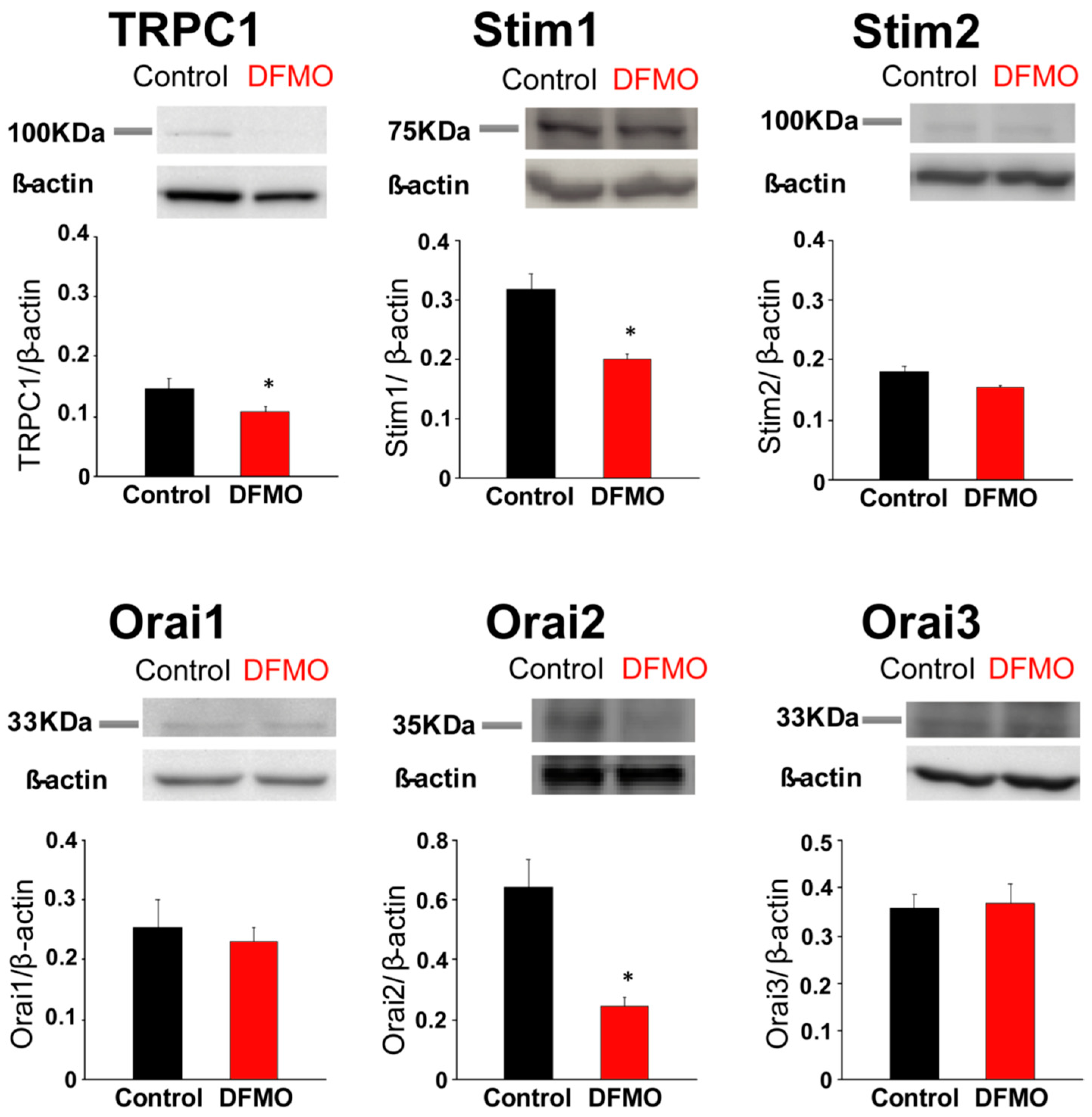
31], likely contributing to sustained signaling in cancer cells. We report now that this TRPC1-dependent component observed only in CRC cells is selectively removed by treatment with DFMO and this effect is fully reversed by an excess of polyamine putrescine indicating that DFMO removes TRPC1 by preventing polyamine biosynthesis. Consistently, we show also that DFMO treatment decreases expression of both TRPC1 channels and STIM1, the sensor that gates Orai1/TRPC1 channel complexes present in CRC cells. Therefore, these results indicate that DFMO reverses the remodeling of SOCs in CRC and the mechanism involves changes in expression of TRPC1 and STIM1 (
Figure 9).
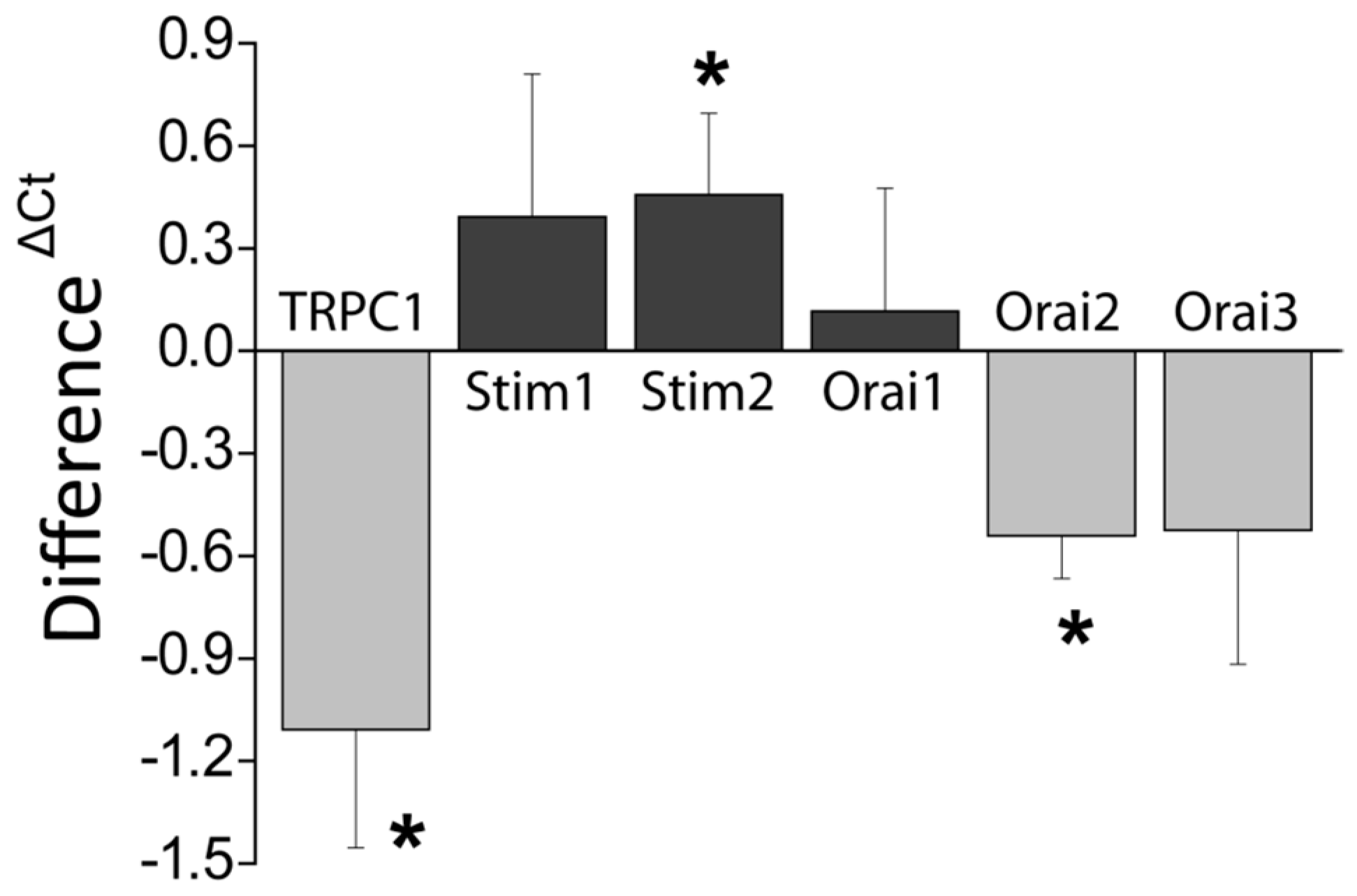
Consistently with the changes in Isoc, we show also that DFMO treatment decreases expression of both TRPC1 channels and STIM1, the sensor that gates Orai1/TRPC1 channel complexes present in CRC cells. DFMO also inhibits expression of ORAI2. However, this isoform does not seem to play any role in SOCE or SOCs in HT29 cells [
22]. In contrast, DFMO has no effect on STIM2 expression at the protein level, in spite of increasing expression at the messenger level. Interestingly, we reported that colon cancer HT29 cells display enhanced STIM1 expression along decreased STIM2 expression relative to normal NCM460 colonic cells [
22], thus, resulting in enhanced STIM1 to STIM2 ratio in cancer cells. In spite that STIM2 protein expression does not change with DFMO, the ratio STIM1/STIM2 is indeed reduced by DFMO treatment consistently with reversal of the tumor phenotype. In addition, our results suggest that inhibition of TRPC1 by DFMO may occur at the transcriptional level while inhibition of STIM1 expression is post-transcriptional. Although further studies are needed to elucidate the DFMO mechanism, our results indicate that it reverses the remodeling of I
SOC in CRC and the mechanism involves changes in expression of TRPC1 and STIM1 (
Figure 9).
Our results indicate not only the mechanism of CRC chemoprevention by polyamine synthesis inhibition with DFMO but also suggest that Ca
2+ channel remodeling is an important step in CRC carcinogenesis and that this process is likely mediated by an excess of polyamine biosynthesis linked to ODC overexpression. It is well known that mutations in the
adenomatous polyposis coli (APC) gene are responsible for
familial adenomatous polyposis (FAP). In addition, APC play a rate-limiting role in most sporadic colorectal cancers, particularly by impinging on the WNT pathway [
2]. In normal conditions, the WNT receptor is activated and transmits the signal intracellularly to the APC, glycogen synthase kinase-3β (GSK-3β) and β-catenin complex. GSK-3β phosphorylates β-catenin, marking it for proteosomal degradation [
2]. In carcinogenesis, the WNT receptor may be activated, and transmission of the signal intracellularly may occur, but inactivation of β-catenin by GSK-3β does not. Cytoplasmic accumulation of β-catenin leads to its nuclear translocation and binding with its cognate partner TCF/LEF. This heterodimerization regulates genes through transcription, notably of C-MYC. MYC and K-RAS transcriptionally activate a number of polyamine metabolic genes, including ODC that converts ornithine to putrescine and other polyamines. Our present findings showing that polyamine synthesis inhibition reverses remodeling of SOCs from TRPC1/Orai1 channels characteristic of cancer cells to the classic Orai1 channels reported in normal cells suggest that channel remodeling is critical to the tumorigenic pathway induced by polyamines. This view is supported by the fact that the effect of DFMO on channel remodeling is reversed by putrescine. In addition, this possibility is also supported by a previous report showing that polyamines may regulate intestinal epithelial restitution through TRPC1-mediated Ca
2+ signaling by altering the ratio of STIM1 to STIM2. As stated above, epithelial restitution is a physiological mechanism activated for tissue repair after the damage that involves the transient activation of cell migration and proliferation [
11,
12]. Polyamines are essential for epithelial restitution. As ODC is highly overexpressed in cancer, CRC can be envisioned as the result of activation of a program of chronic epithelial restitution process in the absence of damage.
DFMO is considered one of the best candidates for CRC chemoprevention. DFMO is particularly effective when combined with the NSAID sulindac. In fact, several clinical trials are testing the combination of sulindac and DFMO [
6,
7,
8]. For example, this combination has been tested in people who had already had adenomas removed within the preceding 5 years. In that study, the development of high-risk adenomas and multiple adenomas was reduced by more than 90 percent in people who took the combination (
n = 191 patients) compared with people who took the placebo (
n = 184 patients) [
6]. This preliminary study is the base of an ongoing phase III, Double-Blind Placebo-Controlled Trial of DFMO and sulindac to prevent the recurrence of high-risk adenomas and second primary colorectal cancers in patients with stage 0 to III colon cancer [
9]. Interestingly, we have reported recently that sulindac is able to inactivate store-operated channels in colonic cells by mitochondria-dependent mechanism [
13]. The mechanism is based on the ability of sulindac and other NSAIDs of acting as mild mitochondrial uncouplers that depolarize mitochondria partially, thus, limiting mitochondrial Ca
2+ uptake. Consequently, Ca
2+ entering through SOCE is not taken by mitochondria leading to Ca
2+-dependent inactivation of SOCs [
29,
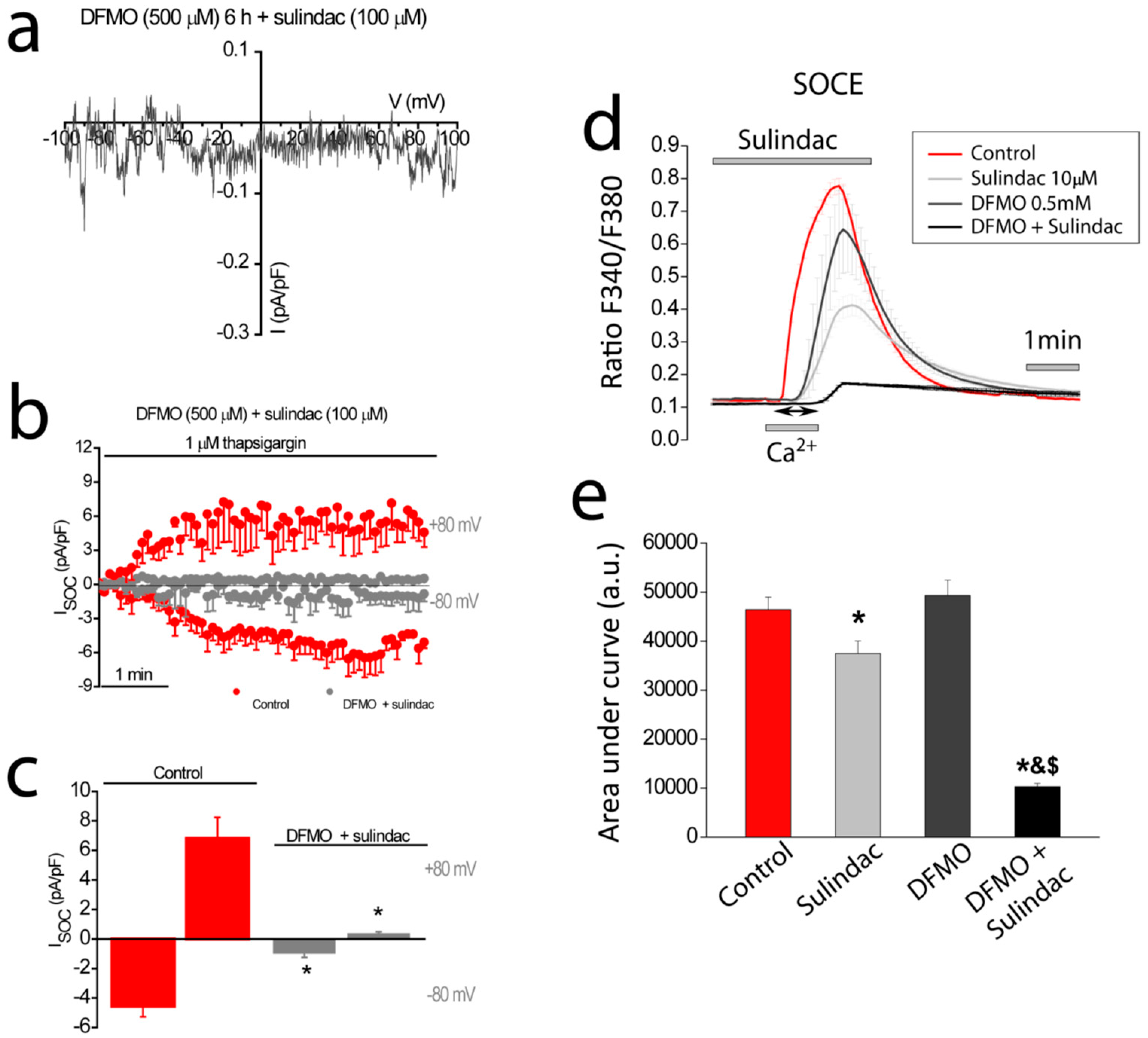
30]. This mechanism is highly efficient in limiting SOCE in normal cells but not as effective in CRC cells where TRPC1 channels do not inactivate. Here we show that DFMO treatment removes this component in CRC cells but not the Ca
2+-selective component that is sensitive to sulindac. Consistently, we show that DFMO and sulindac are able to remove both components of SOCs in CRC cells, thus, abolishing SOCE in CRC cells and providing a mechanism for increased chemoprevention efficiency of the combination vs. therapy with a single compound.
In summary, our results suggest that Ca2+ channel remodeling that is associated to CRC and cancer hallmarks may be linked to excess polyamine biosynthesis associated with APC and/or K-ras mutations. In addition, they provide evidence that polyamine biosynthesis inhibition reverses Ca2+ channel remodeling in colon cancer cells, thus, providing a mechanistic basis for CRC chemoprevention. Finally, our results also provide evidence that the combination of both DFMO and sulindac inhibit both components of SOCs, the inward and outward components driven by both Orai1 and TRPC1 channels leading to efficient inhibition of SOCs and CRC chemoprevention.
4. Materials and Methods
4.1. Materials
HT29 cells were donated by Dr. J.C. Fernández-Checa (CSIC, Barcelona, Spain). Dulbecco’s Modified Eagle’s Medium (DMEM), Penicillin, streptomycin, L-glutamine, and fetal bovine serum is from Lonza (Basel, Switzerland). NCM460 were obtained after a material transfer agreement with INCELL Corporation (San Antonio, TX, USA). M3:10TM medium is from INCELL Corporation (San Antonio, TX, USA). Detachin is from Gelantis (San Diego, CA, USA). Fura-2/AM is from Invitrogen (Carlsbad, CA, USA). Thapsigargin is from Alomone Labs (Jerusalem, Israel). Primers were obtained from Thermo Scientific (Ulm, Germany). Antibodies against TRPC1, Orai1, Orai2, and STIM1 were from Alomone Labs (Jerusalem, Israel). Antibodies against STIM2 and ORAI3 were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-β-actin was from Abcam (Cambridge, UK)
4.2. Cell Culture
Cells were cultured in DMEM 1 g/L glucose (HT29) or in M3:10TM medium (NCM460) as reported previously [
22] and supplemented with 1% Penicillin-Streptomycin, 1% L-glutamine and 10% fetal bovine serum. Cells were maintained in standard conditions (37 °C, 10% CO
2) and subcultured once a week. All cells were used for experiments at passages 3 to 10.
4.3. Apoptosis Assays
Cell survival assay was performed by flow citometry using FITC Annexin V (BD Biosciences, Franklin Lakes, NJ, USA) and propidium iodide (Sigma, Steinheim, Germany). Cells were cultured with media containing solvent or DFMO for several days. Then cells were treated with solvent or 2 mM H2O2 for 150 min and then detached with trypsin-EDTA, centrifuged at 290 g and washed with cold PBS. Then, cells were suspended in binding buffer (0.1 M Hepes, pH 7.4, NaCl 1.4 M and 25 mM CaCl2) at a density of 1 × 106 cells/ml. After that, 1 × 105 cells were incubated with 5 µl of Annexin V and 10 µl of propidium iodide (50 µg/ml) for 15 min at room temperature in the dark. Cells were analyzed using Gallios Flow Cytometer (Beckman Coulter, Brea, CA, USA) and the results were processed with Kaluza Analysis Software (Beckman Coulter, Brea, CA, USA).
4.4. Cell Proliferation
Cells were plated in 6 well plates at a density of 10 × 105 cells and incubated with supplemented DMEM or the same medium containing DFMO. Cells in wells were counted in triplicate at time 0 h and after 72, or 96 h using a hemocytometer. Cell viability was estimated using trypan blue staining.
4.5. Fluorescence Imaging of Cytosolic Ca2+
Cytosolic Ca
2+ concentration ((Ca
2+)
cyt) was monitored as reported previously [
22] by fluorescence imaging of cells using an inverted Zeiss Axiovert microscope equipped with a OrcaER Hamamatsu digital camera (Hamamatsu, Photonics, France). Cells were loaded with fura-2/AM (4 µM, 60 min) in external saline solution containing (in mM): 145 NaCl, 5 KCl, 1 CaCl
2, 1 MgCl
2, glucose 10, Hepes/Na 10 (pH 7.42). For SOCE, cells were washed twice and treated with CPA or thapsigargin (1 µM, 10 min) in the same medium except that it was devoid of Ca
2+ and contained also 0.5 mM EGTA. Then cells were located in the stage of an inverted microscope and subjected to fluorescence imaging whereas continuously perfused with external medium at 37 °C. Cells are epi-illuminated alternately at 340 and 380 nm using band pass filters and light emitted above 520 nm at both excitation lights was filtered by the dichroic mirror, collected every 5 to 10 s with a 40×, 1.4 NA, oil objective.
4.6. Electrophysiological Recordings
I
SOC were recorded using a Port-a-patch planar patch-clamp system (Nanion Technologies, Munich, Germany) in the whole-cell, voltage-clamp configuration, at room temperature (20 ± 2 °C) as reported previously [
22]. Cultured cells (3–5 days after plating) were detached with Detachin and suspended at a cell density of 1–5 × 10
6 cells/mL in external recording solution containing (in mM): 145 NaCl, 2.8 KCl, 2 MgCl
2, 10 CaCl
2, 10 HEPES, 10 D-glucose (pH = 7.4). Suspended cells were placed on the NPC©1 chip surface, and the whole cell configuration was achieved. Internal recording solution containing (in mM): 50 CsCl, 60 CsF, 10 NaCl, 20 EGTA, 10 HEPES, 2 Na
+-ATP (pH = 7.2, adjusted with CsOH) was deposited in recording chips, having resistances of 3–5 MΩ. I
SOC were activated with thapsigargin. I
SOC were assessed using voltage ramps (−100 to +100 mV in 200 ms) applied every 5 s, from a holding potential of 0 mV and acquired with an EPC-10 patch-clamp amplifier (HEKA). Immediately after the whole-cell configuration was established, the cell capacitance and the series resistances (<10 MΩ) were measured. During records, these two parameters were measured, and if exceed by 10% in respect to the initial value, the experiment was discontinued. Resting membrane potentials were estimated by reading the potential of the recorded cell immediately after rupturing the membrane in the current-clamp configuration. Leak currents were eliminated by subtracting the average of the first five ramp currents (obtained just after whole-cell configuration was reached) to all subsequent currents. Inward and outward current amplitude were measured at −80 mV and +80 mV, respectively. Data were normalized respect to cell capacitance. Liquid junction potential and capacitive currents were cancelled using the automatic compensation of the EPC-10. Data were filtered at 10 kHz and sampled at 5 kHz.
4.7. Quantitative RT-PCR
Total cellular RNA was isolated from cells using Trizol reagent (Invitrogen, Carlbads, CA, USA). Extracted RNA integrity was tested by electrophoresis on agarose gels, and the purity and concentration were determined by spectrophotometry. RNA was reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) and the cDNA diluted before PCR amplification. Primers were designed with Primer-BLAST (28). Primer sequences used were reported previously [
22]. Qualitative PCR was performed on a TGradient system (Biometra, Goettingen, Germany) using a Taq polymerase (Fermentas). The reactions protocol consisted in 3 min at 94 °C, 35 cycles of 1 min at 94 °C, 1 min at 57 °C, and 30 s at 72 °C and finished at 72 °C for 10 min. Real-time, quantitative PCR was performed using an SYBR green I detection in a LightCycler rapid thermal cycler (Roche, Mannheim, Germany). The PCR protocol started with 5 min at 95 °C followed by 45 cycles of 15 s at 95 °C, 20 s at 57 °C or at 60 °C and 5 s at 72 °C.
β-actin was used as a housekeeping gene. The data were normalized by PCR analysis of
β-actin. Melting curves were used to determine the specificity of PCR products [
22]).
4.8. Western Blotting
Total protein was extracted from cells and used to quantify expression of TRPC1, STIM1, STIM2, Orai1, Orai2 and Orai3 as reported previously [
22]. Whole-cell lysate was obtained using RIPA buffer (20 mM Tris–HCl, pH 7.8, 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, 1 mM EDTA, 0.05% SDS) supplemented with the Halt™ Protease and Phosphatase Inhibitor Cocktail (100×) from ThermoFisher Scientific (ref #1861281) (Waltham, MC, USA). Protein concentrations were determined by a Bradford protein assay. Proteins were fractionated by SDS-PAGE, electroblotted onto PVDF membranes and probed with the antibodies at dilution 1/200 except the anti-
β-actin that was used at dilution 1/5000. Antibodies were visualized by addition of goat anti-rabbit IgG or rabbit anti-mouse IgG. Detection was performed using Pierce ECL Western Blotting Substrate (Thermo Scientific) and VersaDoc Imaging System (BioRad, Munich, Germany). Quantification of protein expression was carried out using Quantity One software (BioRad, Munich, Germany).
4.9. Statistics
When only 2 means were compared, Student’s t-test was used. For more than 2 groups, the statistical significance of the data was assessed by ANOVA and compared using Bonferroni’s multiple comparison tests. Differences were considered significant at p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}