IGFBP7 Drives Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition in Lung Cancer

 , , and
, , and 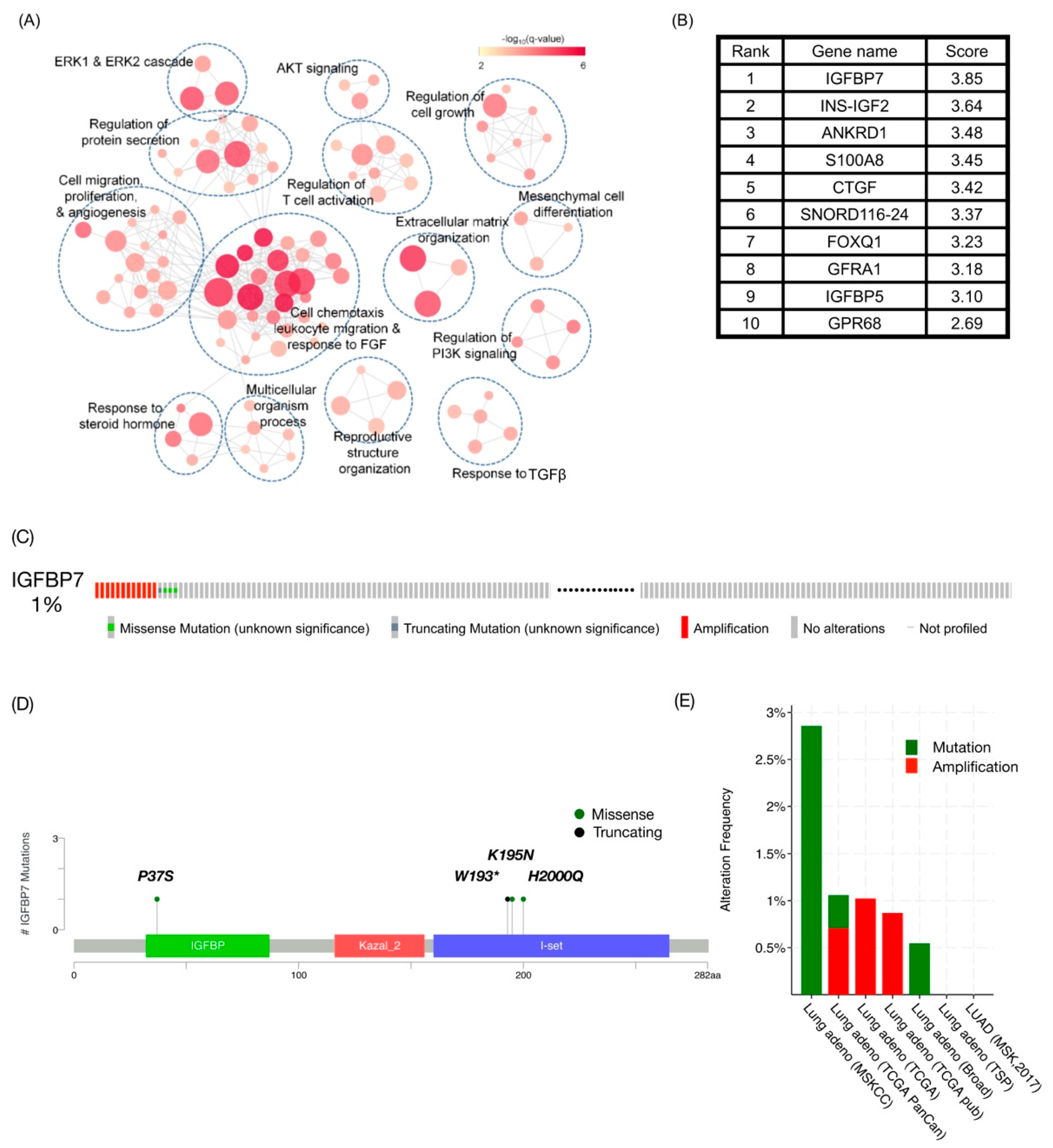{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
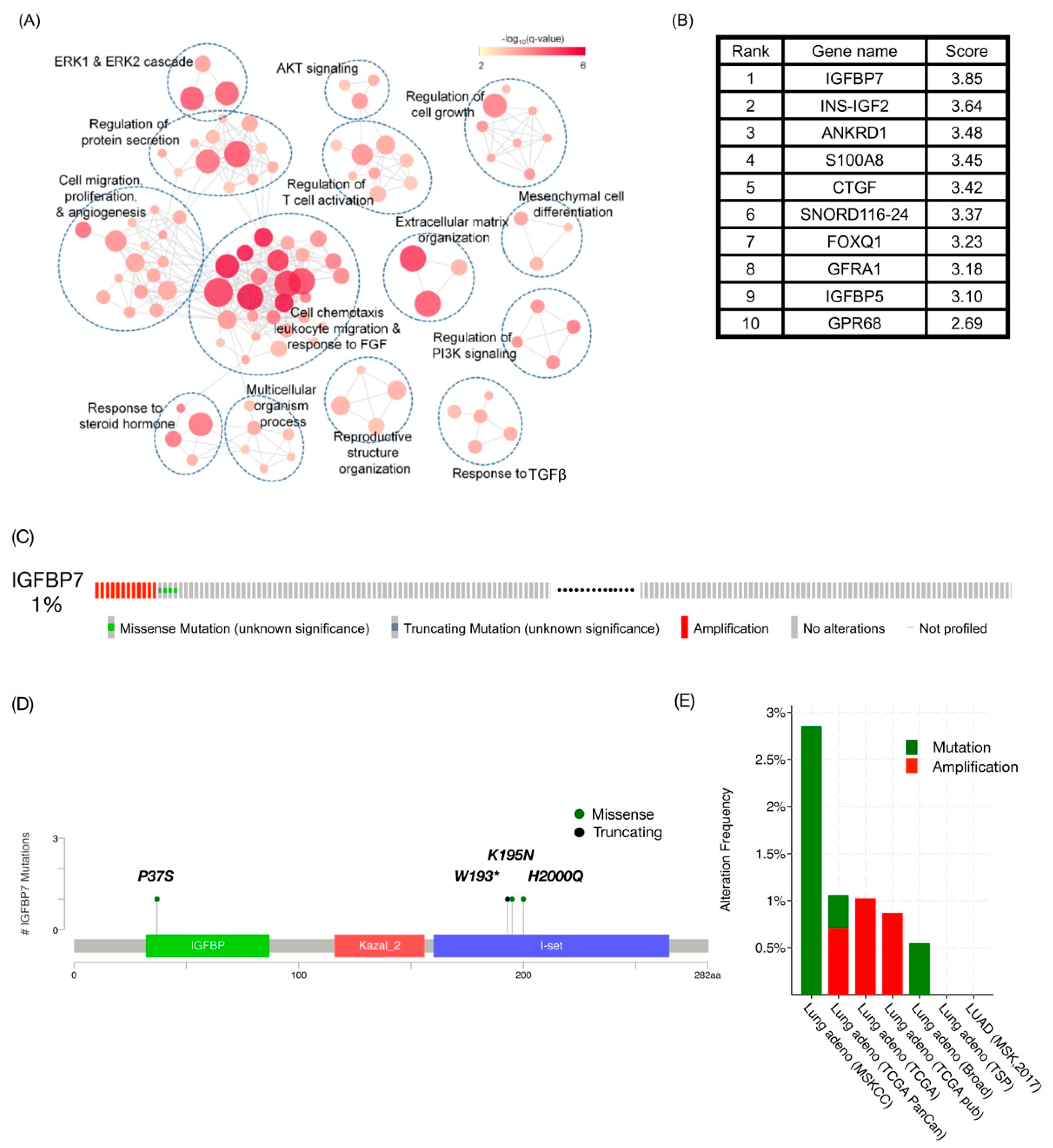
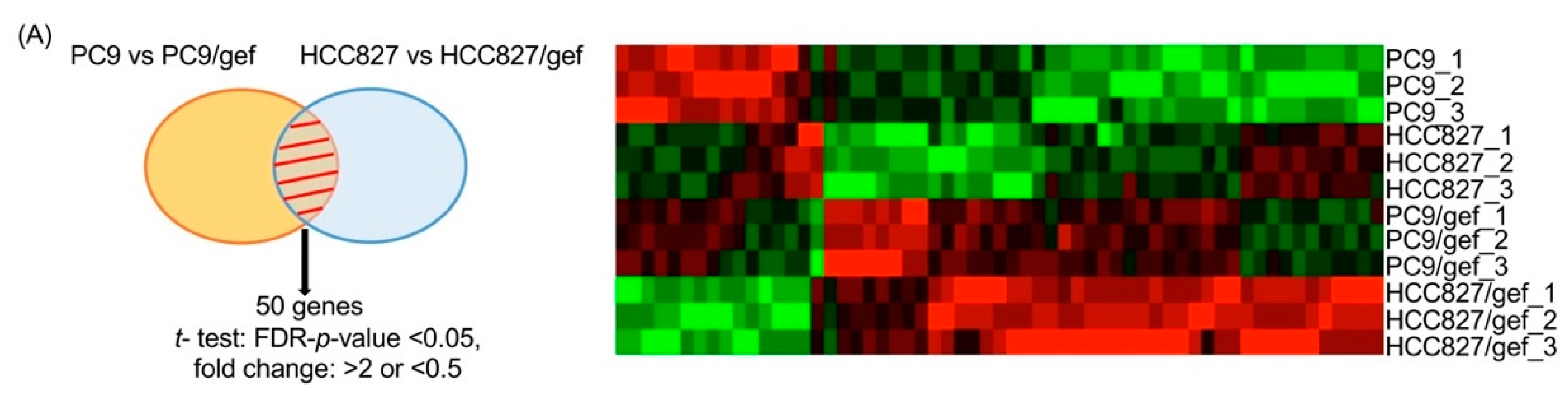
2.1. IGFBP7 Was the Highest-Ranking Gene Related to TKI Resistance in Public Datasets
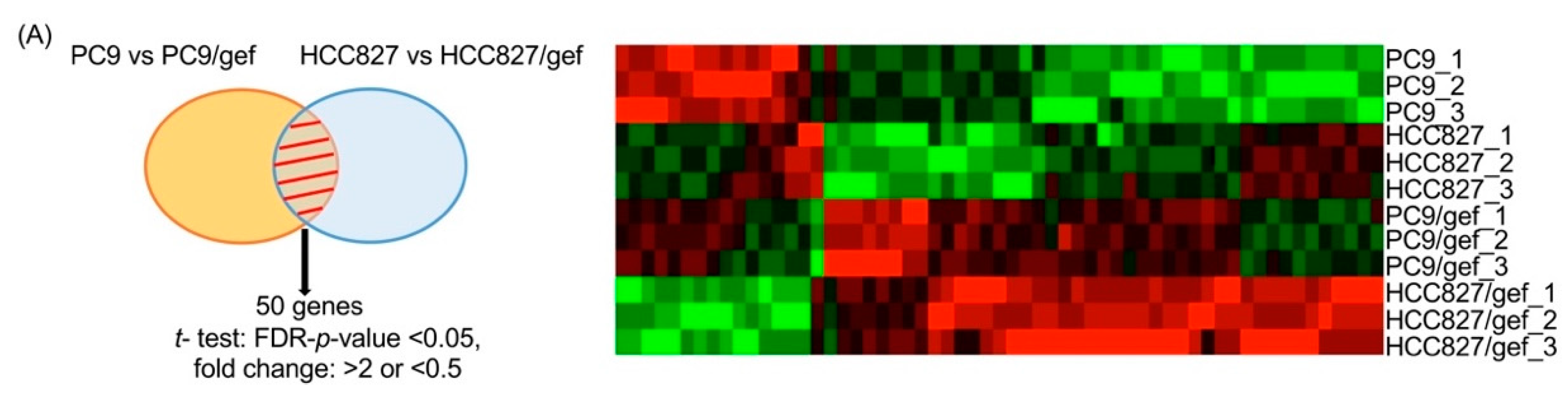
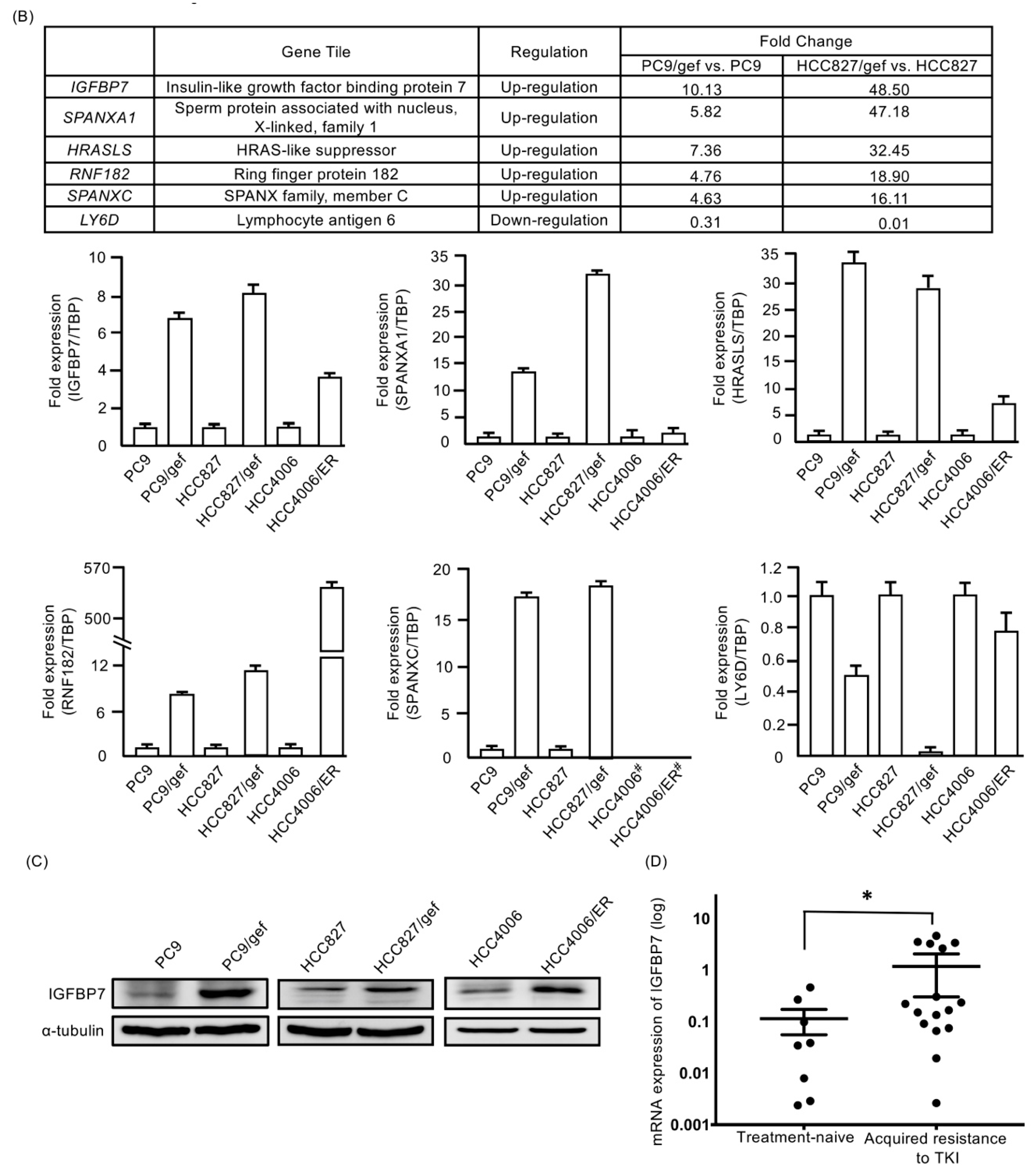
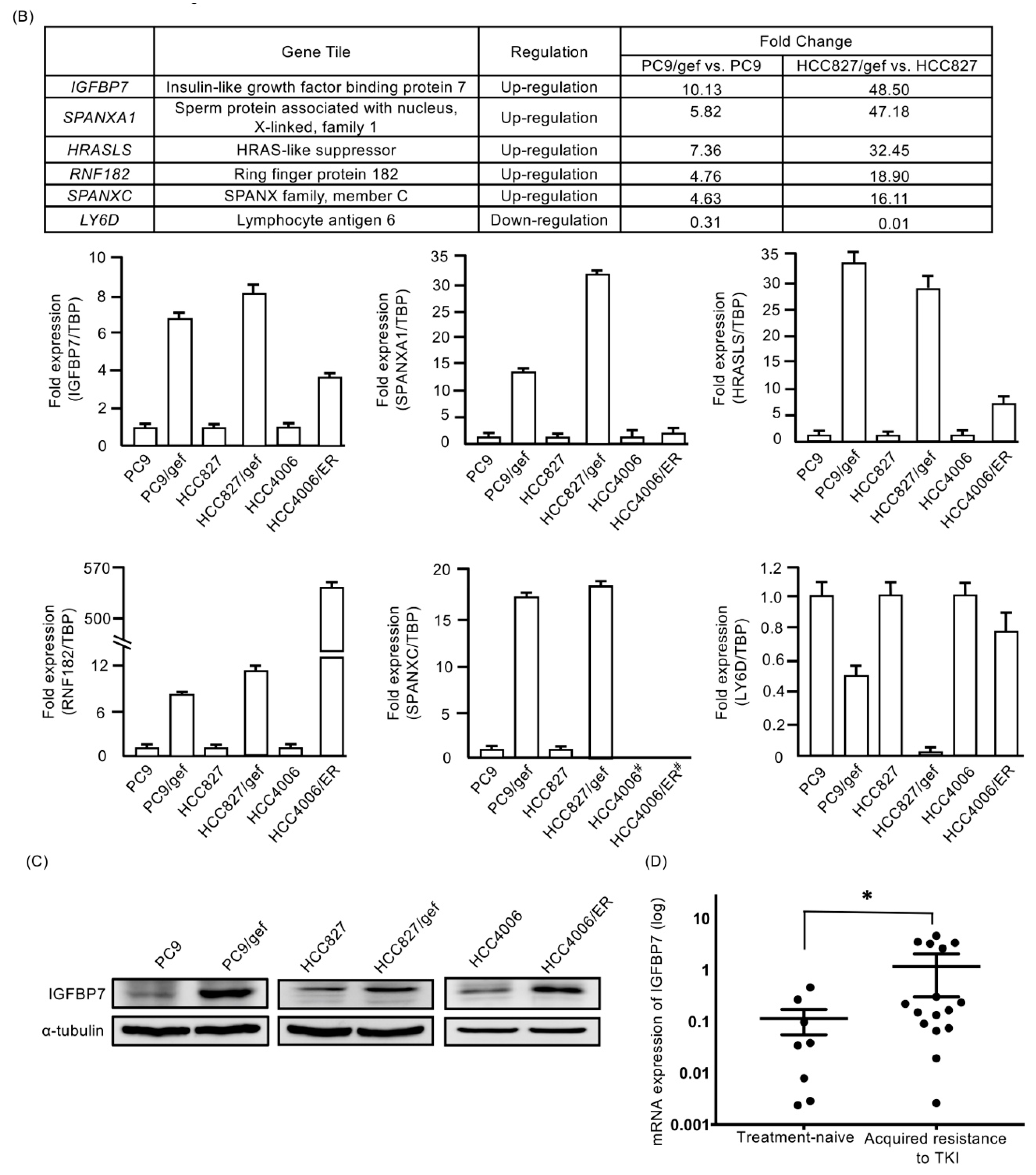
2.2. IGFBP7 Was Significantly Elevated in EGFR-TKI-Resistant Cells
2.3. Higher IGFBP7 Levels in Malignant Pleural Effusion of Lung Adenocarcinoma After Acquired Resistance to EGFR-TKIs
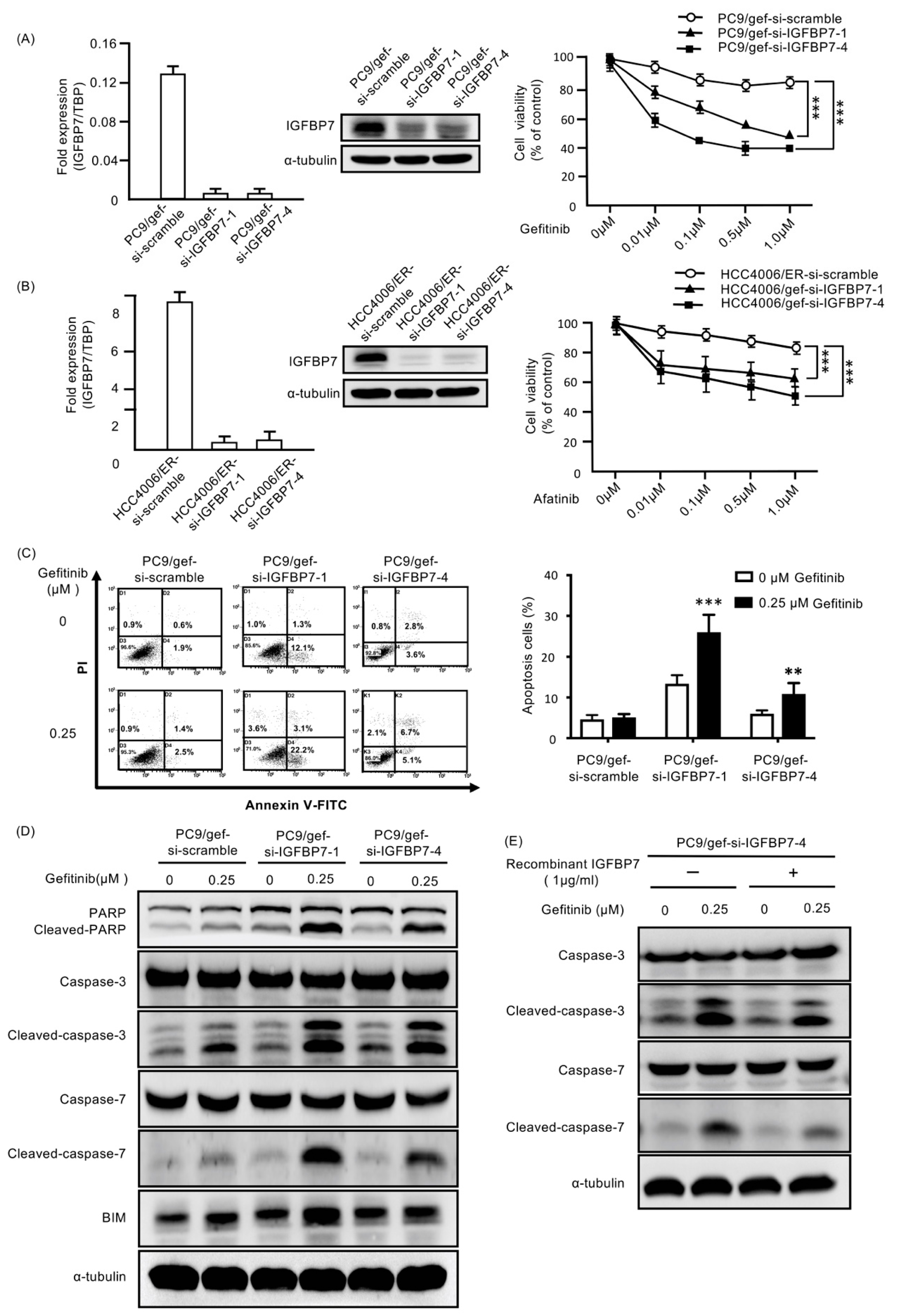
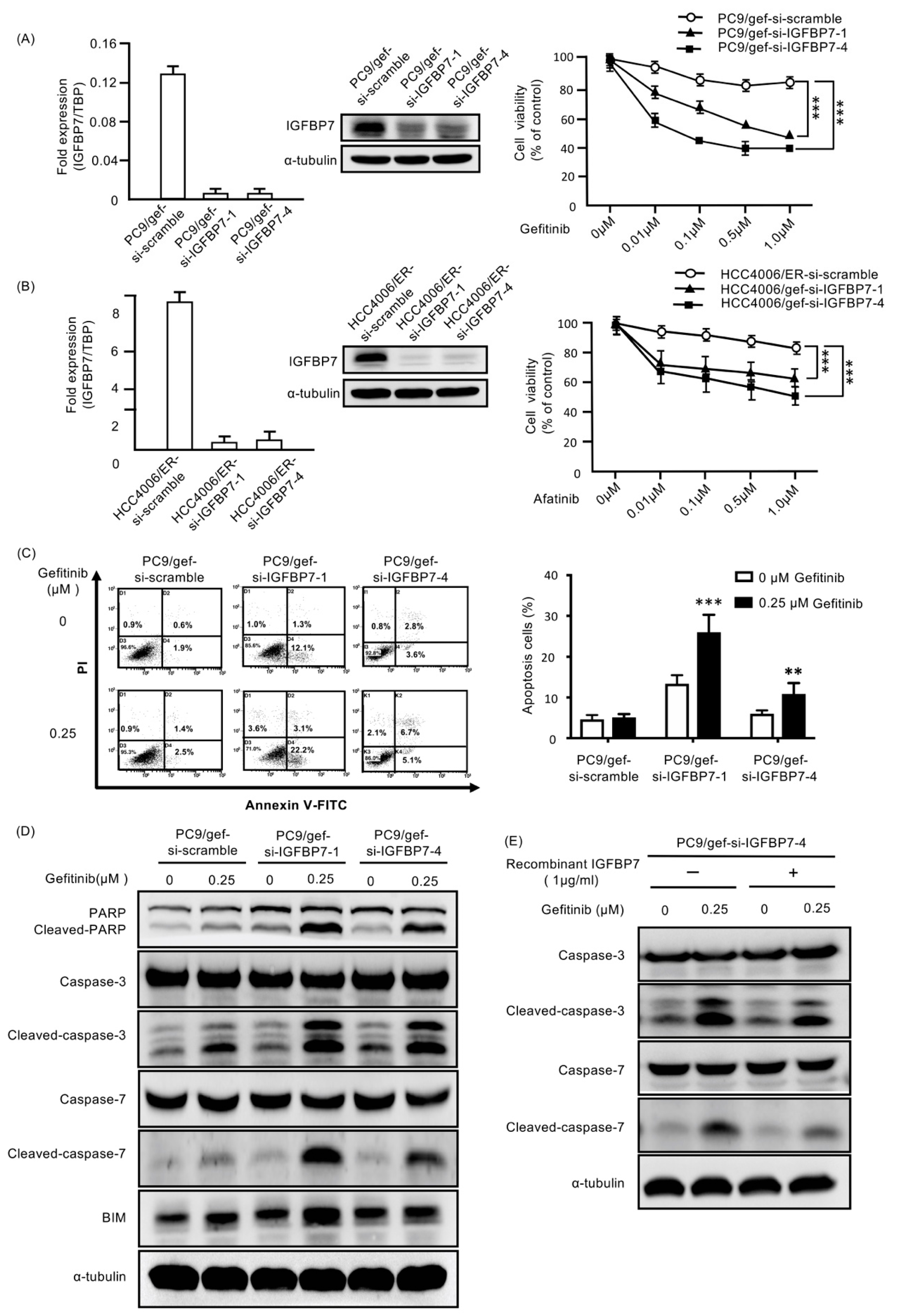
2.4. Suppression of IGFBP7 in EGFR-TKI-Resistant Cells Enhanced Gefitinib-Induced Cell Death
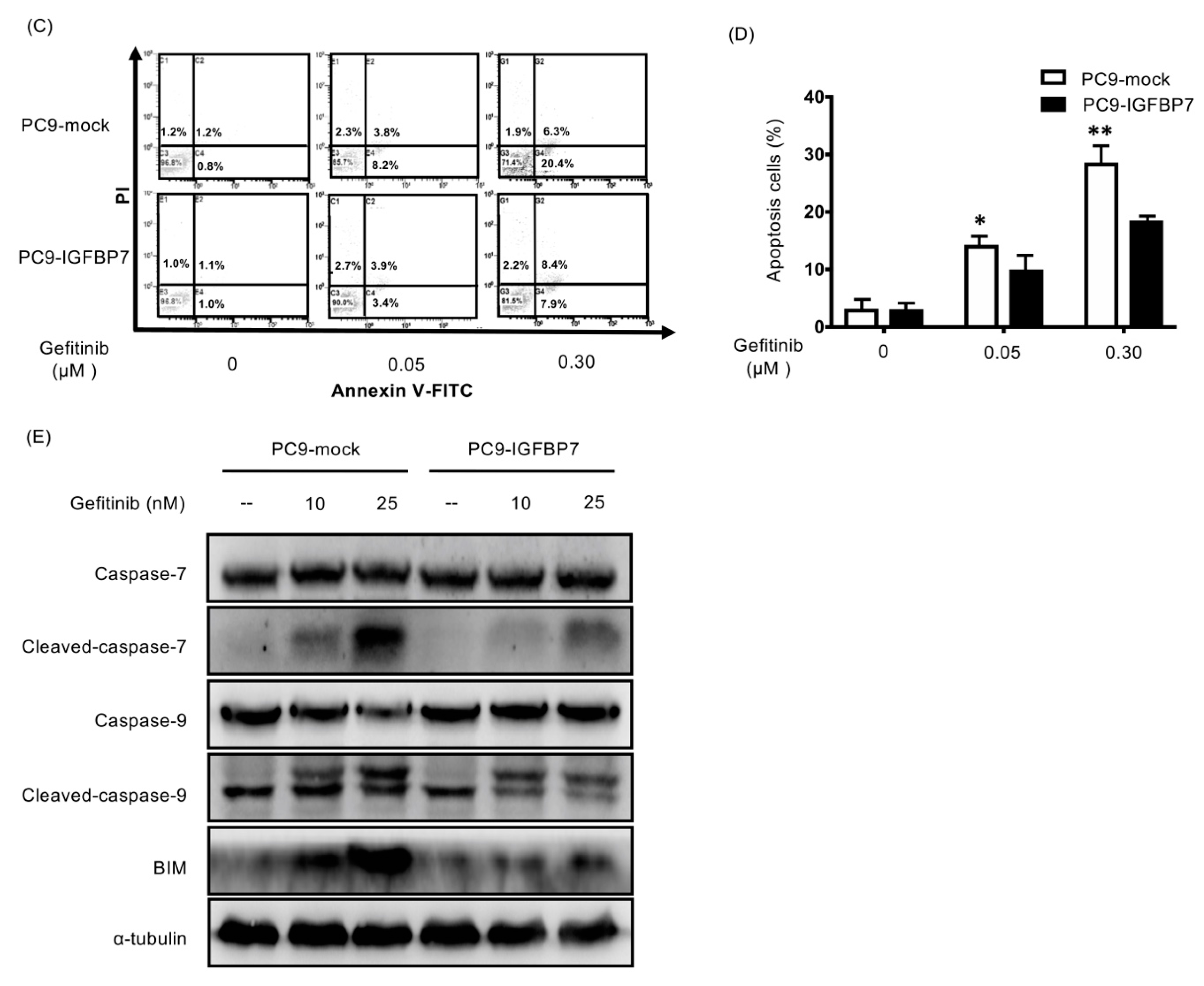
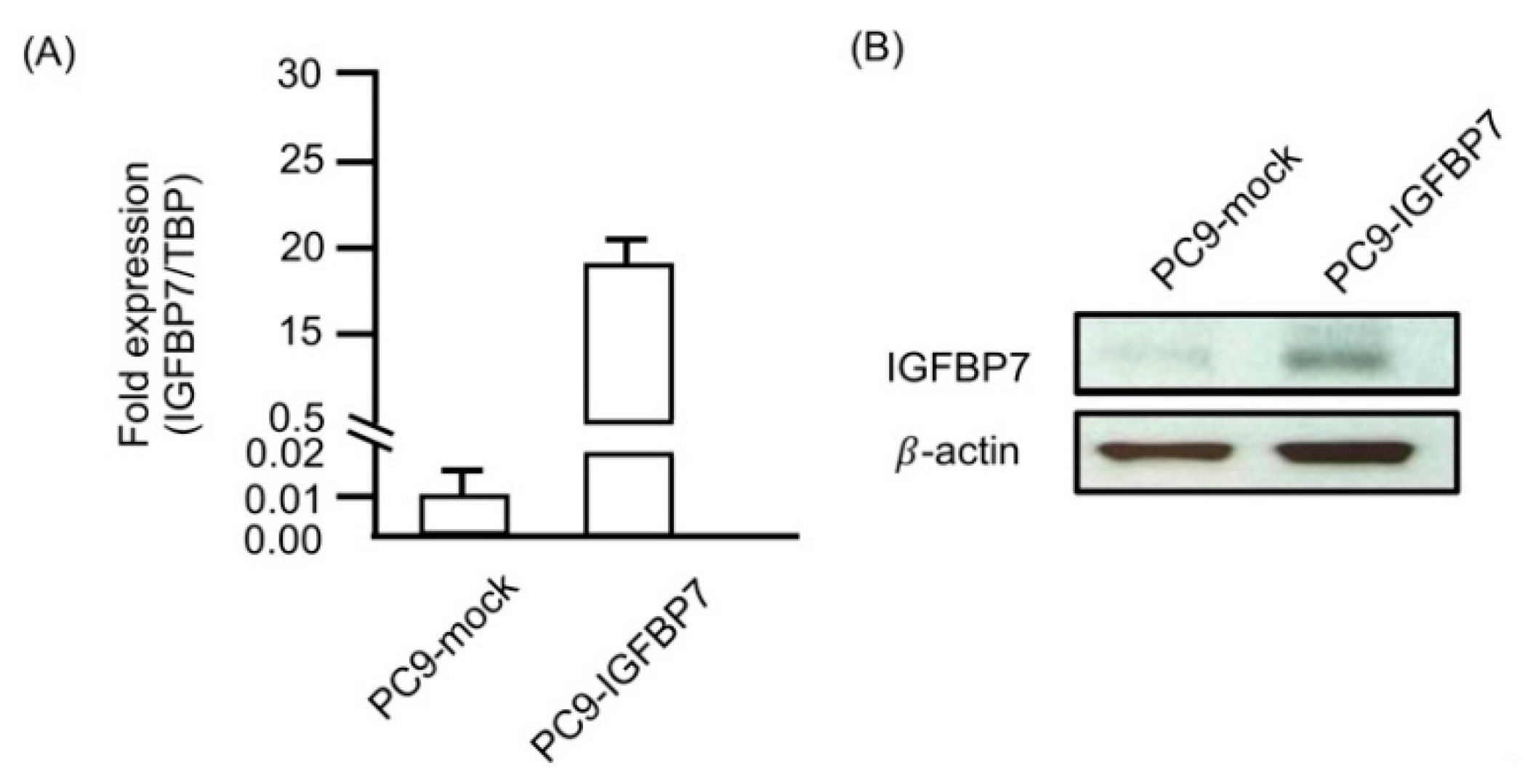
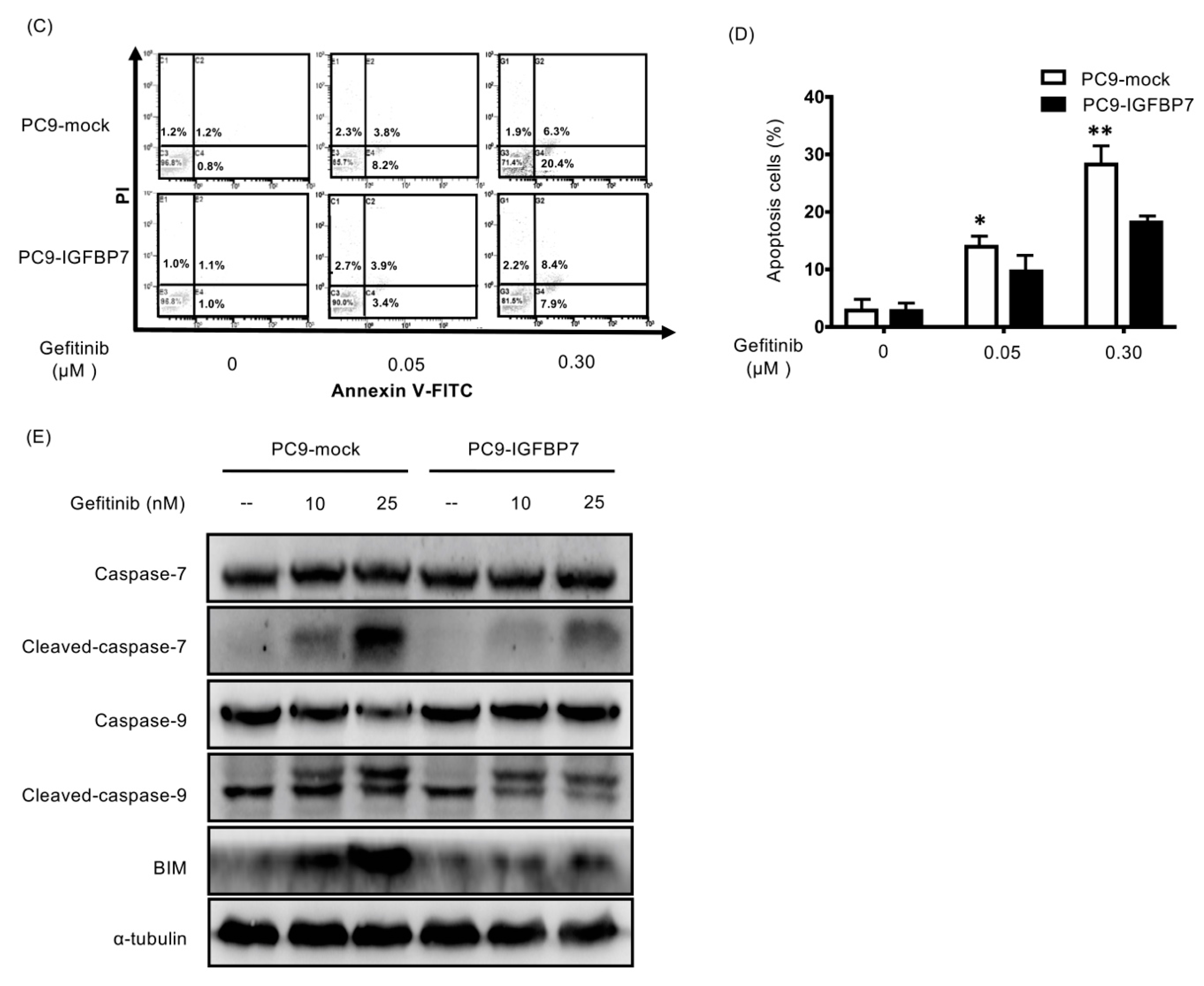
2.5. Enhanced IGFBP7 Expression Prevents EGFR-TKI-Induced Apoptosis
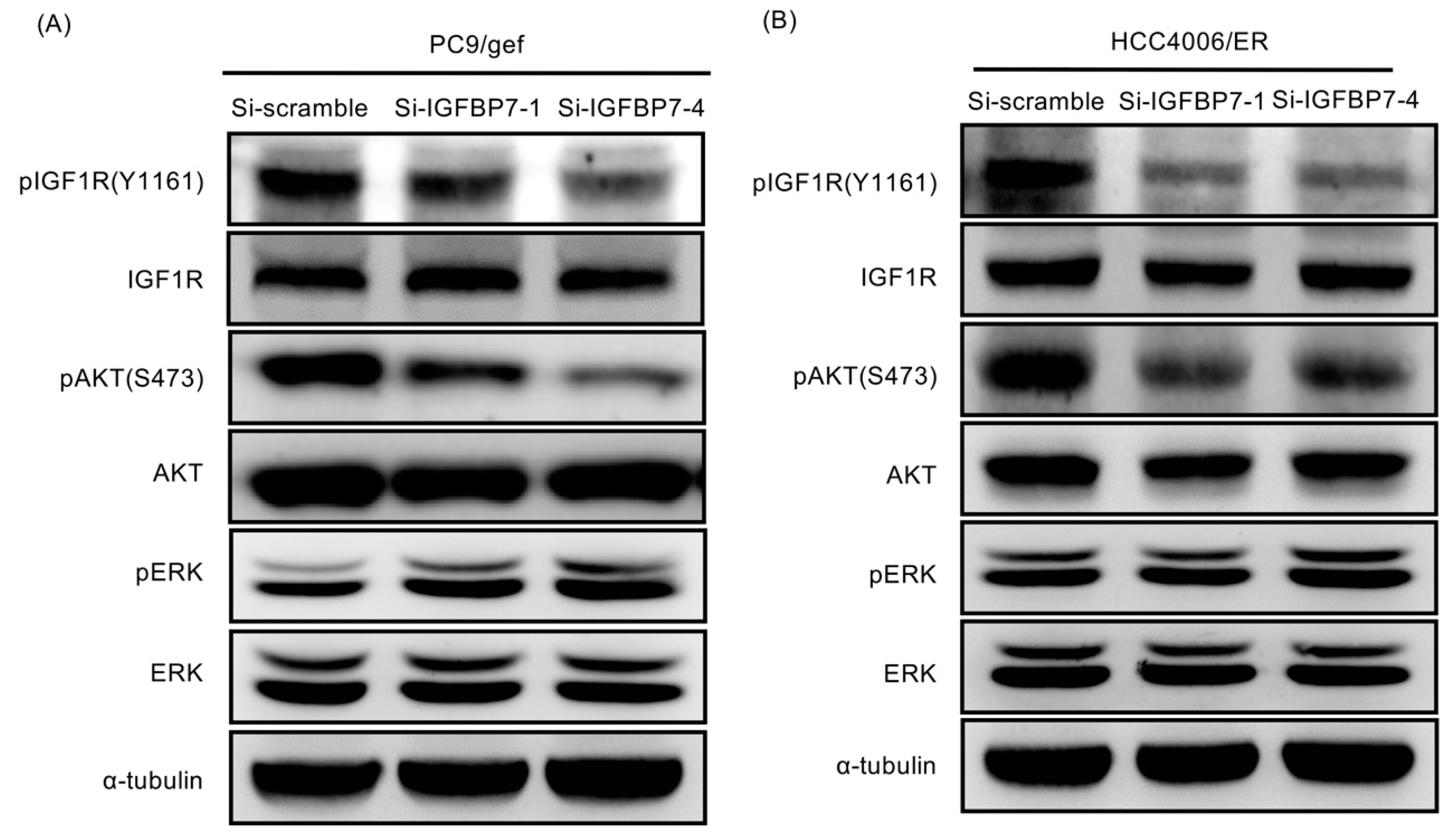
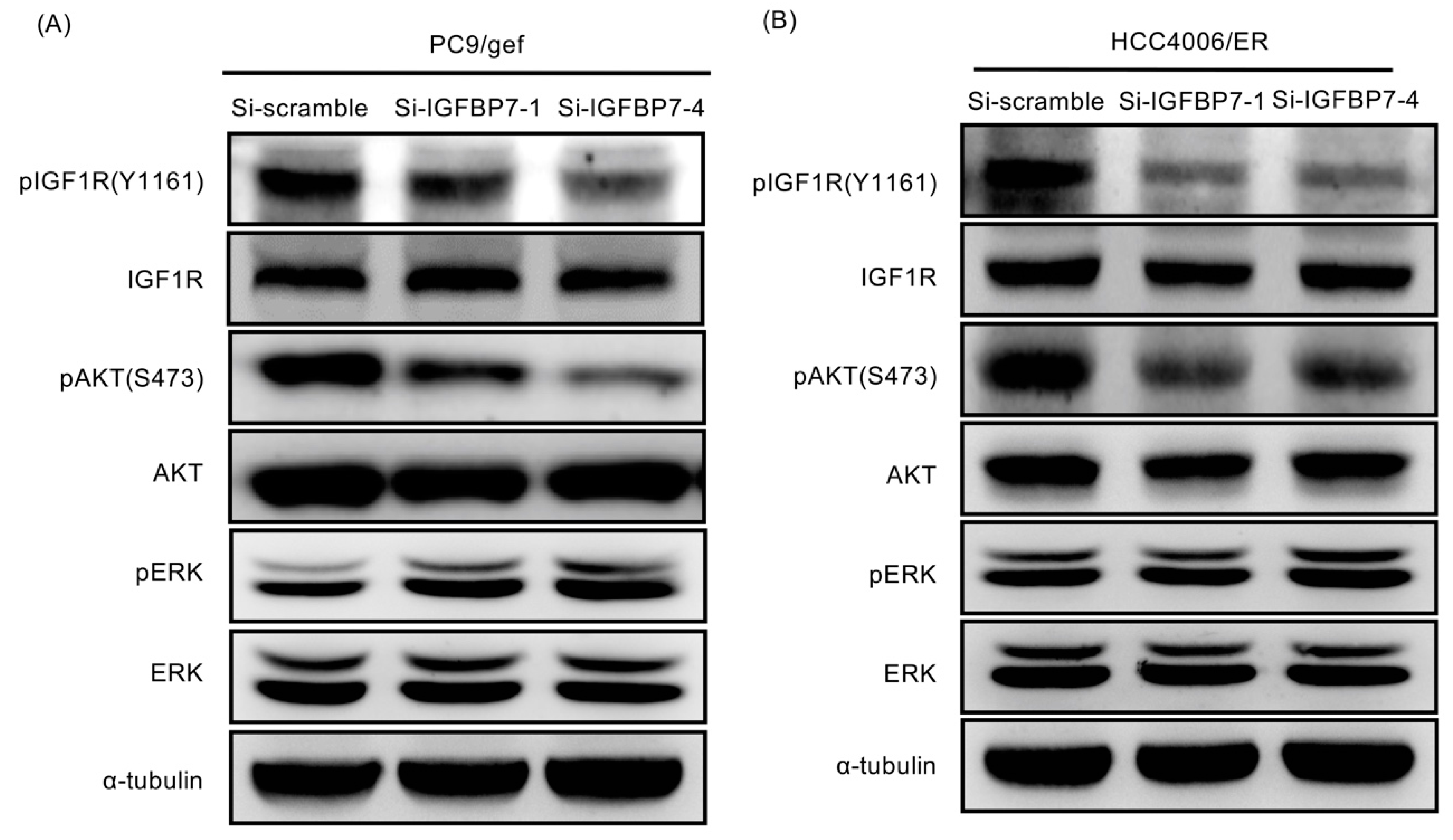
2.6. Suppression of IGFBP7 Attenuates Phosphorylation of IGF-1R and Downstream AKT
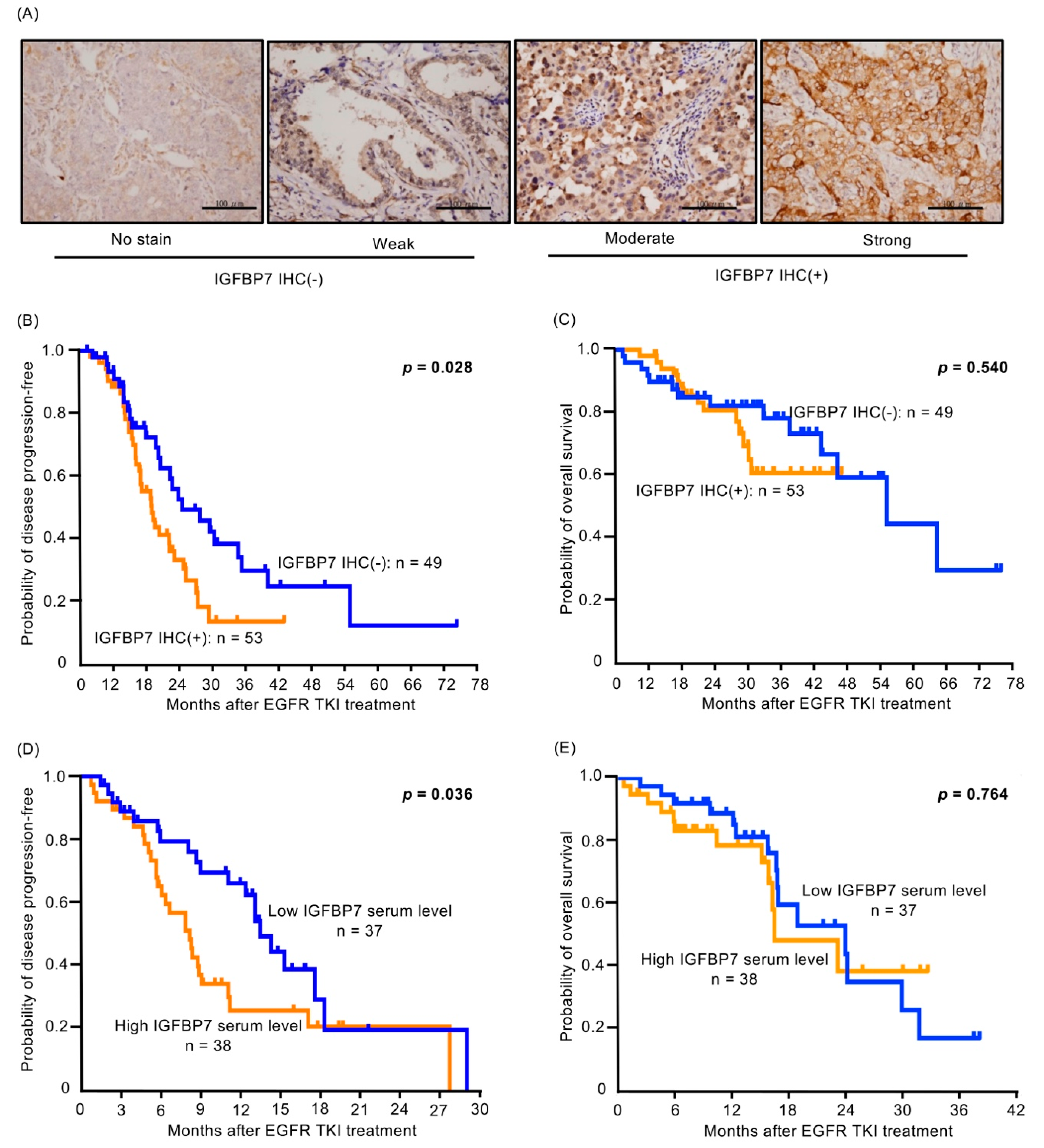
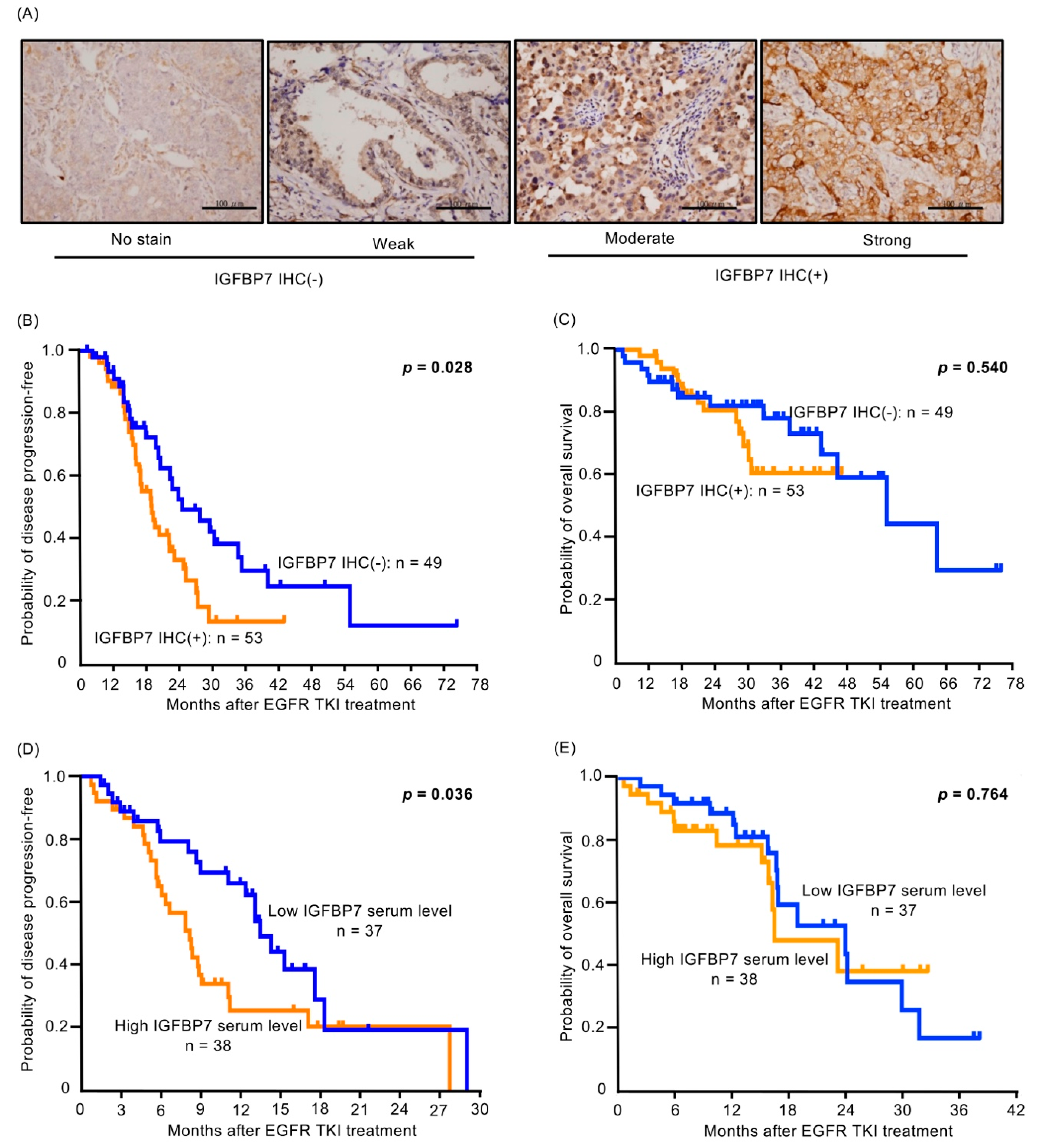
2.7. Higher IGFBP7 Level Is Associated with Shorter Progression-Free Survival of EGFR-TKI-Treated Lung Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Data Integration for TKI-Associated Gene Prioritization and Candidate Gene Alteration Exploration
4.2. Bioinformatics Analysis
4.3. Cell Culture, Small Interfering RNA (siRNA), Recombinant Protein and Drugs
4.4. Gene Expression Microarray Profiling
4.5. Establishment of IGFBP7-Expressing Stable Cell Lines
4.6. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
4.7. ELISA
4.8. Cytotoxicity Assay
4.9. Apoptosis Assay
4.10. Western Blotting
4.11. Patients and Sample Collection
4.12. Malignant Pleural Effusion Isolation
4.13. Tumor Specimens for Immunohistochemical Staining (IHC)
4.14. Peripheral Blood Sample Collection
4.15. Statistical analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Janne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.H.; Tsai, M.F.; Su, K.Y.; Wu, S.G.; Huang, C.P.; Yu, S.L.; Yu, Y.L.; Lan, C.C.; Yang, C.H.; Lin, S.B.; et al. Slug Confers Resistance to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. Am. J. Respir Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Chang, T.H.; Tsai, M.F.; Wu, S.G.; Tsai, T.H.; Chen, H.Y.; Yu, S.L.; Yang, J.C.; Shih, J.Y. IL-8 confers resistance to EGFR inhibitors by inducing stem cell properties in lung cancer. Oncotarget 2015, 6, 10415–10431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fustaino, V.; Presutti, D.; Colombo, T.; Cardinali, B.; Papoff, G.; Brandi, R.; Bertolazzi, P.; Felici, G.; Ruberti, G. Characterization of epithelial-mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non-small cell lung cancer cell lines. Oncotarget 2017, 8, 103340–103363. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Seike, M.; Chiba, M.; Takahashi, S.; Nakamichi, S.; Matsumoto, M.; Takeuchi, S.; Minegishi, Y.; Noro, R.; Kunugi, S.; et al. Ankyrin Repeat Domain 1 Overexpression is Associated with Common Resistance to Afatinib and Osimertinib in EGFR-mutant Lung Cancer. Sci. Rep. 2018, 8, e14896. [Google Scholar] [CrossRef] [PubMed]
- Kani, K.; Garri, C.; Tiemann, K.; Malihi, P.D.; Punj, V.; Nguyen, A.L.; Lee, J.; Hughes, L.D.; Alvarez, R.M.; Wood, D.M.; et al. JUN-Mediated Downregulation of EGFR Signaling Is Associated with Resistance to Gefitinib in EGFR-mutant NSCLC Cell Lines. Mol. Cancer Ther. 2017, 16, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patra, J.C. Integration of multiple data sources to prioritize candidate genes using discounted rating system. BMC 2010, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, e1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef] [Green Version]
- Peled, N.; Wynes, M.W.; Ikeda, N.; Ohira, T.; Yoshida, K.; Qian, J.; Ilouze, M.; Brenner, R.; Kato, Y.; Mascaux, C.; et al. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol. 2013, 36, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2015, 55, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [PubMed]
- Kelley, K.M.; Oh, Y.; Gargosky, S.E.; Gucev, Z.; Matsumoto, T.; Hwa, V.; Ng, L.; Simpson, D.M.; Rosenfeld, R.G. Insulin-like growth factor-binding proteins (IGFBPs) and their regulatory dynamics. Int. J. Biochem. Cell Biol. 1996, 28, 619–637. [Google Scholar] [CrossRef]
- Lu, H.; Wang, L.; Gao, W.; Meng, J.; Dai, B.; Wu, S.; Minna, J.; Roth, J.A.; Hofstetter, W.L.; Swisher, S.G.; et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Mol. Cancer Ther. 2013, 12, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Kishino, Y.; Murata, Y.; Kusumoto, S.; Ishida, H.; Shirai, T.; Hirose, T.; et al. Acquired Resistance Mechanisms to Combination Met-TKI/EGFR-TKI Exposure in Met-Amplified EGFR-TKI-Resistant Lung Adenocarcinoma Harboring an Activating EGFR Mutation. Mol. Cancer Ther. 2016, 15, 3040–3054. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct Afatinib Resistance Mechanisms Identified in Lung Adenocarcinoma Harboring an EGFR Mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; McCusker, R.H.; Vanderhoof, J.A.; Mohammadpour, H.; Harty, R.F.; MacDonald, R.G. Secretion of insulin-like growth factor II (IGF-II) and IGF-binding protein-2 by intestinal epithelial (IEC-6) cells: Implications for autocrine growth regulation. Endocrinology 1992, 131, 1359–1368. [Google Scholar] [CrossRef]
- Corkins, M.R.; Vanderhoof, J.A.; Slentz, D.H.; MacDonald, R.G.; Park, J.H. Growth stimulation by transfection of intestinal epithelial cells with an antisense insulin-like growth factor binding protein-2 construct. Biochem. Biophys. Res. Commun. 1995, 211, 707–713. [Google Scholar] [CrossRef]
- Burger, A.M.; Zhang, X.; Li, H.; Ostrowski, J.L.; Beatty, B.; Venanzoni, M.; Papas, T.; Seth, A. Down-regulation of T1A12/mac25, a novel insulin-like growth factor binding protein related gene, is associated with disease progression in breast carcinomas. Oncogene 1998, 16, 2459–2467. [Google Scholar] [CrossRef] [Green Version]
- Landberg, G.; Ostlund, H.; Nielsen, N.H.; Roos, G.; Emdin, S.; Burger, A.M.; Seth, A. Downregulation of the potential suppressor gene IGFBP-rP1 in human breast cancer is associated with inactivation of the retinoblastoma protein, cyclin E overexpression and increased proliferation in estrogen receptor negative tumors. Oncogene 2001, 20, 3497–3505. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Pacyna-Gengelbach, M.; Ye, F.; Knosel, T.; Lund, P.; Deutschmann, N.; Schluns, K.; Kotb, W.F.; Sers, C.; Yasumoto, H.; et al. Insulin-like growth factor binding protein-related protein 1 (IGFBP-rP1) has potential tumour-suppressive activity in human lung cancer. J. Pathol. 2007, 211, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Smith, P.; Nicholson, L.J.; Syed, N.; Payne, A.; Hiller, L.; Garrone, O.; Occelli, M.; Gasco, M.; Crook, T. Epigenetic inactivation implies independent functions for insulin-like growth factor binding protein (IGFBP)-related protein 1 and the related IGFBPL1 in inhibiting breast cancer phenotypes. Clin. Cancer Res. 2007, 13, 4061–4068. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Ben, Q.W.; Chen, H.T.; Zheng, J.M.; Huang, L.; Li, G.X.; Li, Z.S. Low expression of IGFBP7 is associated with poor outcome of pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2012, 19, 3971–3978. [Google Scholar] [CrossRef] [PubMed]
- Tomimaru, Y.; Eguchi, H.; Wada, H.; Kobayashi, S.; Marubashi, S.; Tanemura, M.; Umeshita, K.; Kim, T.; Wakasa, K.; Doki, Y.; et al. IGFBP7 downregulation is associated with tumor progression and clinical outcome in hepatocellular carcinoma. Int. J. Cancer 2012, 130, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Degeorges, A.; Wang, F.; Frierson, H.F., Jr.; Seth, A.; Chung, L.W.; Sikes, R.A. Human prostate cancer expresses the low affinity insulin-like growth factor binding protein IGFBP-rP1. Cancer Res. 1999, 59, 2787–2790. [Google Scholar] [PubMed]
- Bolomsky, A.; Hose, D.; Schreder, M.; Seckinger, A.; Lipp, S.; Klein, B.; Heintel, D.; Ludwig, H.; Zojer, N. Insulin like growth factor binding protein 7 (IGFBP7) expression is linked to poor prognosis but may protect from bone disease in multiple myeloma. J. Hematol. Oncol. 2015, 8, e10. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Ruszkiewicz, A.R.; Jamieson, G.G.; Drew, P.A. IGFBP7 is associated with poor prognosis in oesophageal adenocarcinoma and is regulated by promoter DNA methylation. Br. J. Cancer 2014, 110, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Sepiashvili, L.; Hui, A.; Ignatchenko, V.; Shi, W.; Su, S.; Xu, W.; Huang, S.H.; O’Sullivan, B.; Waldron, J.; Irish, J.C.; et al. Potentially novel candidate biomarkers for head and neck squamous cell carcinoma identified using an integrated cell line-based discovery strategy. Mol. Cell Proteomics 2012, 11, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Pen, A.; Moreno, M.J.; Durocher, Y.; Deb-Rinker, P.; Stanimirovic, D.B. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-beta signaling. Oncogene 2008, 27, 6834–6844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Zhu, B.; Duan, Y.; Chen, F.; Nian, W.; Sun, J.; Zhang, B.; Tong, Z.; Chen, Z. IGFBP7 functions as a potential lymphangiogenesis inducer in non-small cell lung carcinoma. Oncol. Rep. 2016, 35, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Shersher, D.D.; Vercillo, M.S.; Fhied, C.; Basu, S.; Rouhi, O.; Mahon, B.; Coon, J.S.; Warren, W.H.; Faber, L.P.; Hong, E.; et al. Biomarkers of the insulin-like growth factor pathway predict progression and outcome in lung cancer. Ann. Thorac. Surg. 2011, 92, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, D.; Maier, P.; Laufs, S.; Li, L.; Sleeman, J.P.; Trunk, M.J.; Leupold, J.H.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; et al. The disparate twins: A comparative study of CXCR4 and CXCR7 in SDF-1alpha-induced gene expression, invasion and chemosensitivity of colon cancer. Clin. Cancer Res. 2014, 20, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, A.B.; de Vasconcellos, J.F.; Sodek, L.; Spago, M.C.; Fornazim, M.C.; Tone, L.G.; Brandalise, S.R.; Nowill, A.E.; Yunes, J.A. IGFBP7 participates in the reciprocal interaction between acute lymphoblastic leukemia and BM stromal cells and in leukemia resistance to asparaginase. Leukemia 2011, 26, 1001–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Huang, M.H.; Lee, J.H.; Chang, Y.J.; Tsai, H.H.; Lin, Y.L.; Lin, A.M.; Yang, J.C. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol. Oncol. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Crowley, J.; Chansky, K.; Giroux, D.J.; Groome, P.A.; Rami-Porta, R.; Postmus, P.E.; Rusch, V.; Sobin, L. The IASLC Lung Cancer Staging Project: Proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J. Thorac. Oncol. 2007, 2, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.M.; Dublin, E.A.; Fisher, C.J.; Levison, D.A.; Millis, R.R. Immunohistochemical detection of p53 protein in mammary carcinoma: An important new independent indicator of prognosis? Hum. Pathol. 1993, 24, 469–476. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-G.; Chang, T.-H.; Tsai, M.-F.; Liu, Y.-N.; Hsu, C.-L.; Chang, Y.-L.; Yu, C.-J.; Shih, J.-Y. IGFBP7 Drives Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition in Lung Cancer. Cancers 2019, 11, 36. https://doi.org/10.3390/cancers11010036
Wu S-G, Chang T-H, Tsai M-F, Liu Y-N, Hsu C-L, Chang Y-L, Yu C-J, Shih J-Y. IGFBP7 Drives Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition in Lung Cancer. Cancers. 2019; 11(1):36. https://doi.org/10.3390/cancers11010036
Chicago/Turabian StyleWu, Shang-Gin, Tzu-Hua Chang, Meng-Feng Tsai, Yi-Nan Liu, Chia-Lang Hsu, Yih-Leong Chang, Chong-Jen Yu, and Jin-Yuan Shih. 2019. "IGFBP7 Drives Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition in Lung Cancer" Cancers 11, no. 1: 36. https://doi.org/10.3390/cancers11010036
APA StyleWu, S.-G., Chang, T.-H., Tsai, M.-F., Liu, Y.-N., Hsu, C.-L., Chang, Y.-L., Yu, C.-J., & Shih, J.-Y. (2019). IGFBP7 Drives Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition in Lung Cancer. Cancers, 11(1), 36. https://doi.org/10.3390/cancers11010036