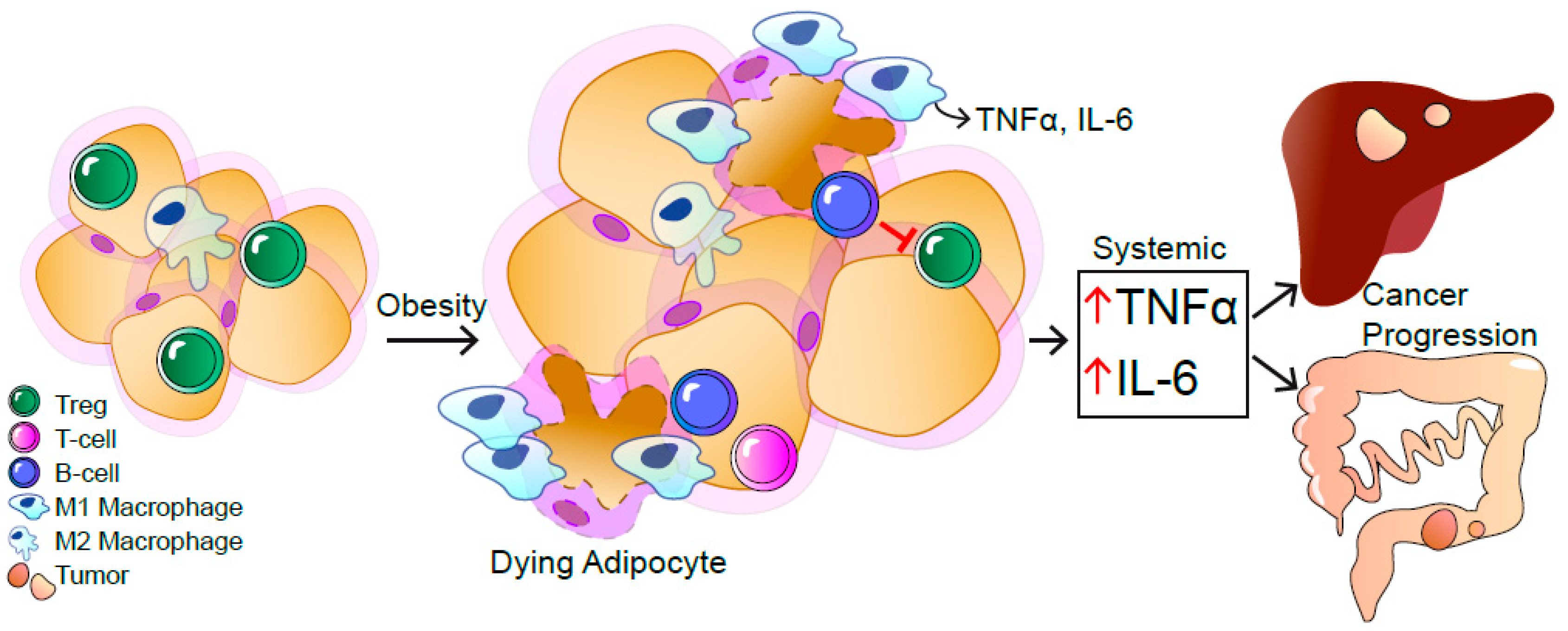
Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers

{kind=link}
{kind=link}
Abstract
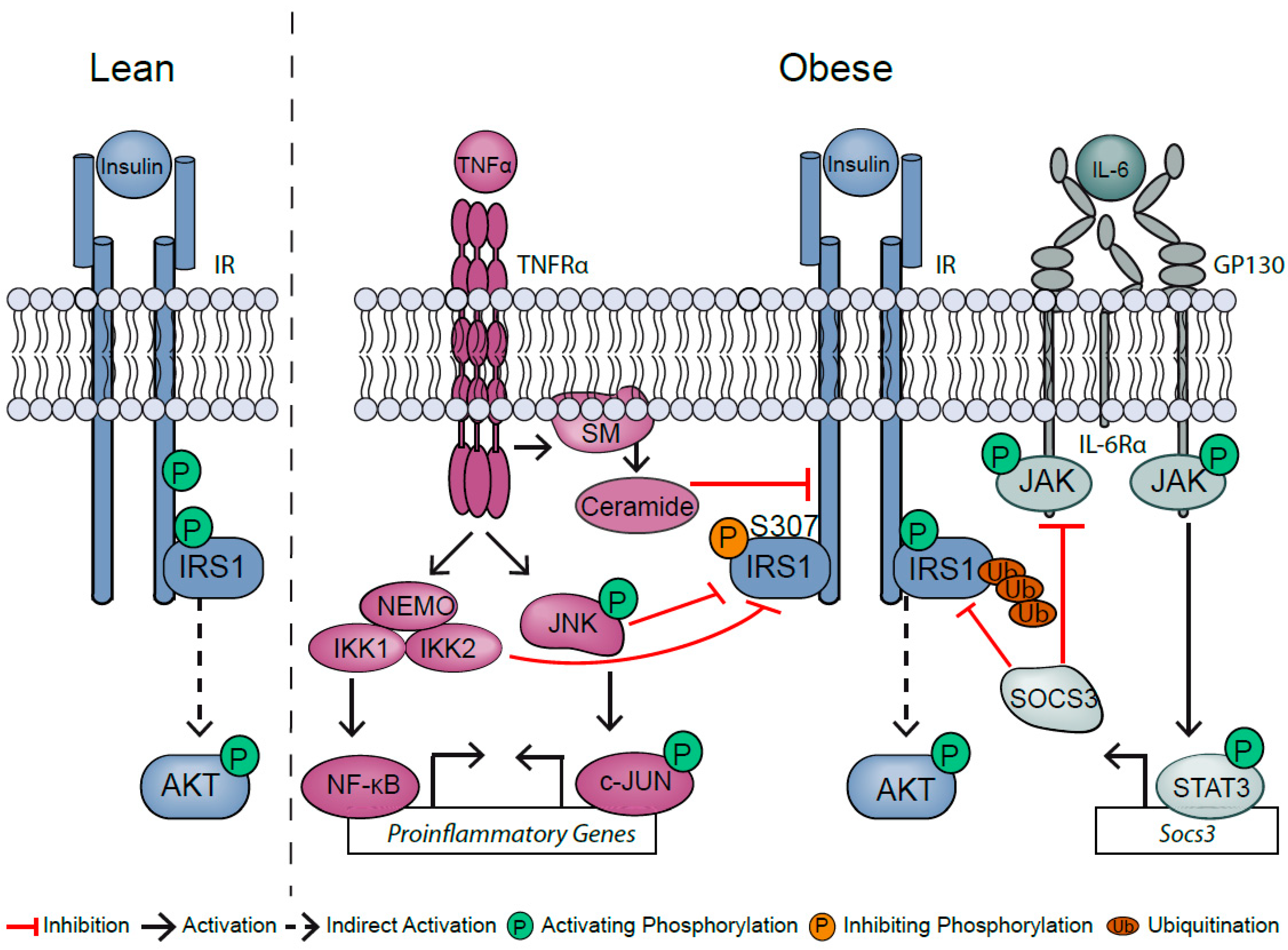
1. Obesity-Induced Inflammation Impairs Insulin Action
2. TNFα and Its Bivalent Role in Liver Homeostasis
3. TNFα: A Key Driver of Hepatocellular Carcinoma (HCC) Development
4. Hepatic IL-6 Signaling: Cytokine with Versatile Functions
5. IL-6 Signaling: A Fatal Player in Obesity-Induced HCC
6. Obesity Disturbs the Balance of Inflammatory Effectors and Inhibitors
7. Obesity-Induced Inflammation and CRC
8. Conclusions
Funding
Conflicts of Interest
References
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Jais, A.; Bruning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Bar Hen, A.; Bedossa, P.; et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Caspar-Bauguil, S.; Cousin, B.; Bour, S.; Casteilla, L.; Penicaud, L.; Carpene, C. Adipose tissue lymphocytes: Types and roles. J. Physiol. Biochem. 2009, 65, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflugers Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Revelo, X.S.; Luck, H.; Winer, S.; Winer, D.A. Morphological and inflammatory changes in visceral adipose tissue during obesity. Endocr. Pathol. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelencic, V.; Valentic, S.; Sestan, M.; Wensveen, T.T.; Theurich, S.; Glasner, A.; Mendrila, D.; Stimac, D.; Wunderlich, F.T.; et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 2015, 16, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Theurich, S.; Tsaousidou, E.; Hanssen, R.; Lempradl, A.M.; Mauer, J.; Timper, K.; Schilbach, K.; Folz-Donahue, K.; Heilinger, C.; Sexl, V.; et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 2017, 26, 171–184.e6. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef]
- Lee, M.K.; Yvan-Charvet, L.; Masters, S.L.; Murphy, A.J. The modern interleukin-1 superfamily: Divergent roles in obesity. Semin. Immunol. 2016, 28, 441–449. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Thomas, B.J.; Feng, A.C.; Hong, C.; Wang, J.; Vergnes, L.; Sallam, T.; Wang, B.; Sandhu, J.; Seldin, M.M.; et al. IL-10 Signaling Remodels Adipose Chromatin Architecture to Limit Thermogenesis and Energy Expenditure. Cell 2018, 172, 218–233.e7. [Google Scholar] [CrossRef]
- Garidou, L.; Pomie, C.; Klopp, P.; Waget, A.; Charpentier, J.; Aloulou, M.; Giry, A.; Serino, M.; Stenman, L.; Lahtinen, S.; et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORgammat and Controls Metabolic Disease. Cell Metab. 2015, 22, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.; Guo, B.; Doyle, S.E.; Cheng, G. Cutting edge: Involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J. Immunol. 2007, 178, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Nish, S.A.; Schenten, D.; Wunderlich, F.T.; Pope, S.D.; Gao, Y.; Hoshi, N.; Yu, S.; Yan, X.; Lee, H.K.; Pasman, L.; et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife 2014, 3, e01949. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Hotamisligil, G.S.; Buurman, W.A.; White, M.F.; Spiegelman, B.M. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J. Biol. Chem. 1996, 271, 13018–13022. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Declercq, W.; Beyaert, R.; Fiers, W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995, 5, 392–399. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Copps, K.D.; Hancer, N.J.; Opare-Ado, L.; Qiu, W.; Walsh, C.; White, M.F. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010, 11, 84–92. [Google Scholar] [CrossRef]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef]
- Sachithanandan, N.; Fam, B.C.; Fynch, S.; Dzamko, N.; Watt, M.J.; Wormald, S.; Honeyman, J.; Galic, S.; Proietto, J.; Andrikopoulos, S.; et al. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 2010, 52, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Bronneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, F.T.; Strohle, P.; Konner, A.C.; Gruber, S.; Tovar, S.; Bronneke, H.S.; Juntti-Berggren, L.; Li, L.S.; van Rooijen, N.; Libert, C.; et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010, 12, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Pereira, M.M.A.; Karakasilioti, I.; Theurich, S.; Al-Maarri, M.; Rappl, G.; Waisman, A.; Wunderlich, F.T.; Bruning, J.C. Temporal and tissue-specific requirements for T-lymphocyte IL-6 signalling in obesity-associated inflammation and insulin resistance. Nat. Commun. 2017, 8, 14803. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Van, T.M.; Polykratis, A.; Straub, B.K.; Kondylis, V.; Papadopoulou, N.; Pasparakis, M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J. Clin. Investig. 2017, 127, 2662–2677. [Google Scholar] [CrossRef]
- Ehlken, H.; Krishna-Subramanian, S.; Ochoa-Callejero, L.; Kondylis, V.; Nadi, N.E.; Straub, B.K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014, 21, 1721–1732. [Google Scholar] [CrossRef]
- Lang, I.; Fullsack, S.; Wyzgol, A.; Fick, A.; Trebing, J.; Arana, J.A.; Schafer, V.; Weisenberger, D.; Wajant, H. Binding Studies of TNF Receptor Superfamily (TNFRSF) Receptors on Intact Cells. J. Biol. Chem. 2016, 291, 5022–5037. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Lin, A. Activation of the JNK signaling pathway: Breaking the brake on apoptosis. Bioessays 2003, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Karin, M. Inflammation and liver tumorigenesis. Front. Med. 2013, 7, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Hashimoto, E.; Yatsuji, S.; Kaneda, H.; Taniai, M.; Tokushige, K.; Shiratori, K. Nonalcoholic steatohepatitis: Cirrhosis, hepatocellular carcinoma, and burnt-out NASH. J. Gastroenterol. 2004, 39, 1215–1218. [Google Scholar] [CrossRef]
- Fausto, N. Mouse liver tumorigenesis: Models, mechanisms, and relevance to human disease. Semin. Liver Dis. 1999, 19, 243–252. [Google Scholar] [CrossRef]
- Knight, B.; Yeoh, G.C.; Husk, K.L.; Ly, T.; Abraham, L.J.; Yu, C.; Rhim, J.A.; Fausto, N. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J. Exp. Med. 2000, 192, 1809–1818. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Kucharczak, J.; Simmons, M.J.; Fan, Y.; Gelinas, C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene 2003, 22, 8961–8982. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Singh, A.; Borkham-Kamphorst, E.; Nevzorova, Y.A.; Al Masaoudi, M.; Haas, U.; Boekschoten, M.V.; Gassler, N.; Weiskirchen, R.; Muller, M.; et al. TNFR1 determines progression of chronic liver injury in the IKKgamma/Nemo genetic model. Cell Death Differ. 2013, 20, 1580–1592. [Google Scholar] [CrossRef]
- Wunderlich, F.T.; Luedde, T.; Singer, S.; Schmidt-Supprian, M.; Baumgartl, J.; Schirmacher, P.; Pasparakis, M.; Bruning, J.C. Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 1297–1302. [Google Scholar] [CrossRef]
- Kondylis, V.; Polykratis, A.; Ehlken, H.; Ochoa-Callejero, L.; Straub, B.K.; Krishna-Subramanian, S.; Van, T.M.; Curth, H.M.; Heise, N.; Weih, F.; et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell 2015, 28, 830. [Google Scholar] [CrossRef]
- Wesemann, D.R.; Benveniste, E.N. STAT-1 alpha and IFN-gamma as modulators of TNF-alpha signaling in macrophages: Regulation and functional implications of the TNF receptor 1:STAT-1 alpha complex. J. Immunol. 2003, 171, 5313–5319. [Google Scholar] [CrossRef]
- Li, N.; Wang, J.; Zhang, N.; Zhuang, M.; Zong, Z.; Zou, J.; Li, G.; Wang, X.; Zhou, H.; Zhang, L.; et al. Cross-talk between TNF-alpha and IFN-gamma signaling in induction of B7-H1 expression in hepatocellular carcinoma cells. Cancer Immunol. Immunother. CII 2018, 67, 271–283. [Google Scholar] [CrossRef]
- Chen, H.; Hu, L.; Luo, Z.; Zhang, J.; Zhang, C.; Qiu, B.; Dong, L.; Tan, Y.; Ding, J.; Tang, S.; et al. A20 suppresses hepatocellular carcinoma proliferation and metastasis through inhibition of Twist1 expression. Mol. Cancer 2015, 14, 186. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Farhang Ghahremani, M.; Vereecke, L.; Youssef, S.A.; Mc Guire, C.; Sze, M.; Weber, A.; Heikenwalder, M.; de Bruin, A.; Beyaert, R.; et al. A20 prevents chronic liver inflammation and cancer by protecting hepatocytes from death. Cell Death Dis. 2016, 7, e2250. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation Mediated by JNK in Myeloid Cells Promotes the Development of Hepatitis and Hepatocellular Carcinoma. Cell Rep. 2016, 15, 19–26. [Google Scholar] [CrossRef]
- Bettermann, K.; Vucur, M.; Haybaeck, J.; Koppe, C.; Janssen, J.; Heymann, F.; Weber, A.; Weiskirchen, R.; Liedtke, C.; Gassler, N.; et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell 2010, 17, 481–496. [Google Scholar] [CrossRef]
- Wen, J.; Wu, Y.; Wei, W.; Li, Z.; Wang, P.; Zhu, S.; Dong, W. Protective effects of recombinant human cytoglobin against chronic alcohol-induced liver disease in vivo and in vitro. Sci. Rep. 2017, 7, 41647. [Google Scholar] [CrossRef]
- Kastl, L.; Sauer, S.W.; Ruppert, T.; Beissbarth, T.; Becker, M.S.; Suss, D.; Krammer, P.H.; Gulow, K. TNF-alpha mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-kappaB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [PubMed]
- Themanns, M.; Mueller, K.M.; Kessler, S.M.; Golob-Schwarzl, N.; Mohr, T.; Kaltenecker, D.; Bourgeais, J.; Paier-Pourani, J.; Friedbichler, K.; Schneller, D.; et al. Hepatic Deletion of Janus Kinase 2 Counteracts Oxidative Stress in Mice. Sci. Rep. 2016, 6, 34719. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E. The biological effects of interleukin 6. Med. Res. Rev. 1996, 16, 87–109. [Google Scholar] [CrossRef]
- Yasukawa, K.; Hirano, T.; Watanabe, Y.; Muratani, K.; Matsuda, T.; Nakai, S.; Kishimoto, T. Structure and expression of human B cell stimulatory factor-2 (BSF-2/IL-6) gene. EMBO J. 1987, 6, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, K.; Nakagawa, T.; Fukunaga, K.; Tseng, L.T.; Yamamura, Y.; Kishimoto, T. Isolation and characterization of B cell differentiation factor (BCDF) secreted from a human B lymphoblastoid cell line. J. Immunol. 1984, 132, 2948–2954. [Google Scholar] [PubMed]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Denson, J.L.; Bruning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the exercise factor: Is IL-6 a candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Kammoun, H.L.; Allen, T.L.; Deswaerte, V.; Henstridge, D.C.; Estevez, E.; Matthews, V.B.; Neill, B.; White, D.A.; Murphy, A.J.; et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab. 2015, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Elewaut, D.; McInnes, I.B.; Dayer, J.M.; Neurath, M.F. How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Kopf, M.; Lassmann, H.; Fontana, A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur. J. Immunol. 1998, 28, 2178–2187. [Google Scholar] [CrossRef]
- Alonzi, T.; Fattori, E.; Lazzaro, D.; Costa, P.; Probert, L.; Kollias, G.; De Benedetti, F.; Poli, V.; Ciliberto, G. Interleukin 6 is required for the development of collagen-induced arthritis. J. Exp. Med. 1998, 187, 461–468. [Google Scholar] [CrossRef]
- Ohshima, S.; Saeki, Y.; Mima, T.; Sasai, M.; Nishioka, K.; Nomura, S.; Kopf, M.; Katada, Y.; Tanaka, T.; Suemura, M.; et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8222–8226. [Google Scholar] [CrossRef]
- Richards, H.B.; Satoh, M.; Shaw, M.; Libert, C.; Poli, V.; Reeves, W.H. Interleukin 6 dependence of anti-DNA antibody production: Evidence for two pathways of autoantibody formation in pristane-induced lupus. J. Exp. Med. 1998, 188, 985–990. [Google Scholar] [CrossRef]
- Lattanzio, G.; Libert, C.; Aquilina, M.; Cappelletti, M.; Ciliberto, G.; Musiani, P.; Poli, V. Defective development of pristane-oil-induced plasmacytomas in interleukin-6-deficient BALB/c mice. Am. J. Pathol. 1997, 151, 689–696. [Google Scholar]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300 Pt 2, 281–290. [Google Scholar] [CrossRef]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.; Straub, B.K.; Ackermann, P.J.; Wunderlich, C.M.; Mauer, J.; Seeger, J.M.; Buning, H.; Heukamp, L.; Kashkar, H.; Schirmacher, P.; et al. Obesity promotes liver carcinogenesis via Mcl-1 stabilization independent of IL-6Ralpha signaling. Cell Rep. 2013, 4, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Han, Y.; Hughart, N.; McCarra, J.; Alpini, G.; Meng, F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl. Gastrointest. Cancer 2012, 1, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Lutticken, C.; Wegenka, U.M.; Yuan, J.; Buschmann, J.; Schindler, C.; Ziemiecki, A.; Harpur, A.G.; Wilks, A.F.; Yasukawa, K.; Taga, T.; et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994, 263, 89–92. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.E., Jr.; Yancopoulos, G.D. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Gerhartz, C.; Heesel, B.; Sasse, J.; Hemmann, U.; Landgraf, C.; Schneider-Mergener, J.; Horn, F.; Heinrich, P.C.; Graeve, L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J. Biol. Chem. 1996, 271, 12991–12998. [Google Scholar] [CrossRef]
- Ihle, J.N. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv. Immunol. 1995, 60, 1–35. [Google Scholar] [PubMed]
- Wegiel, B.; Bjartell, A.; Culig, Z.; Persson, J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 2008, 122, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Nakajima, K.; Hibi, M. Signaling mechanisms through gp130: A model of the cytokine system. Cytokine Growth Factor Rev. 1997, 8, 241–252. [Google Scholar] [CrossRef]
- Novick, D.; Engelmann, H.; Wallach, D.; Rubinstein, M. Soluble cytokine receptors are present in normal human urine. J. Exp. Med. 1989, 170, 1409–1414. [Google Scholar] [CrossRef]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Mullberg, J.; Schooltink, H.; Stoyan, T.; Gunther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef]
- Blindenbacher, A.; Wang, X.; Langer, I.; Savino, R.; Terracciano, L.; Heim, M.H. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology 2003, 38, 674–682. [Google Scholar] [CrossRef]
- Dienz, O.; Rud, J.G.; Eaton, S.M.; Lanthier, P.A.; Burg, E.; Drew, A.; Bunn, J.; Suratt, B.T.; Haynes, L.; Rincon, M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012, 5, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hoge, J.; Yan, I.; Janner, N.; Schumacher, V.; Chalaris, A.; Steinmetz, O.M.; Engel, D.R.; Scheller, J.; Rose-John, S.; Mittrucker, H.W. IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J. Immunol. 2013, 190, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Kohler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.P.; Wright, K.; Lauder, S.N.; Nowell, M.A.; Jones, G.W.; Godkin, A.J.; Jones, S.A.; Gallimore, A.M. Interleukin-6 is crucial for recall of influenza-specific memory CD4 T cells. PLoS Pathog. 2008, 4, e1000006. [Google Scholar] [CrossRef] [PubMed]
- Neveu, W.A.; Allard, J.B.; Dienz, O.; Wargo, M.J.; Ciliberto, G.; Whittaker, L.A.; Rincon, M. IL-6 is required for airway mucus production induced by inhaled fungal allergens. J. Immunol. 2009, 183, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Waage, A.; Brandtzaeg, P.; Halstensen, A.; Kierulf, P.; Espevik, T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 1989, 169, 333–338. [Google Scholar] [CrossRef]
- Kado, S.; Nagase, T.; Nagata, N. Circulating levels of interleukin-6, its soluble receptor and interleukin-6/interleukin-6 receptor complexes in patients with type 2 diabetes mellitus. Acta Diabetol. 1999, 36, 67–72. [Google Scholar] [CrossRef]
- Van Greevenbroek, M.M.; Schalkwijk, C.G.; Stehouwer, C.D. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Boeing, H.; Nothlings, U.; Jenab, M.; Fedirko, V.; Kaaks, R.; Lukanova, A.; Trichopoulou, A.; Trichopoulos, D.; Boffetta, P.; et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014, 60, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; De Amici, M.; Quaglini, S.; Paglino, C.; Tagliani, F.; Boncimino, A.; Moratti, R.; Corazza, G.R. Circulating interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann. Oncol. 2008, 19, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Oh, B.S.; Kwon, J.H.; You, C.R.; Chung, K.W.; Kay, C.S.; Jung, H.S. Serum interleukin-6 and C-reactive protein as a prognostic indicator in hepatocellular carcinoma. Cytokine 2012, 60, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Spannbauer, M.; Peng, J.; Al Masaoudi, M.; Sellge, G.; Nevzorova, Y.A.; Gassler, N.; Liedtke, C.; Cubero, F.J.; Trautwein, C. Lack of gp130 expression in hepatocytes attenuates tumor progression in the DEN model. Cell Death Dis. 2015, 6, e1667. [Google Scholar] [CrossRef] [PubMed]
- Mittenbuhler, M.J.; Sprenger, H.G.; Gruber, S.; Wunderlich, C.M.; Kern, L.; Bruning, J.C.; Wunderlich, F.T. Hepatic leptin receptor expression can partially compensate for IL-6Ralpha deficiency in DEN-induced hepatocellular carcinoma. Mol. Metab. 2018, 17, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306 e1220. [Google Scholar] [CrossRef]
- Bergmann, J.; Muller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lucke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef]
- Wang, C.Q.; Sun, H.T.; Gao, X.M.; Ren, N.; Sheng, Y.Y.; Wang, Z.; Zheng, Y.; Wei, J.W.; Zhang, K.L.; Yu, X.X.; et al. Interleukin-6 enhances cancer stemness and promotes metastasis of hepatocellular carcinoma via up-regulating osteopontin expression. Am. J. Cancer Res. 2016, 6, 1873–1889. [Google Scholar]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983.e11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F translation initiation complex: A critical nexus for cancer development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef]
- Wendel, H.G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; Martineau, Y.; Sato, T.A.; Larsson, O.; Rajasekhar, V.K.; Sonenberg, N. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS ONE 2007, 2, e242. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar] [CrossRef]
- Fan, L.C.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Chu, P.Y.; Hsieh, F.S.; Lin, H.; Yu, H.C.; Chen, K.F. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2017, 36, 5768–5769. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J. Negative regulators of cytokine signal transduction. Cell. Mol. Life Sci. 1999, 55, 1568–1577. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, P.J.; Chu, P.Y.; Huang, H.P.; Liu, C.Y.; Huang, J.W.; Chen, K.F. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 2014, 59, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; Willson, T.A.; Farley, A.; Starr, R.; Zhang, J.G.; Baca, M.; Alexander, W.S.; Metcalf, D.; Hilton, D.J.; Nicola, N.A. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999, 18, 375–385. [Google Scholar] [CrossRef]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Forster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef]
- Alexander, W.S.; Starr, R.; Fenner, J.E.; Scott, C.L.; Handman, E.; Sprigg, N.S.; Corbin, J.E.; Cornish, A.L.; Darwiche, R.; Owczarek, C.M.; et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999, 98, 597–608. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, e24053. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; De Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Kroemer, R.T. A three-dimensional model of Suppressor Of Cytokine Signalling 1 (SOCS-1). Protein Eng. 2003, 16, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.J.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA 2015, 112, E2979. [Google Scholar] [CrossRef]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef]
- Haan, S.; Ferguson, P.; Sommer, U.; Hiremath, M.; McVicar, D.W.; Heinrich, P.C.; Johnston, J.A.; Cacalano, N.A. Tyrosine phosphorylation disrupts elongin interaction and accelerates SOCS3 degradation. J. Biol. Chem. 2003, 278, 31972–31979. [Google Scholar] [CrossRef]
- Starr, R.; Metcalf, D.; Elefanty, A.G.; Brysha, M.; Willson, T.A.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. USA 1998, 95, 14395–14399. [Google Scholar] [CrossRef]
- Marine, J.C.; McKay, C.; Wang, D.; Topham, D.J.; Parganas, E.; Nakajima, H.; Pendeville, H.; Yasukawa, H.; Sasaki, A.; Yoshimura, A.; et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell 1999, 98, 617–627. [Google Scholar] [CrossRef]
- Roberts, A.W.; Robb, L.; Rakar, S.; Hartley, L.; Cluse, L.; Nicola, N.A.; Metcalf, D.; Hilton, D.J.; Alexander, W.S. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA 2001, 98, 9324–9329. [Google Scholar] [CrossRef]
- Ogata, H.; Chinen, T.; Yoshida, T.; Kinjyo, I.; Takaesu, G.; Shiraishi, H.; Iida, M.; Kobayashi, T.; Yoshimura, A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 2006, 25, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Hovelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAKSTAT 2013, 2, e23878. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Rubin, D.C.; Shaker, A.; Levin, M.S. Chronic intestinal inflammation: Inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 2012, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Dignam, J.J.; Polite, B.N.; Yothers, G.; Raich, P.; Colangelo, L.; O’Connell, M.J.; Wolmark, N. Body mass index and outcomes in patients who receive adjuvant chemotherapy for colon cancer. J. Natl. Cancer Inst. 2006, 98, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.; Davis, J.M.; Carson, J.A. The interaction of a high-fat diet and regular moderate intensity exercise on intestinal polyp development in Apc Min/+ mice. Cancer Prev. Res. 2009, 2, 641–649. [Google Scholar] [CrossRef]
- Yang, K.; Edelmann, W.; Fan, K.; Lau, K.; Leung, D.; Newmark, H.; Kucherlapati, R.; Lipkin, M. Dietary modulation of carcinoma development in a mouse model for human familial adenomatous polyposis. Cancer Res. 1998, 58, 5713–5717. [Google Scholar]
- Schulz, M.D.; Atay, C.; Heringer, J.; Romrig, F.K.; Schwitalla, S.; Aydin, B.; Ziegler, P.K.; Varga, J.; Reindl, W.; Pommerenke, C.; et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 2014, 514, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Newmark, H.L.; Yang, K.; Kurihara, N.; Fan, K.; Augenlicht, L.H.; Lipkin, M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: A preclinical model for human sporadic colon cancer. Carcinogenesis 2009, 30, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Kurihara, N.; Fan, K.; Newmark, H.; Rigas, B.; Bancroft, L.; Corner, G.; Livote, E.; Lesser, M.; Edelmann, W.; et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008, 68, 7803–7810. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2012, 23, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation that is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Rah, B.; Bastola, D.; Dhawan, P.; Singh, A.B. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci. Rep. 2017, 7, 5125. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Manco, M.; Ibanez, J.; Garcia-Fuentes, E.; Ortega, F.; Gorostiaga, E.; Vendrell, J.; Izquierdo, M.; Martinez, C.; Nolfe, G.; et al. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int. J. Obes. 2010, 34, 240–249. [Google Scholar] [CrossRef]
- Reinecker, H.C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MacDermott, R.P.; Raedler, A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 1993, 94, 174–181. [Google Scholar] [CrossRef]
- Harper, J.W.; Zisman, T.L. Interaction of obesity and inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 7868–7881. [Google Scholar] [CrossRef]
- Izano, M.; Wei, E.K.; Tai, C.; Swede, H.; Gregorich, S.; Harris, T.B.; Klepin, H.; Satterfield, S.; Murphy, R.; Newman, A.B.; et al. Chronic inflammation and risk of colorectal and other obesity-related cancers: The health, aging and body composition study. Int. J. Cancer 2016, 138, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Ackermann, P.J.; Ostermann, A.L.; Adams-Quack, P.; Vogt, M.C.; Tran, M.L.; Nikolajev, A.; Waisman, A.; Garbers, C.; Theurich, S.; et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat. Commun. 2018, 9, 1646. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Yamada, Y.; Shimizu, M.; Saito, K.; Moriwaki, H.; Hara, A. Genetic ablation of Tnfalpha demonstrates no detectable suppressive effect on inflammation-related mouse colon tumorigenesis. Chem. Biol. Interact. 2010, 184, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569. [Google Scholar] [CrossRef]
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Männel, D.N.; Andus, T.; Schölmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997, 107, 353–358. [Google Scholar] [CrossRef]
- Pendyala, S.; Neff, L.M.; Suarez-Farinas, M.; Holt, P.R. Diet-induced weight loss reduces colorectal inflammation: Implications for colorectal carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef]
- Olivo-Marston, S.E.; Hursting, S.D.; Perkins, S.N.; Schetter, A.; Khan, M.; Croce, C.; Harris, C.C.; Lavigne, J. Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and microRNA expression. PLoS ONE 2014, 9, e94765. [Google Scholar] [CrossRef]
- Aboura, I.; Nani, A.; Belarbi, M.; Murtaza, B.; Fluckiger, A.; Dumont, A.; Benammar, C.; Tounsi, M.S.; Ghiringhelli, F.; Rialland, M.; et al. Protective effects of polyphenol-rich infusions from carob (Ceratonia siliqua) leaves and cladodes of Opuntia ficus-indica against inflammation associated with diet-induced obesity and DSS-induced colitis in Swiss mice. Biomed. Pharmacother. 2017, 96, 1022–1035. [Google Scholar] [CrossRef]
- Saadatdoust, Z.; Pandurangan, A.K.; Ananda Sadagopan, S.K.; Mohd Esa, N.; Ismail, A.; Mustafa, M.R. Dietary cocoa inhibits colitis associated cancer: A crucial involvement of the IL-6/STAT3 pathway. J. Nutr. Biochem. 2015, 26, 1547–1558. [Google Scholar] [CrossRef]
- Yook, J.S.; Kim, K.A.; Kim, M.; Cha, Y.S. Black Adzuki Bean (Vigna angularis) Attenuates High-Fat Diet-Induced Colon Inflammation in Mice. J. Med. Food 2017, 20, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; de Sousa Moraes, L.F.; Lebow, N.; Zhu, M.J. Dietary Green Pea Protects against DSS-Induced Colitis in Mice Challenged with High-Fat Diet. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- Setayesh, T.; Nersesyan, A.; Misik, M.; Noorizadeh, R.; Haslinger, E.; Javaheri, T.; Lang, E.; Grusch, M.; Huber, W.; Haslberger, A.; et al. Gallic acid, a common dietary phenolic protects against high fat diet induced DNA damage. Eur. J. Nutr. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Tabrizian, T.; Novaj, A.; Nakanishi, M.; Rosenberg, D.W.; Huffman, D.M. Dietary Walnuts Protect Against Obesity-Driven Intestinal Stem Cell Decline and Tumorigenesis. Front. Nutr. 2018, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, L.; Li, J.; Lai, M.; Wen, X. The Effects of Berberine on the Gut Microbiota in Apc (min/+) Mice Fed with a High Fat Diet. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Kang, Y.; Du, M.; Zhu, M.J. Maternal high-fat diet consumption enhances offspring susceptibility to DSS-induced colitis in mice. Obesity 2017, 25, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Sun, Y.M. The IL-6/JAK/STAT3 pathway: Potential therapeutic strategies in treating colorectal cancer (Review). Int. J. Oncol. 2014, 44, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Kantola, T.; Klintrup, K.; Vayrynen, J.P.; Vornanen, J.; Bloigu, R.; Karhu, T.; Herzig, K.H.; Napankangas, J.; Makela, J.; Karttunen, T.J.; et al. Stage-dependent alterations of the serum cytokine pattern in colorectal carcinoma. Br. J. Cancer 2012, 107, 1729–1736. [Google Scholar] [CrossRef]
- Knupfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients--a summary of published results. Int. J. Color. Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef]
- Lee, W.S.; Baek, J.H.; You, D.H.; Nam, M.J. Prognostic value of circulating cytokines for stage III colon cancer. J. Surg. Res. 2013, 182, 49–54. [Google Scholar] [CrossRef]
- Sasaki, Y.; Takeda, H.; Sato, T.; Orii, T.; Nishise, S.; Nagino, K.; Iwano, D.; Yaoita, T.; Yoshizawa, K.; Saito, H.; et al. Serum Interleukin-6, insulin, and HOMA-IR in male individuals with colorectal adenoma. Clin. Cancer Res. 2012, 18, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Neurath, M.F. TGF-beta as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev. 2006, 17, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Lehr, H.A.; Galle, P.R.; Rose-John, S.; Neurath, M.F. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle 2005, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef]
- Aden, K.; Breuer, A.; Rehman, A.; Geese, H.; Tran, F.; Sommer, J.; Waetzig, G.H.; Reinheimer, T.M.; Schreiber, S.; Rose-John, S.; et al. Classic IL-6R signalling is dispensable for intestinal epithelial proliferation and repair. Oncogenesis 2016, 5, e270. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kern, L.; Mittenbühler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers. Cancers 2019, 11, 24. https://doi.org/10.3390/cancers11010024
Kern L, Mittenbühler MJ, Vesting AJ, Ostermann AL, Wunderlich CM, Wunderlich FT. Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers. Cancers. 2019; 11(1):24. https://doi.org/10.3390/cancers11010024
Chicago/Turabian StyleKern, Lara, Melanie J. Mittenbühler, Anna Juliane Vesting, Anna Lena Ostermann, Claudia Maria Wunderlich, and F. Thomas Wunderlich. 2019. "Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers" Cancers 11, no. 1: 24. https://doi.org/10.3390/cancers11010024
APA StyleKern, L., Mittenbühler, M. J., Vesting, A. J., Ostermann, A. L., Wunderlich, C. M., & Wunderlich, F. T. (2019). Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation—Driven Liver and Colorectal Cancers. Cancers, 11(1), 24. https://doi.org/10.3390/cancers11010024