Hypoxic Radioresistance: Can ROS Be the Key to Overcome It?



Abstract
1. Introduction
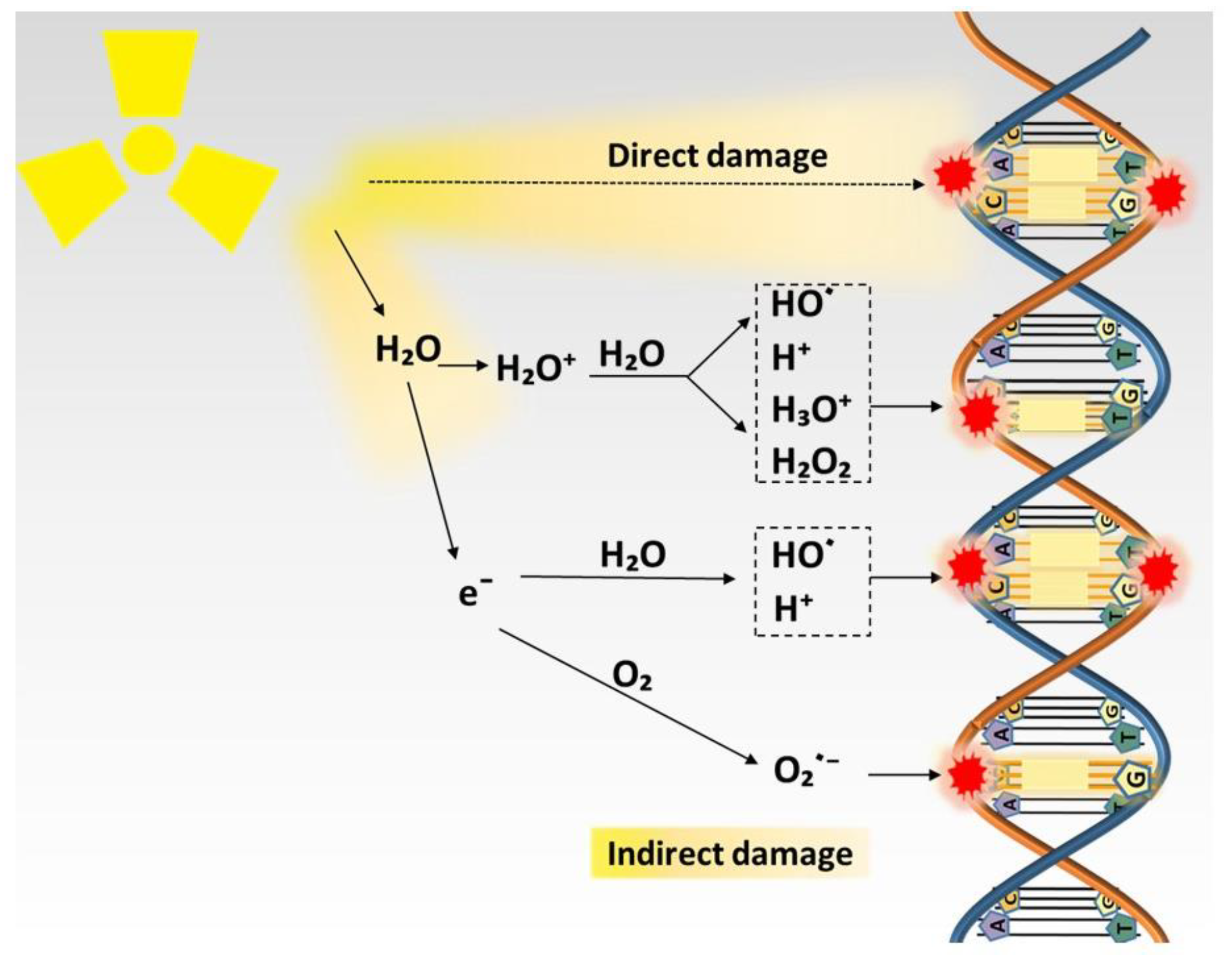
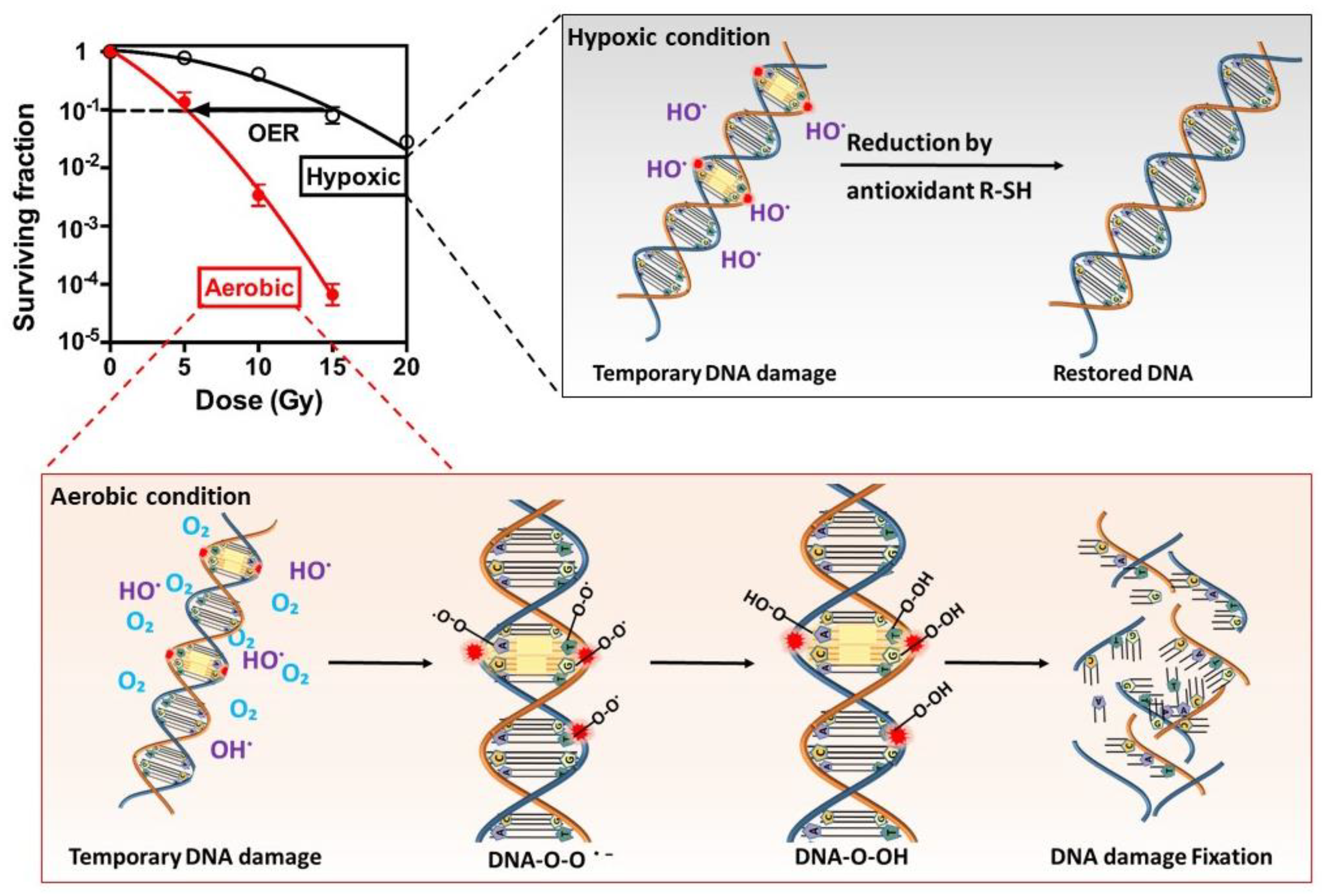
2. Biological Action of Radiation
3. Hypoxia
3.1. Tumor Vasculature and Hypoxia
3.2. Mechanisms of Hypoxic Radioresistance
3.3. Tumor Hypoxia and Radiotherapy Outcome
4. ROS
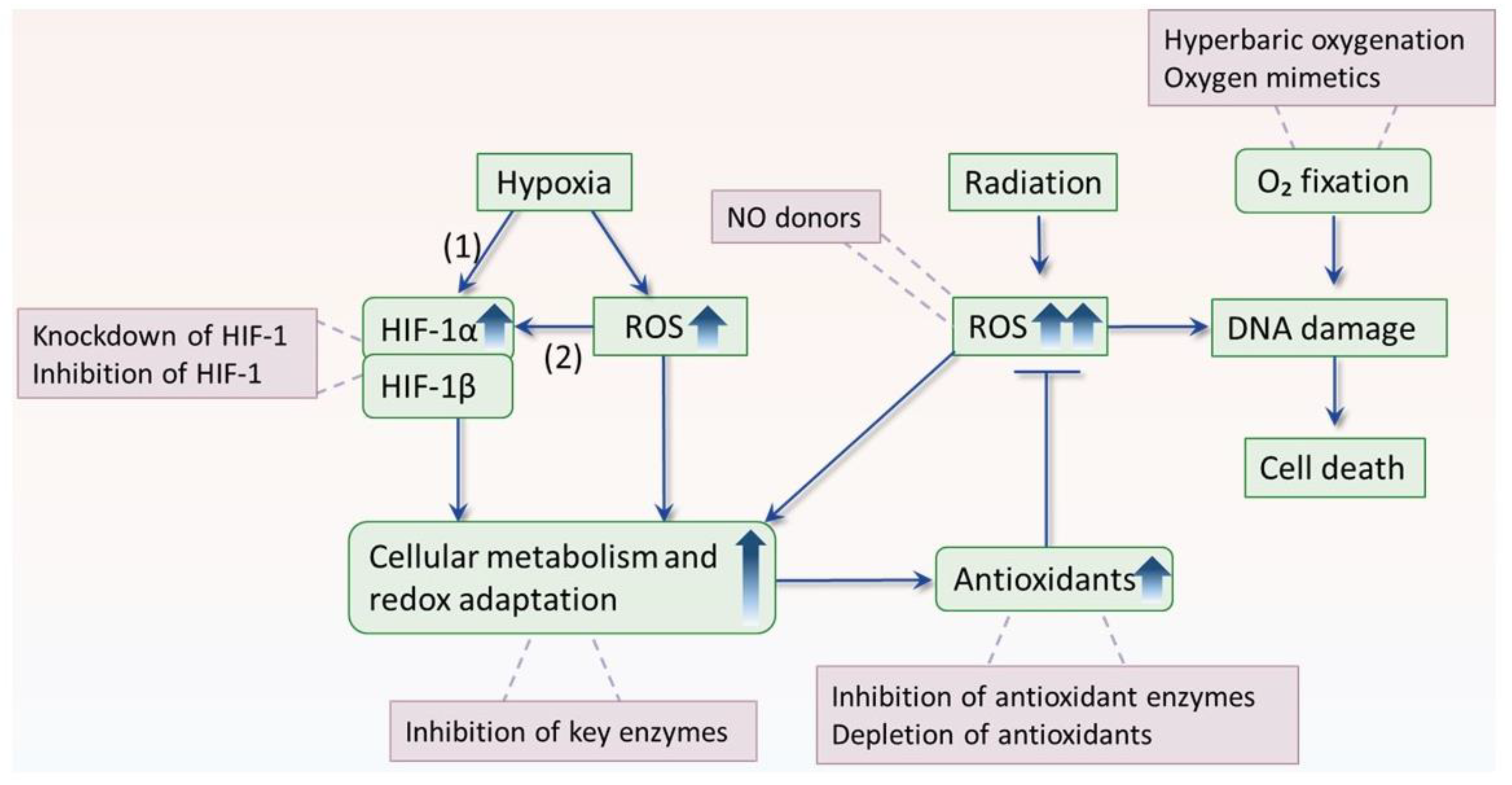
5. The Interplay between Hypoxia and ROS
5.1. Hypoxia Enhances ROS Production
5.2. ROS Mediates Hypoxia Adaptation
6. Overcoming Hypoxic Radioresistance by Disruption of ROS Homeostasis
6.1. Hypoxic Radiosensitization by NO
6.2. Hypoxic Radiosensitization by Inhibition of Antioxidant Enzymes
6.3. Hypoxic Radiosensitization by Inhibition of HIF-1
6.4. Hypoxic Radiosensitization by Inhibition of Tumor Metabolism
6.5. Hypoxic Radiosensitization via Reduction in Oxygen Demand
6.6. Others
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Ohno, T. Particle radiotherapy with carbon ion beams. EPMA J. 2013, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Kase, Y. Biological characteristics of carbon-ion therapy. Int. J. Radiat. Biol. 2009, 85, 715–728. [Google Scholar] [CrossRef]
- Ogata, T.; Teshima, T.; Kagawa, K.; Hishikawa, Y.; Takahashi, Y.; Kawaguchi, A.; Suzumoto, Y.; Nojima, K.; Furusawa, Y.; Matsuura, N. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005, 65, 113–120. [Google Scholar] [PubMed]
- Cui, X.; Oonishi, K.; Tsujii, H.; Yasuda, T.; Matsumoto, Y.; Furusawa, Y.; Akashi, M.; Kamada, T.; Okayasu, R. Effects of carbon ion beam on putative colon cancer stem cells and its comparison with X-rays. Cancer Res. 2011, 71, 3676–3687. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J. Hypoxic radiosensitization: Adored and ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef] [PubMed]
- McParland, B.J. Nuclear Medicine Radiation Dosimetry: Advanced Theoretical Principles; Springer: London, UK, 2010. [Google Scholar]
- Pouget, J.P.; Frelon, S.; Ravanat, J.L.; Testard, I.; Odin, F.; Cadet, J. Formation of modified DNA bases in cells exposed either to gamma radiation or to high-LET particles. Radiat. Res. 2002, 157, 589–595. [Google Scholar] [CrossRef]
- Sachs, R.K.; Chen, P.L.; Hahnfeldt, P.J.; Hlatky, L.R. DNA damage caused by ionizing radiation. Math. Biosci. 1992, 112, 271–303. [Google Scholar] [CrossRef]
- Zhang, Y.; Martin, S.G. Redox proteins and radiotherapy. Clin. Oncol. 2014, 26, 289–300. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, M.; Verellen, D.; Verovski, V.; Storme, G. Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide 2008, 19, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Russo, A.; Biaglow, J.E.; McPherson, S. Cellular glutathione depletion by diethyl maleate or buthionine sulfoximine: No effect of glutathione depletion on the oxygen enhancement ratio. Radiat. Res. 1983, 96, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Biaglow, J.E.; Clark, E.P.; Epp, E.R.; Morse-Guadio, M.; Varnes, M.E.; Mitchell, J.B. Nonprotein thiols and the radiation response of A549 human lung carcinoma cells. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1983, 44, 489–495. [Google Scholar] [CrossRef]
- Held, K.D.; Epp, E.R.; Clark, E.P.; Biaglow, J.E. Effect of dimethyl fumarate on the radiation sensitivity of mammalian cells in vitro. Radiat. Res. 1988, 115, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Bump, E.A.; Yu, N.Y.; Brown, J.M. Radiosensitization of hypoxic tumor cells by depletion of intracellular glutathione. Science 1982, 217, 544–545. [Google Scholar] [CrossRef]
- Matschke, J.; Riffkin, H.; Klein, D.; Handrick, R.; Ludemann, L.; Metzen, E.; Shlomi, T.; Stuschke, M.; Jendrossek, V. Targeted Inhibition of Glutamine-Dependent Glutathione Metabolism Overcomes Death Resistance Induced by Chronic Cycling Hypoxia. Antioxid. Redox Signal. 2016, 25, 89–107. [Google Scholar] [CrossRef]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat. Res. 2016, 186, 385–395. [Google Scholar] [CrossRef]
- Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Auranofin radiosensitizes tumor cells through targeting thioredoxin reductase and resulting overproduction of reactive oxygen species. Oncotarget 2017, 8, 35728–35742. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, D.; Shen, L.; Dong, K.; Wu, M.; Ou, Z.; Shi, D. Redox homeostasis protects mitochondria through accelerating ROS conversion to enhance hypoxia resistance in cancer cells. Sci. Rep. 2016, 6, 22831. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, M.; Wouters, B.G. The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin. Radiat. Oncol. 2013, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-modulated phenomena and radiation therapy: The central role of superoxide dismutases. Antioxid. Redox Signal. 2014, 20, 1567–1589. [Google Scholar] [CrossRef]
- Horsman, M.R. Measurement of tumor oxygenation. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 701–704. [Google Scholar] [CrossRef]
- Brown, J.M. The hypoxic cell: A target for selective cancer therapy—Eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br. J. Radiol. 1979, 52, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Rey, S.; Schito, L.; Koritzinsky, M.; Wouters, B.G. Molecular targeting of hypoxia in radiotherapy. Adv. Drug Deliv. Rev. 2017, 109, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016, 76, 6458–6462. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 3–23. [Google Scholar]
- Wu, S.B.; Wei, Y.H. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: Implication of the cell survival in mitochondrial diseases. Biochim. Biophys. Acta 2012, 1822, 233–247. [Google Scholar] [CrossRef]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar] [PubMed]
- Knocke, T.H.; Weitmann, H.D.; Feldmann, H.J.; Selzer, E.; Potter, R. Intratumoral pO2-measurements as predictive assay in the treatment of carcinoma of the uterine cervix. Radiother. Oncol. 1999, 53, 99–104. [Google Scholar] [CrossRef]
- Sundfor, K.; Lyng, H.; Trope, C.G.; Rofstad, E.K. Treatment outcome in advanced squamous cell carcinoma of the uterine cervix: Relationships to pretreatment tumor oxygenation and vascularization. Radiother. Oncol. 2000, 54, 101–107. [Google Scholar] [CrossRef]
- Fyles, A.; Milosevic, M.; Hedley, D.; Pintilie, M.; Levin, W.; Manchul, L.; Hill, R.P. Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J. Clin. Oncol. 2002, 20, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Nordsmark, M.; Loncaster, J.; Aquino-Parsons, C.; Chou, S.C.; Gebski, V.; West, C.; Lindegaard, J.C.; Havsteen, H.; Davidson, S.E.; Hunter, R.; et al. The prognostic value of pimonidazole and tumour pO2 in human cervix carcinomas after radiation therapy: A prospective international multi-center study. Radiother. Oncol. 2006, 80, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Kessler, H.B.; Rosenblum, J.S.; Coia, L.R.; Moldofsky, P.J.; Hartz, W.H.; Broder, G.J. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1988, 14, 831–838. [Google Scholar] [CrossRef]
- Brizel, D.M.; Dodge, R.K.; Clough, R.W.; Dewhirst, M.W. Oxygenation of head and neck cancer: Changes during radiotherapy and impact on treatment outcome. Radiother. Oncol. 1999, 53, 113–117. [Google Scholar] [CrossRef]
- Stadler, P.; Becker, A.; Feldmann, H.J.; Hansgen, G.; Dunst, J.; Wurschmidt, F.; Molls, M. Influence of the hypoxic subvolume on the survival of patients with head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 1999, 44, 749–754. [Google Scholar] [CrossRef]
- Rudat, V.; Stadler, P.; Becker, A.; Vanselow, B.; Dietz, A.; Wannenmacher, M.; Molls, M.; Dunst, J.; Feldmann, H.J. Predictive value of the tumor oxygenation by means of pO2 histography in patients with advanced head and neck cancer. Strahlenther. Onkol. 2001, 177, 462–468. [Google Scholar] [CrossRef]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Brizel, D.M.; Scully, S.P.; Harrelson, J.M.; Layfield, L.J.; Bean, J.M.; Prosnitz, L.R.; Dewhirst, M.W. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996, 56, 941–943. [Google Scholar] [PubMed]
- Nordsmark, M.; Alsner, J.; Keller, J.; Nielsen, O.S.; Jensen, O.M.; Horsman, M.R.; Overgaard, J. Hypoxia in human soft tissue sarcomas: Adverse impact on survival and no association with p53 mutations. Br. J. Cancer 2001, 84, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Lopci, E.; Grassi, I.; Chiti, A.; Nanni, C.; Cicoria, G.; Toschi, L.; Fonti, C.; Lodi, F.; Mattioli, S.; Fanti, S. PET radiopharmaceuticals for imaging of tumor hypoxia: A review of the evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384. [Google Scholar] [PubMed]
- Rajendran, J.G.; Mankoff, D.A.; O’Sullivan, F.; Peterson, L.M.; Schwartz, D.L.; Conrad, E.U.; Spence, A.M.; Muzi, M.; Farwell, D.G.; Krohn, K.A. Hypoxia and glucose metabolism in malignant tumors: Evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin. Cancer Res. 2004, 10, 2245–2252. [Google Scholar] [CrossRef]
- Koh, W.J.; Bergman, K.S.; Rasey, J.S.; Peterson, L.M.; Evans, M.L.; Graham, M.M.; Grierson, J.R.; Lindsley, K.L.; Lewellen, T.K.; Krohn, K.A.; et al. Evaluation of oxygenation status during fractionated radiotherapy in human nonsmall cell lung cancers using [F-18]fluoromisonidazole positron emission tomography. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 391–398. [Google Scholar] [CrossRef]
- Rajendran, J.G.; Schwartz, D.L.; O’Sullivan, J.; Peterson, L.M.; Ng, P.; Scharnhorst, J.; Grierson, J.R.; Krohn, K.A. Tumor hypoxia imaging with [F-18] fluoromisonidazole positron emission tomography in head and neck cancer. Clin. Cancer Res. 2006, 12, 5435–5441. [Google Scholar] [CrossRef]
- Horsman, M.R.; Mortensen, L.S.; Petersen, J.B.; Busk, M.; Overgaard, J. Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol. 2012, 9, 674–687. [Google Scholar] [CrossRef]
- Loncaster, J.A.; Carrington, B.M.; Sykes, J.R.; Jones, A.P.; Todd, S.M.; Cooper, R.; Buckley, D.L.; Davidson, S.E.; Logue, J.P.; Hunter, R.D.; et al. Prediction of radiotherapy outcome using dynamic contrast enhanced MRI of carcinoma of the cervix. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 759–767. [Google Scholar] [CrossRef]
- Mayr, N.A.; Wang, J.Z.; Zhang, D.; Grecula, J.C.; Lo, S.S.; Jaroura, D.; Montebello, J.; Zhang, H.; Li, K.; Lu, L.; et al. Longitudinal changes in tumor perfusion pattern during the radiation therapy course and its clinical impact in cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 502–508. [Google Scholar] [CrossRef]
- Andersen, E.K.; Hole, K.H.; Lund, K.V.; Sundfor, K.; Kristensen, G.B.; Lyng, H.; Malinen, E. Dynamic contrast-enhanced MRI of cervical cancers: Temporal percentile screening of contrast enhancement identifies parameters for prediction of chemoradioresistance. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, e485–e492. [Google Scholar] [CrossRef]
- Hompland, T.; Hole, K.H.; Ragnum, H.B.; Aarnes, E.K.; Vlatkovic, L.; Lie, A.K.; Patzke, S.; Brennhovd, B.; Seierstad, T.; Lyng, H. Combined MR Imaging of Oxygen Consumption and Supply Reveals Tumor Hypoxia and Aggressiveness in Prostate Cancer Patients. Cancer Res. 2018, 78, 4774–4785. [Google Scholar] [CrossRef]
- Airley, R.E.; Phillips, R.M.; Evans, A.E.; Double, J.; Burger, A.M.; Feibig, H.H.; West, C.M.; Stratford, I.J. Hypoxia-regulated glucose transporter Glut-1 may influence chemosensitivity to some alkylating agents: Results of EORTC (First Translational Award) study of the relevance of tumour hypoxia to the outcome of chemotherapy in human tumour-derived xenografts. Int. J. Oncol. 2005, 26, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Heijn, M.; di Tomaso, E.; Griffon-Etienne, G.; Ancukiewicz, M.; Koike, C.; Park, K.R.; Ferrara, N.; Jain, R.K.; Suit, H.D.; et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000, 60, 5565–5570. [Google Scholar] [PubMed]
- Mauceri, H.J.; Sutton, H.G.; Darga, T.E.; Kocherginsky, M.; Kochanski, J.; Weichselbaum, R.R.; Vokes, E.E. Everolimus exhibits efficacy as a radiosensitizer in a model of non-small cell lung cancer. Oncol. Rep. 2012, 27, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Bangoura, G.; Liu, Z.S.; Qian, Q.; Jiang, C.Q.; Yang, G.F.; Jing, S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 3176–3182. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.J.; Jirstrom, K.; Kronblad, A.; Millikan, R.C.; Landberg, G.; Duffy, M.J.; Ryden, L.; Gallagher, W.M.; O’Brien, S.L. CA IX is an independent prognostic marker in premenopausal breast cancer patients with one to three positive lymph nodes and a putative marker of radiation resistance. Clin. Cancer Res. 2006, 12, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Laude, K.; Cai, H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M. Handbook of Models for Human Aging; Academic Press, Elsevier: Cambridge, MA, USA, 2006; ISBN 9780123693914. [Google Scholar]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Woolston, C.M.; Al-Attar, A.; Storr, S.J.; Ellis, I.O.; Morgan, D.A.; Martin, S.G. Redox protein expression predicts radiotherapeutic response in early-stage invasive breast cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Su, Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett. 2009, 286, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Woolston, C.M.; Storr, S.J.; Ellis, I.O.; Morgan, D.A.; Martin, S.G. Expression of thioredoxin system and related peroxiredoxin proteins is associated with clinical outcome in radiotherapy treated early stage breast cancer. Radiother. Oncol. 2011, 100, 308–313. [Google Scholar] [CrossRef]
- Hardmeier, R.; Hoeger, H.; Fang-Kircher, S.; Khoschsorur, A.; Lubec, G. Transcription and activity of antioxidant enzymes after ionizing irradiation in radiation-resistant and radiation-sensitive mice. Proc. Natl. Acad. Sci. USA 1997, 94, 7572–7576. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Luccioni, C.; Moustacchi, E.; Rigaud, O. Contribution of antioxidant enzymes to the adaptive response to ionizing radiation of human lymphoblasts. Int. J. Radiat. Biol. 1999, 75, 639–645. [Google Scholar] [CrossRef]
- Sabitha, K.E.; Shyamaladevi, C.S. Oxidant and antioxidant activity changes in patients with oral cancer and treated with radiotherapy. Oral Oncol. 1999, 35, 273–277. [Google Scholar] [CrossRef]
- Terakado, N.; Shintani, S.; Nakahara, Y.; Mihara, M.; Tomizawa, K.; Suzuki, K.; Taniguchi, N.; Matsumura, T. Expression of Cu,Zn-SOD, Mn-SOD and GST-pi in oral cancer treated with preoperative radiation therapy. Oncol. Rep. 2000, 7, 1113–1117. [Google Scholar] [CrossRef]
- Megan K Johnson, R.R.V. Eunice S. Wang, Hypoxia-Associated Effects on Reactive Oxygen Species Generation by Human Acute Myeloid Leukemia Cells. Blood 2011, 118, 4998. [Google Scholar]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998, 273, 11619–11624. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, P.A.; Meltzer, S.; Sonstevold, L.; Esbensen, Y.; Dueland, S.; Flatmark, K.; Sitter, B.; Bathen, T.F.; Seierstad, T.; Redalen, K.R.; et al. Markers of Mitochondrial Metabolism in Tumor Hypoxia, Systemic Inflammation, and Adverse Outcome of Rectal Cancer. Transl. Oncol. 2019, 12, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Song, S.Y.; Park, S.G.; Song, S.U.; Xia, Y.; Sung, J.H. Primary involvement of NADPH oxidase 4 in hypoxia-induced generation of reactive oxygen species in adipose-derived stem cells. Stem Cells Dev. 2012, 21, 2212–2221. [Google Scholar] [CrossRef]
- Li, S.; Tabar, S.S.; Malec, V.; Eul, B.G.; Klepetko, W.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F.; Hanze, J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008, 10, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Garcia-Lecea, M.; Cadenas, S.; Hernandez, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376 Pt 2, 537–544. [Google Scholar] [CrossRef]
- Robin, E.; Guzy, R.D.; Loor, G.; Iwase, H.; Waypa, G.B.; Marks, J.D.; Hoek, T.L.; Schumacker, P.T. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J. Biol. Chem. 2007, 282, 19133–19143. [Google Scholar] [CrossRef] [PubMed]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Littauer, A.; de Groot, H. Release of reactive oxygen by hepatocytes on reoxygenation: Three phases and role of mitochondria. Am. J. Physiol. 1992, 262 Pt 1, G1015–G1020. [Google Scholar] [CrossRef]
- Du, H.; Yang, W.; Chen, L.; Shen, B.; Peng, C.; Li, H.; Ann, D.K.; Yen, Y.; Qiu, W. Emerging role of autophagy during ischemia-hypoxia and reperfusion in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 2049–2057. [Google Scholar]
- Karlenius, T.C.; Shah, F.; Di Trapani, G.; Clarke, F.M.; Tonissen, K.F. Cycling hypoxia up-regulates thioredoxin levels in human MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 419, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Hayashi, J.; Nakagawara, A.; Takenaga, K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 2009, 284, 33185–33194. [Google Scholar] [CrossRef]
- Du, J.; Xu, R.; Hu, Z.; Tian, Y.; Zhu, Y.; Gu, L.; Zhou, L. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1alpha expression in MCF-7 breast cancer cells. PLoS ONE 2011, 6, e25213. [Google Scholar] [CrossRef]
- Palmer, L.A.; Gaston, B.; Johns, R.A. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: Redox-dependent effect of nitrogen oxides. Mol. Pharmacol. 2000, 58, 1197–1203. [Google Scholar] [CrossRef]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brune, B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Yasinska, I.M.; Sumbayev, V.V. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Cook, J.A.; Krishna, M.C.; DeGraff, W.; Gamson, J.; Fisher, J.; Christodoulou, D.; Wink, D.A. Radiation sensitisation by nitric oxide releasing agents. Br. J. Cancer Suppl. 1996, 27, S181–S184. [Google Scholar] [PubMed]
- Mitchell, J.B.; Wink, D.A.; DeGraff, W.; Gamson, J.; Keefer, L.K.; Krishna, M.C. Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res. 1993, 53, 5845–5848. [Google Scholar] [PubMed]
- Illum, H.; Wang, D.H.; Dowell, J.E.; Hittson, W.J.; Torrisi, J.R.; Meyer, J.; Huerta, S. Phase I dose escalation trial of nitroglycerin in addition to 5-fluorouracil and radiation therapy for neoadjuvant treatment of operable rectal cancer. Surgery 2015, 158, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Makepeace, C.M.; Hur, W.J.; Song, C.W. Radiosensitization of hypoxic tumor cells in vitro by nitric oxide. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 377–383. [Google Scholar] [CrossRef]
- Verovski, V.N.; Van den Berge, D.L.; Soete, G.A.; Bols, B.L.; Storme, G.A. Intrinsic radiosensitivity of human pancreatic tumour cells and the radiosensitising potency of the nitric oxide donor sodium nitroprusside. Br. J. Cancer 1996, 74, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Janssens, M.Y.; Van den Berge, D.L.; Verovski, V.N.; Monsaert, C.; Storme, G.A. Activation of inducible nitric oxide synthase results in nitric oxide-mediated radiosensitization of hypoxic EMT-6 tumor cells. Cancer Res. 1998, 58, 5646–5648. [Google Scholar] [PubMed]
- Jordan, B.F.; Gregoire, V.; Demeure, R.J.; Sonveaux, P.; Feron, O.; O’Hara, J.; Vanhulle, V.P.; Delzenne, N.; Gallez, B. Insulin increases the sensitivity of tumors to irradiation: Involvement of an increase in tumor oxygenation mediated by a nitric oxide-dependent decrease of the tumor cells oxygen consumption. Cancer Res. 2002, 62, 3555–3561. [Google Scholar]
- Bump, E.A.; Brown, J.M. Role of glutathione in the radiation response of mammalian cells in vitro and in vivo. Pharmacol. Ther. 1990, 47, 117–136. [Google Scholar] [CrossRef]
- Kinsella, T.J.; Dobson, P.P.; Russo, A.; Mitchell, J.B.; Fornace, A.J., Jr. Modulation of X ray DNA damage by SR-2508 +/- buthionine sulfoximine. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1127–1130. [Google Scholar] [CrossRef]
- Biaglow, J.E.; Varnes, M.E.; Astor, M.; Hall, E.J. Non-protein thiols and cellular response to drugs and radiation. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 719–723. [Google Scholar] [CrossRef]
- Yao, J.X.; Yao, Z.F.; Li, Z.F.; Liu, Y.B. Radio-sensitization by Piper longumine of human breast adenoma MDA-MB-231 cells in vitro. Asian Pac. J. Cancer Prev. 2014, 15, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, T.; Cao, J.; Fan, S. Hypoxia-inducible factor-1alpha downregulation by small interfering RNA inhibits proliferation, induces apoptosis, and enhances radiosensitivity in chemical hypoxic human hepatoma SMMC-7721 cells. Cancer Biother. Radiopharm. 2011, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, J.; Yan, C.; Hou, J.; Pu, J.; Zhang, G.; Fu, Z.; Wang, X. Effect of small interfering RNA targeting hypoxia-inducible factor-1alpha on radiosensitivity of PC3 cell line. Urology 2012, 79, 744.e17–744.e24. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Kawai, K.; Hata, K.; Murono, K.; Emoto, S.; Kaneko, M.; Sasaki, K.; Nishikawa, T.; Tanaka, T.; Nozawa, H. SN-38 Acts as a Radiosensitizer for Colorectal Cancer by Inhibiting the Radiation-induced Up-regulation of HIF-1alpha. Anticancer Res. 2018, 38, 3323–3331. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, M.; Xing, D.; Feng, Y. Atorvastatin enhances radiosensitivity in hypoxia-induced prostate cancer cells related with HIF-1alpha inhibition. Biosci. Rep. 2017, 37, BSR20170340. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, X.; Zhang, Q.; Guo, Q.; He, J.; Qin, Q.; Zhu, H.; Liu, J.; Zhan, L.; Lu, J.; et al. STAT3 inhibitor NSC74859 radiosensitizes esophageal cancer via the downregulation of HIF-1alpha. Tumour Biol. 2014, 35, 9793–9799. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, C.; Yang, X.; Yang, B.; Wang, J.; Kang, Y.; Wang, Z.; Li, D.; Huang, G.; Ma, Z.; et al. Berberine inhibits the expression of hypoxia induction factor-1alpha and increases the radiosensitivity of prostate cancer. Diagn. Pathol. 2014, 9, 98. [Google Scholar] [CrossRef]
- Lee, D.E.; Alhallak, K.; Jenkins, S.V.; Vargas, I.; Greene, N.P.; Quinn, K.P.; Griffin, R.J.; Dings, R.P.M.; Rajaram, N. A Radiosensitizing Inhibitor of HIF-1 alters the Optical Redox State of Human Lung Cancer Cells In Vitro. Sci. Rep. 2018, 8, 8815. [Google Scholar] [CrossRef]
- Oike, T.; Suzuki, Y.; Al-Jahdari, W.; Mobaraki, A.; Saitoh, J.I.; Torikai, K.; Shirai, K.; Nakano, T. Suppression of HIF-1alpha expression and radiation resistance in acute hypoxic conditions. Exp. Ther. Med. 2012, 3, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, X.; Li, Z. Combination therapy with local radiofrequency ablation and YC-1 inhibits the proliferation and metastasis of hepatocellular carcinoma through activating beta-catenin signaling. Pharmazie 2016, 71, 524–529. [Google Scholar] [PubMed]
- Moon, S.Y.; Chang, H.W.; Roh, J.L.; Kim, G.C.; Choi, S.H.; Lee, S.W.; Cho, K.J.; Nam, S.Y.; Kim, S.Y. Using YC-1 to overcome the radioresistance of hypoxic cancer cells. Oral Oncol. 2009, 45, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Ghattass, K.; Assah, R.; El-Sabban, M.; Gali-Muhtasib, H. Targeting hypoxia for sensitization of tumors to radio- and chemotherapy. Curr. Cancer Drug Targets 2013, 13, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef]
- Sun, L.; Moritake, T.; Ito, K.; Matsumoto, Y.; Yasui, H.; Nakagawa, H.; Hirayama, A.; Inanami, O.; Tsuboi, K. Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets. PLoS ONE 2017, 12, e0176162. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Shavit, R.; Ilouze, M.; Feinberg, T.; Lawrence, Y.R.; Tzur, Y.; Peled, N. Mitochondrial induction as a potential radio-sensitizer in lung cancer cells—A short report. Cell. Oncol. 2015, 38, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Maggiorella, L.; Wen, B.; Frascogna, V.; Opolon, P.; Bourhis, J.; Deutsch, E. Combined radiation sensitizing and anti-angiogenic effects of ionizing radiation and the protease inhibitor ritonavir in a head and neck carcinoma model. Anticancer Res. 2005, 25, 4357–4362. [Google Scholar]
- Dwarakanath, B.S.; Singh, D.; Banerji, A.K.; Sarin, R.; Venkataramana, N.K.; Jalali, R.; Vishwanath, P.N.; Mohanti, B.K.; Tripathi, R.P.; Kalia, V.K.; et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. J. Cancer Res. Ther. 2009, 5 (Suppl. S1), S21–S26. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.H.; He, S.Q.; Alfieri, A.A.; Young, C.W. Potentiation of radiation effects on multicellular tumor spheroids (MTS) of HeLa cells by lonidamine. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 1277–1280. [Google Scholar] [CrossRef]
- Crokart, N.; Jordan, B.F.; Baudelet, C.; Cron, G.O.; Hotton, J.; Radermacher, K.; Gregoire, V.; Beghein, N.; Martinive, P.; Bouzin, C.; et al. Glucocorticoids modulate tumor radiation response through a decrease in tumor oxygen consumption. Clin. Cancer Res. 2007, 13 Pt 1, 630–635. [Google Scholar] [CrossRef]
- Crokart, N.; Radermacher, K.; Jordan, B.F.; Baudelet, C.; Cron, G.O.; Gregoire, V.; Beghein, N.; Bouzin, C.; Feron, O.; Gallez, B. Tumor radiosensitization by antiinflammatory drugs: Evidence for a new mechanism involving the oxygen effect. Cancer Res. 2005, 65, 7911–7916. [Google Scholar] [CrossRef] [PubMed]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef]
- de Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Gregoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res. 2012, 72, 482–490. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Zhang, X.D.; Luo, Z.; Chen, J.; Shen, X.; Song, S.; Sun, Y.; Fan, S.; Fan, F.; Leong, D.T.; Xie, J. Ultrasmall Au(10-12)(SG)(10-12) nanomolecules for high tumor specificity and cancer radiotherapy. Adv. Mater. 2014, 26, 4565–4568. [Google Scholar] [CrossRef]
- Cui, L.; Her, S.; Borst, G.R.; Bristow, R.G.; Jaffray, D.A.; Allen, C. Radiosensitization by gold nanoparticles: Will they ever make it to the clinic? Radiother. Oncol. 2017, 124, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Luo, Z.; Chen, J.; Song, S.; Yuan, X.; Shen, X.; Wang, H.; Sun, Y.; Gao, K.; Zhang, L.; et al. Ultrasmall glutathione-protected gold nanoclusters as next generation radiotherapy sensitizers with high tumor uptake and high renal clearance. Sci. Rep. 2015, 5, 8669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Chen, J.; Luo, Z.; Wu, D.; Shen, X.; Song, S.S.; Sun, Y.M.; Liu, P.X.; Zhao, J.; Huo, S.; et al. Enhanced tumor accumulation of sub-2 nm gold nanoclusters for cancer radiation therapy. Adv. Healthc. Mater. 2014, 3, 133–141. [Google Scholar] [CrossRef]
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, E.J.; Kim, J.W.; Chung, U.S.; Koh, W.G.; Keum, K.C.; Koom, W.S. Gold nanoparticles enhance anti-tumor effect of radiotherapy to hypoxic tumor. Radiat. Oncol. J. 2016, 34, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.; Zhang, C.; Gu, Z.; Du, J.; Guo, Z.; Dong, X.; Xie, J.; Zhang, G.; Liu, X.; Zhao, Y. Polyoxometalate-Based Radiosensitization Platform for Treating Hypoxic Tumors by Attenuating Radioresistance and Enhancing Radiation Response. ACS Nano 2017, 11, 7164–7176. [Google Scholar] [CrossRef] [PubMed]
- Howard-Flanders, P. Effect of nitric oxide on the radiosensitivity of bacteria. Nature 1957, 180, 1191–1192. [Google Scholar] [CrossRef]
- Reyman, B.; Zegers, M.G.K.; Dubois, L.; Lambin, P. Nitroglycerin as a sensitizer in the treatment of non small cell lung cancer: From cells in vitro to phase 3 trial. Radiother. Oncol. 2015, 115 (Suppl. S1), S290–S291. [Google Scholar] [CrossRef]
- Siemens, D.R.; Heaton, J.P.; Adams, M.A.; Kawakami, J.; Graham, C.H. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology 2009, 74, 878–883. [Google Scholar] [CrossRef]
- Arrieta, O.; Blake, M.; de la Mata-Moya, M.D.; Corona, F.; Turcott, J.; Orta, D.; Alexander-Alatorre, J.; Gallardo-Rincon, D. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014, 111, 311–315. [Google Scholar] [CrossRef]
- Jiang, H.; Verovski, V.N.; Leonard, W.; Law, K.L.; Vermeersch, M.; Storme, G.; Van den Berge, D.; Gevaert, T.; Sermeus, A.; De Ridder, M. Hepatocytes determine the hypoxic microenvironment and radiosensitivity of colorectal cancer cells through production of nitric oxide that targets mitochondrial respiration. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; De Ridder, M.; Verovski, V.N.; Sonveaux, P.; Jordan, B.F.; Law, K.; Monsaert, C.; Van den Berge, D.L.; Verellen, D.; Feron, O.; et al. Activated macrophages as a novel determinant of tumor cell radioresponse: The role of nitric oxide-mediated inhibition of cellular respiration and oxygen sparing. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Kaz, A.M.; Snyder, S.A.; Richardson, R.A.; Cardenas-Navia, L.I.; Braun, R.D.; Pawloski, J.R.; Tozer, G.M.; Bonaventura, J.; McMahon, T.J.; et al. Oxygen regulation of tumor perfusion by S-nitrosohemoglobin reveals a pressor activity of nitric oxide. Circ. Res. 2005, 96, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.F.; Sonveaux, P.; Feron, O.; Gregoire, V.; Beghein, N.; Gallez, B. Nitric oxide-mediated increase in tumor blood flow and oxygenation of tumors implanted in muscles stimulated by electric pulses. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 1066–1073. [Google Scholar] [CrossRef]
- Wang, Z.; Cook, T.; Alber, S.; Liu, K.; Kovesdi, I.; Watkins, S.K.; Vodovotz, Y.; Billiar, T.R.; Blumberg, D. Adenoviral gene transfer of the human inducible nitric oxide synthase gene enhances the radiation response of human colorectal cancer associated with alterations in tumor vascularity. Cancer Res. 2004, 64, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Frerart, F.; Sonveaux, P.; Rath, G.; Smoos, A.; Meqor, A.; Charlier, N.; Jordan, B.F.; Saliez, J.; Noel, A.; Dessy, C.; et al. The acidic tumor microenvironment promotes the reconversion of nitrite into nitric oxide: Towards a new and safe radiosensitizing strategy. Clin. Cancer Res. 2008, 14, 2768–2774. [Google Scholar] [CrossRef]
- Jordan, B.F.; Peeterbroeck, J.; Karroum, O.; Diepart, C.; Magat, J.; Gregoire, V.; Gallez, B. Captopril and S-nitrosocaptopril as potent radiosensitizers: Comparative study and underlying mechanisms. Cancer Lett. 2010, 293, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, V.; Nicklee, T.; Hedley, D.W. Differential effects of buthionine sulphoximine in hypoxic and non-hypoxic regions of human cervical carcinoma xenografts. Radiother. Oncol. 2001, 60, 69–73. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Hamilton, T.C.; LaCreta, F.P.; Gallo, J.M.; Kilpatrick, D.; Halbherr, T.; Brennan, J.; Bookman, M.A.; Hoffman, J.; Young, R.C.; et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J. Clin. Oncol. 1996, 14, 249–256. [Google Scholar] [CrossRef]
- Bailey, H.H.; Mulcahy, R.T.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Tombes, M.B.; Wilding, G.; Pomplun, M.; Spriggs, D.R. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: An attempt at modulation of glutathione. J. Clin. Oncol. 1994, 12, 194–205. [Google Scholar] [CrossRef]
- Bailey, H.H.; Ripple, G.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Feierabend, C.; Mahvi, D.; Schink, J.; Pomplun, M.; Mulcahy, R.T.; et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl. Cancer Inst. 1997, 89, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Xia, Y.; Ji, J.; Chen, W.; Zhang, J.; Chen, X.; Rajamanickam, V.; Chen, G.; Wang, Z.; Chen, L.; et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. 2016, 375, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Folda, A.; Baldoin, M.C.; Scutari, G.; Bindoli, A. Effect of auranofin on the mitochondrial generation of hydrogen peroxide. Role of thioredoxin reductase. Free Radic. Res. 2005, 39, 687–695. [Google Scholar] [CrossRef]
- Ban, H.S.; Uto, Y.; Nakamura, H. Hypoxia-inducible factor inhibitors: A survey of recent patented compounds (2004–2010). Expert Opin. Ther. Pat. 2011, 21, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Cairns, R.A.; Chaudary, N.; Vellanki, R.N.; Kalliomaki, T.; Moriyama, E.H.; Mujcic, H.; Wilson, B.C.; Wouters, B.G.; Hill, R.; et al. Metabolic targeting of HIF-dependent glycolysis reduces lactate, increases oxygen consumption and enhances response to high-dose single-fraction radiotherapy in hypoxic solid tumors. BMC Cancer 2017, 17, 418. [Google Scholar] [CrossRef]
- Shen, L.F.; Zhao, X.; Zhou, S.H.; Lu, Z.J.; Zhao, K.; Fan, J.; Zhou, M.L. In vivo evaluation of the effects of simultaneous inhibition of GLUT-1 and HIF-1alpha by antisense oligodeoxynucleotides on the radiosensitivity of laryngeal carcinoma using micro 18F-FDG PET/CT. Oncotarget 2017, 8, 34709–34726. [Google Scholar]
- Shin, D.H.; Kim, J.H.; Jung, Y.J.; Kim, K.E.; Jeong, J.M.; Chun, Y.S.; Park, J.W. Preclinical evaluation of YC-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett. 2007, 255, 107–116. [Google Scholar] [CrossRef]
- Kim, H.L.; Yeo, E.J.; Chun, Y.S.; Park, J.W. A domain responsible for HIF-1alpha degradation by YC-1, a novel anticancer agent. Int. J. Oncol. 2006, 29, 255–260. [Google Scholar]
- Sun, H.L.; Liu, Y.N.; Huang, Y.T.; Pan, S.L.; Huang, D.Y.; Guh, J.H.; Lee, F.Y.; Kuo, S.C.; Teng, C.M. YC-1 inhibits HIF-1 expression in prostate cancer cells: Contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene 2007, 26, 3941–3951. [Google Scholar] [CrossRef]
- Harada, H.; Itasaka, S.; Zhu, Y.; Zeng, L.; Xie, X.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. Treatment regimen determines whether an HIF-1 inhibitor enhances or inhibits the effect of radiation therapy. Br. J. Cancer 2009, 100, 747–757. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.F.; Wang, X.J.; Kang, H.F.; Bai, M.H.; Guan, H.T.; Wang, Z.W.; Zan, Y.; Song, L.Q.; Min, W.L.; Lin, S.; et al. Saikosaponin-D enhances radiosensitivity of hepatoma cells under hypoxic conditions by inhibiting hypoxia-inducible factor-1alpha. Cell. Physiol. Biochem. 2014, 33, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Palayoor, S.T.; Mitchell, J.B.; Cerna, D.; Degraff, W.; John-Aryankalayil, M.; Coleman, C.N. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int. J. Cancer 2008, 123, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.L.; Powis, G.; Thitai-Kumar, A.; He, Y.; Bankson, J.; Williams, R.; Lemos, R.; Oh, J.; Volgin, A.; Soghomonyan, S.; et al. The selective hypoxia inducible factor-1 inhibitor PX-478 provides in vivo radiosensitization through tumor stromal effects. Mol. Cancer Ther. 2009, 8, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.L.; Bankson, J.A.; Lemos, R. Jr.; Lai, S.Y.; Thittai, A.K.; He, Y.; Hostetter, G.; Demeure, M.J.; Von Hoff, D.D.; Powis, G. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef]
- Lin, A.; Maity, A. Molecular Pathways: A Novel Approach to Targeting Hypoxia and Improving Radiotherapy Efficacy via Reduction in Oxygen Demand. Clin. Cancer Res. 2015, 21, 1995–2000. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de The, H. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2002, 2, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Hori, S.; Nishitoh, H.; Ichijo, H.; Ogawa, O.; Kakehi, Y.; Kakizuka, A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001, 61, 5432–5440. [Google Scholar] [PubMed]
- Sun, R.C.; Board, P.G.; Blackburn, A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol. Cancer 2011, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhao, H.; Nolley, R.; Reese, S.W.; Young, S.R.; Li, X.; Peehl, D.M.; Knox, S.J. Darinaparsin: Solid tumor hypoxic cytotoxin and radiosensitizer. Clin. Cancer Res. 2012, 18, 3366–3376. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fung, S.Y.; Xu, S.; Sutherland, D.P.; Kollmann, T.R.; Liu, M.; Turvey, S.E. Amino Acid-Dependent Attenuation of Toll-like Receptor Signaling by Peptide-Gold Nanoparticle Hybrids. ACS Nano 2015, 9, 6774–6784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Wu, D.; Shen, X.; Liu, P.X.; Fan, F.Y.; Fan, S.J. In vivo renal clearance, biodistribution, toxicity of gold nanoclusters. Biomaterials 2012, 33, 4628–4638. [Google Scholar] [CrossRef]
- Chen, X.; Wang, P.; Guo, F.; Wang, X.; Wang, J.; Xu, J.; Yuan, D.; Zhang, J.; Shao, C. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int. J. Radiat. Biol. 2017, 93, 764–770. [Google Scholar] [CrossRef]
- He, W.S.; Dai, X.F.; Jin, M.; Liu, C.W.; Rent, J.H. Hypoxia-induced autophagy confers resistance of breast cancer cells to ionizing radiation. Oncol. Res. 2012, 20, 251–258. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Ramaekers, C.H.; Schaaf, M.B.; Keulers, T.G.; Savelkouls, K.G.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Publication | No. of Patients | Oxygenation Parameter | Endpoint | p-Value * |
|---|---|---|---|---|
| Cervix Cancer | ||||
| Hockel et al., 1996 [45] | 103 | median pO2 < 10 mm Hg | DFS | =0.009 |
| OS | =0.004 | |||
| Knocke et al., 1999 [46] | 51 | median pO2 ≤ 10 mm Hg | DFS | <0.02 |
| Sundfor et al., 2000 [47] | 40 | subvolume pO2 < 5 mm Hg | DFS | =0.0001 |
| OS | =0.0004 | |||
| LC | =0.0006 | |||
| Fyles et al., 2002 [48] | 106 | fraction pO2 < 5 mm Hg | PFS | <0.004 |
| Nordsmark et al., 2006 [49] | 120 | median pO2 < 4 mm Hg | LC; OS | n.s. |
| Head and Neck Tumors | ||||
| Gatenby et al., 1988 [50] | 31 | pO2 < 5 mm Hg | LC | <0.001 |
| Brizel et al., 1999 [51] | 63 | median pO2 < 10 mm Hg | DFS | =0.005 |
| OS | =0.02 | |||
| LC | =0.01 | |||
| Stadler et al., 1999 [52] | 59 | subvolume pO2 < 5 mm Hg | OS | <0.01 |
| Rudat et al., 2001 [53] | 134 | fraction pO2 < 2.5 mm Hg | OS | =0.004 |
| Nordsmark et al., 2005 [54] | 397 | fraction pO2 ≤ 2.5 mm Hg | OS | =0.006 |
| Soft Tissue Sarcomas | ||||
| Brizel et al., 1996 [55] | 22 | median pO2 ≤ 10 mm Hg | DF | =0.01 |
| Nordsmark et al., 2001 [56] | 31 | median pO2 ≤ 19 mm Hg | OS | =0.01 |
| Name of the Agents | Mechanisms of Action | Cancer Types | References |
|---|---|---|---|
| Hypoxic Radiosensitization by NO | |||
| Diethylamine nonoate | NO donor | Chinese hamster V79 lung fibroblast | [107,108] |
| S-Nitrosoglutathione | NO donor | Chinese hamster V79 lung fibroblast | [107] |
| Nitroglycerin | NO donor | Rectal cancer | [109] |
| Spermine nonoate | NO donor | Murine mammary carcinoma SCK | [110] |
| Sodium nitroprusside | NO donor | Human pancreatic tumor cells | [111] |
| Insulin | Activate eNOS | Liver and fibrosarcoma mouse tumors | [112] |
| Endogenous NO | Activate iNOS | Murine mammary carcinoma EMT6 | [113] |
| Hypoxic Radiosensitization by Inhibition of Antioxidant Enzymes | |||
| Buthionine sulphoximine + Misonidazole | Deplete glutathione and mimic oxygen | Multiple types of cancer cells | [114] |
| Buthionine sulphoximine + SR2508 | Deplete glutathione and mimic oxygen | Multiple types of cancer cells | [114,115] |
| Dimethylfumarate | Deplete glutathione | Chinese hamster ovary cells | [16] |
| Diethylmaleate | Deplete glutathione | murine mammary carcinoma EMT6 | [17] |
| DMF + Misonidazole | Deplete glutathione and mimic oxygen | Ehrlich ascites tumors | [116] |
| DEM + Misonidazole | Deplete glutathione and mimic oxygen | Multiple types of cancer cells | [114,116] |
| Piperlongumine | Inhibit glutathione S-transferase and thioredoxin reductase | Lung cancer cells | [18,117] |
| Auranofin | Inhibit thioredoxin reductase | Breast cancer cells and tumor models | [19,20] |
| Auranofin + BSO | Inhibit thioredoxin reductase and deplete glutathione | Breast cancer cells and tumor models | [19,20] |
| Hypoxic Radiosensitization by Inhibition of HIF-1 | |||
| HIF-1 siRNA | Silence HIF-1α | Hepatoma cells SMMC-7721 and prostate cancer cells PC3 | [118,119] |
| SN-38 | Inhibit radiation-induced HIF-1α | Colorectal cancer cells HT29 and SW480 | [120] |
| Atorvastatin | Inhibit hypoxia-induced HIF-1α | Prostate cancer cells PC3 | [121] |
| NSC74859 | Inhibit HIF-1α and VEGF expression | Esophageal squamous carcinoma cells ECA109 and TE13 | [122] |
| Berberine | Inhibit HIF-1α and VEGF expression | Prostate tumor models | [123] |
| YC-1 | Inhibit HIF-1α translation and degrade HIF-1α | Multiple types of cancer cells | [124,125,126,127] |
| PX-478 | Decrease HIF-1α transcription and translation and degrade HIF-1α | Multiple types of cancer | [128,129] |
| Hypoxic Radiosensitization by Inhibition of Tumor Metabolism | |||
| Dichloroacetate | Inhibit glycolysis | Multiple types of cancer cells | [130,131,132,133] |
| Ritonavir | Inhibit glucose transporter | Head and neck carcinoma model HEP-2 | [134] |
| 2-deoxyglucose | Inhibit hexokinase | Glioblastoma | [135] |
| lonidamine | Inhibit hexokinase | Cervical cancer HeLa cells | [136] |
| Hypoxic Radiosensitization via Reduction in Oxygen Demand | |||
| Glucocorticoids | Decrease oxygen consumption | Liver and fibrosarcoma mouse tumors | [137] |
| NSAIDs | Mediate mitochondrial respiration | Liver and fibrosarcoma mouse tumors | [138] |
| Metformin | Inhibit mitochondrial complex I | Multiple types of cancer | [139,140,141] |
| Others | |||
| Arsenic trioxide | Inhibit mitochondrial complex IV | Liver and Lewis lung carcinoma models | [142] |
| Gold nanoparticles | Donate electrons to form ROS | Multiple types of tumor models | [143,144,145,146,147,148,149,150] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. https://doi.org/10.3390/cancers11010112
Wang H, Jiang H, Van De Gucht M, De Ridder M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers. 2019; 11(1):112. https://doi.org/10.3390/cancers11010112
Chicago/Turabian StyleWang, Hui, Heng Jiang, Melissa Van De Gucht, and Mark De Ridder. 2019. "Hypoxic Radioresistance: Can ROS Be the Key to Overcome It?" Cancers 11, no. 1: 112. https://doi.org/10.3390/cancers11010112
APA StyleWang, H., Jiang, H., Van De Gucht, M., & De Ridder, M. (2019). Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers, 11(1), 112. https://doi.org/10.3390/cancers11010112