SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer

 , ,
, ,
Abstract
1. Introduction
2. Results
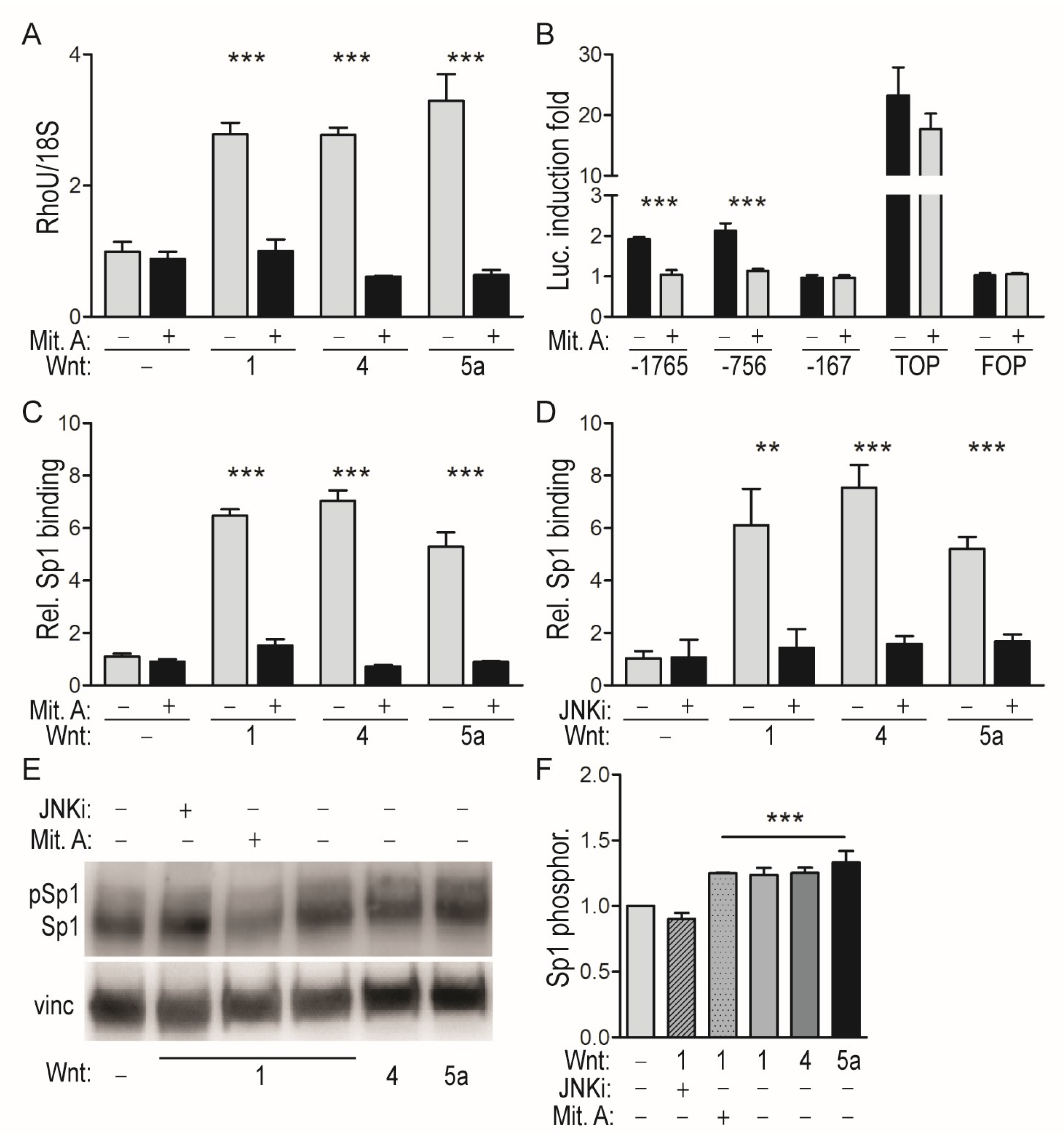
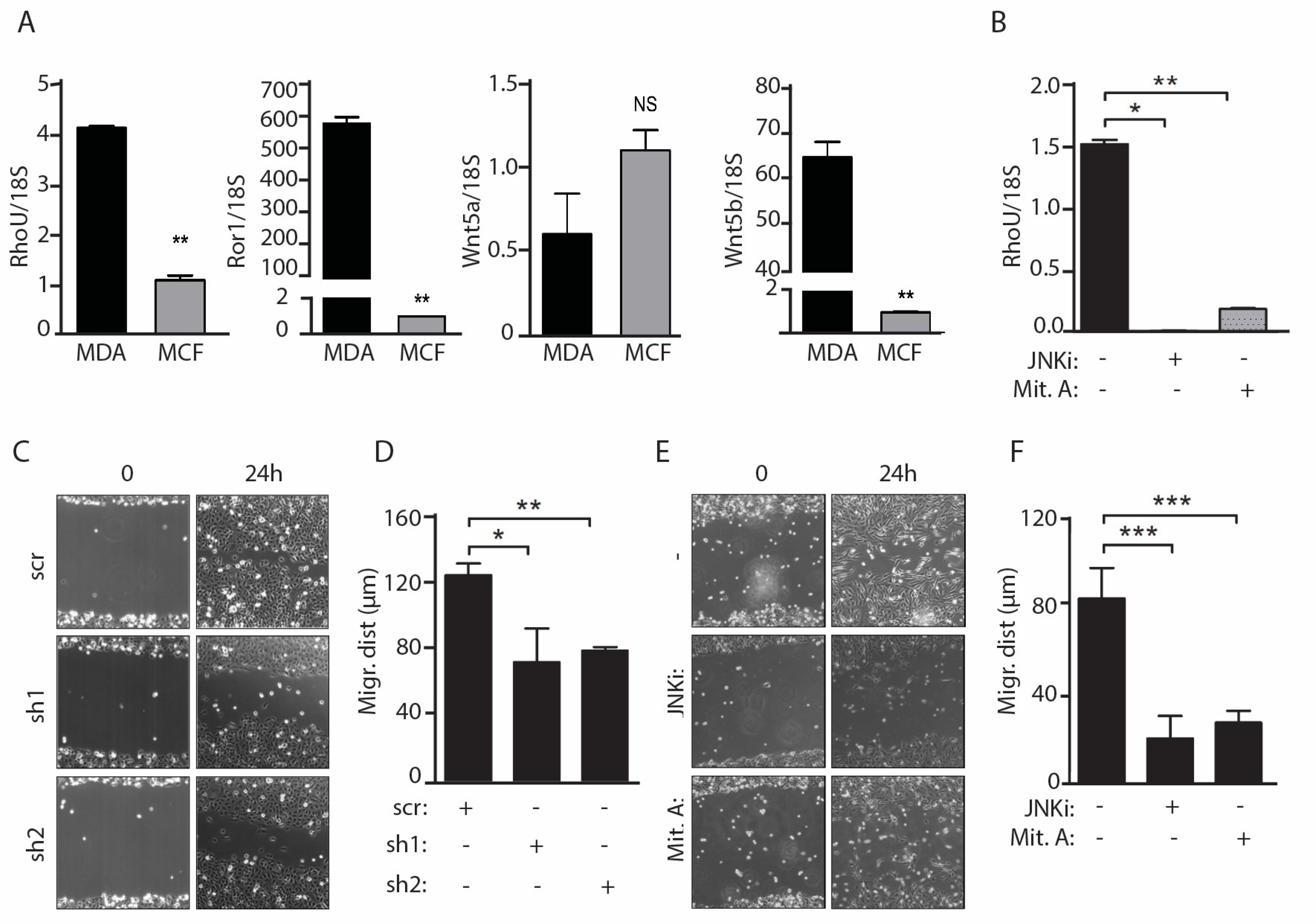
2.1. RhoU Expression Can Be Induced by both Canonical and Non-Canonical WNT Ligands
2.2. WNT-Mediated Induction of the RhoU Promoter Rquires JNK-Dependent SP1 Recruitment
2.3. Migration of MDA-MB-231 Cells Downstream of Constitutively Active WNT5a Requires both JNK and SP1 Activities and RhoU Expression
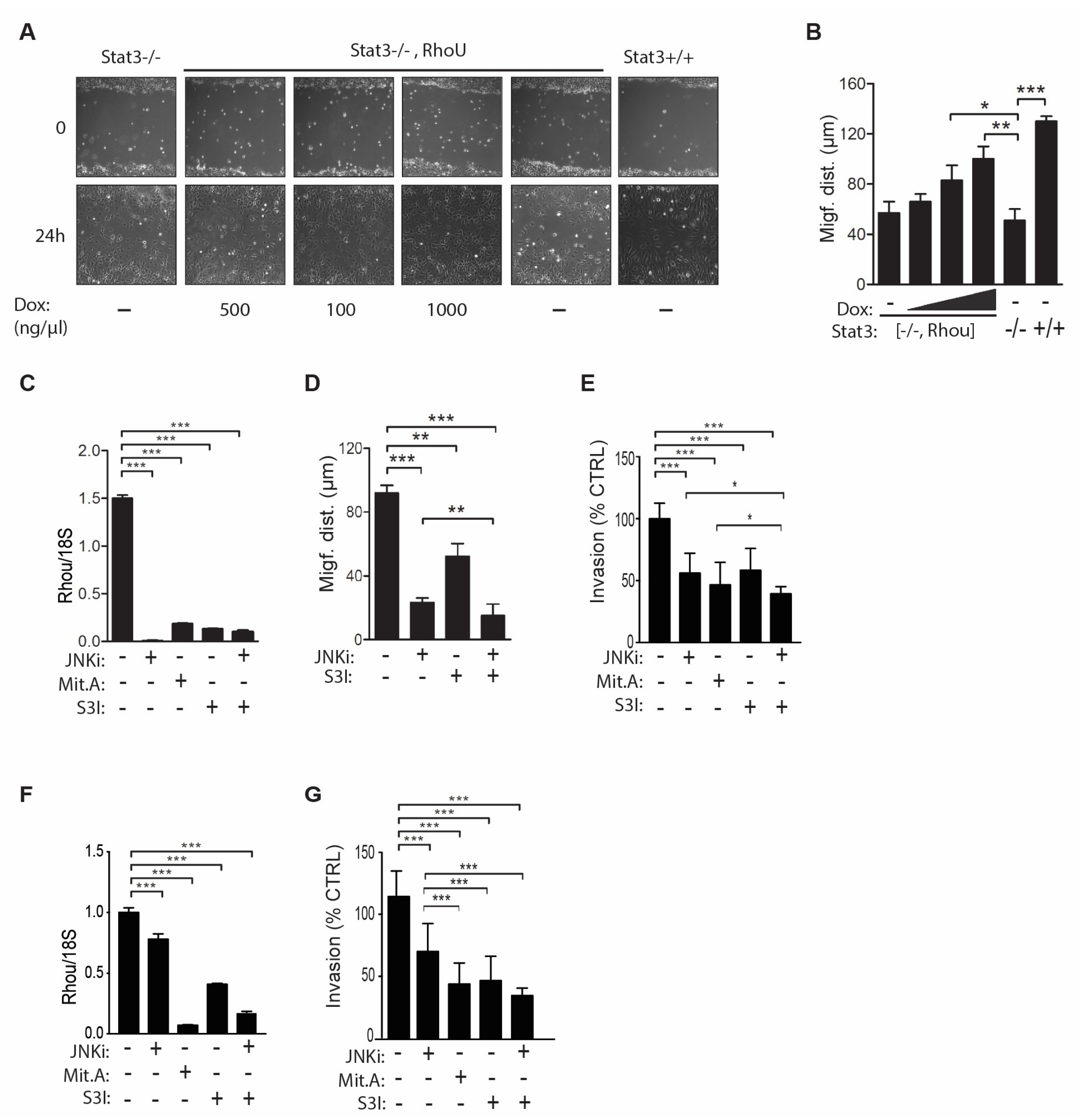
2.4. RhoU is also Involved in Mediating Cell Migration Downstream of STAT3 Signaling
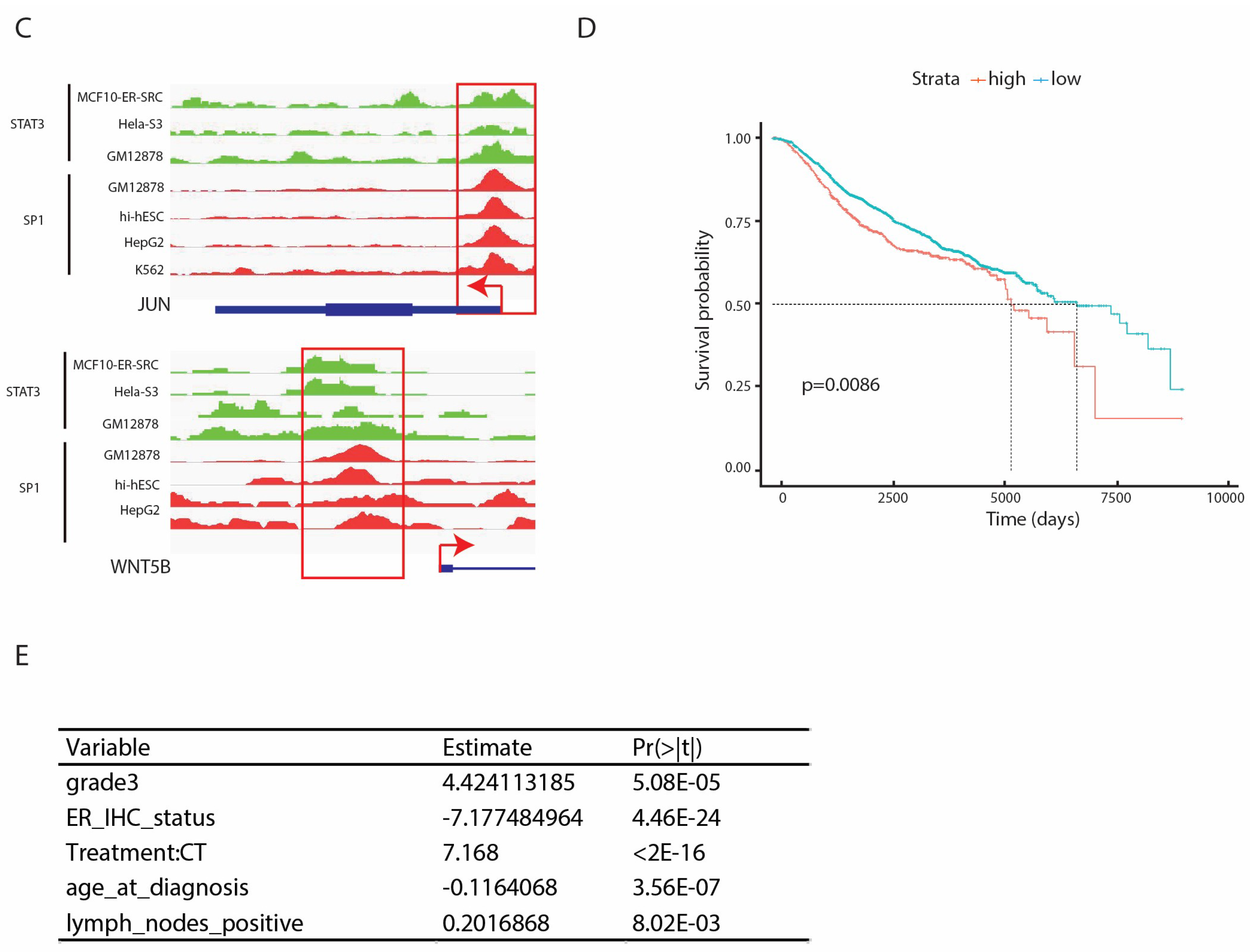
2.5. SP1 and STAT3 In Vivo Binding Defines a Subclass of Genes Belonging to Non-Canonical WNT or IL-6 Pathway
2.6. High Expression of SP1 and STAT3 Dual Targets is Associated with Low Survival in Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Cell lines and Treatments
4.2. Cell Transfection and Transduction, Co-Culture and Luciferase Assays
4.3. Chromatin Immunoprecipitation Assays
4.4. Wound Healing Migration Assay
4.5. Matrigel Invasion Assay
4.6. Protein Extracts and Immunoblotting
4.7. RNA Extraction, Retrotranscription and Quantitative Real-Time PCR (qRT-PCR)
4.8. Statistical Analysis
4.9. Bioinformatics Analysis
4.9.1. Total Binding Affinity (TBA)
4.9.2. Source Data from Database
4.9.3. ChIP-Seq Data Analysis
4.9.4. Gene set Data
4.9.5. Survival Analysis
4.9.6. Univariate Analysis
4.9.7. Plots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldhirsch, A.; Ingle, J.N.; Gelber, R.D.; Coates, A.S.; Thürlimann, B.; Senn, H.J. Thresholds for therapies: Highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2009. Ann. Oncol. 2009, 20, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Viale, G. The current state of breast cancer classification. Ann. Oncol. 2012, 23 (Suppl. 10). [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Ellis, M.J.; Perou, C.M. Practical implications of gene-expression-based assays for breast oncologists. Nat. Rev. Clin. Oncol. 2012, 9, 48–57. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. the Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon: Functions and mechanisms of β-catenin-independent Wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef]
- Peradziryi, H.; Kaplan, N.A.; Podleschny, M.; Liu, X.; Wehner, P.; Borchers, A.; Tolwinski, N.S. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 2011, 30, 3729–3740. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.P.; Beckett, K. Off-track takes Frizzled off the canonical path. EMBO J. 2011, 30, 3665–3666. [Google Scholar] [CrossRef]
- Ilyas, M. Wnt signalling and the mechanistic basis of tumour development. J. Pathol. 2005, 205, 130–144. [Google Scholar] [CrossRef]
- Wang, Y. Wnt/Planar cell polarity signaling: A new paradigm for cancer therapy. Mol. Cancer Ther. 2009, 8, 2103–2109. [Google Scholar] [CrossRef]
- Klemm, F.; Bleckmann, L.; Siam, H.N.; Chuang, E.; Rietktter, D.; Behme, M.; Schulz, M.; Schaffrinski, S.; Schindler, L.; Trümper, F.; et al. β-catenin-independent WNT signaling in basal-like breast cancer and brain metastasis. Carcinogenesis 2011, 32, 434–442. [Google Scholar] [CrossRef]
- MacMillan, C.D.; Leong, H.S.; Dales, D.W.; Robertson, A.E.; Lewis, J.D.; Chambers, A.F.; Tuck, A.B. Stage of Breast Cancer Progression Influences Cellular Response to Activation of the WNT/Planar Cell Polarity Pathway. Sci. Rep. 2014, 3, 6315. [Google Scholar] [CrossRef]
- Lien, W.H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef]
- Schambony, A.; Wedlich, D. Wnt-5A/Ror2 Regulate Expression of XPAPC through an Alternative Noncanonical Signaling Pathway. Dev. Cell 2007, 12, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Ory, S.; Brazier, H.; Blangy, A. Identification of a bipartite focal adhesion localization signal in RhoU/Wrch-1, a Rho family GTPase that regulates cell adhesion and migration. Biol. Cell Auspices Eur. Cell Biol. Organ. 2007, 99, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Pennica, D.; Xu, L.; Kalejta, R.F.; Levine, A.J. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes Dev. 2001, 15, 1796–1807. [Google Scholar] [CrossRef]
- Saras, J.; Wollberg, P.; Aspenström, P. Wrch1 is a GTPase-deficient Cdc42-like protein with unusual binding characteristics and cellular effects. Exp. Cell Res. 2004, 299, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Berzat, A.C.; Chenette, E.J.; Cox, A.D.; Der, C.J. Biochemical analyses of the Wrch atypical Rho family GTPases. Methods Enzymol. 2006, 406, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Dewilde, S.; Vallania, F.; Turkson, J.; di Cunto, F.; Poli, V. The RhoU/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem. J. 2009, 421, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The stats of cancer-New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis. Jak-Stat 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.R.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Foat, B.C.; Morozov, A.V.; Bussemaker, H.J. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics 2006, 22, e141–e149. [Google Scholar] [CrossRef] [PubMed]
- Grassi, E.; Zapparoli, E.; Molineris, I.; Provero, P. Total binding affinity profiles of regulatory regions predict transcription factor binding and gene expression in human cells. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Molineris, I.; Grassi, E.; Ala, U.; di Cunto, F.; Provero, P. Evolution of promoter affinity for transcription factors in the human lineage. Mol. Biol. Evol. 2011, 28, 2173–2183. [Google Scholar] [CrossRef]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef]
- Benasciutti, E.; Pagès, G.; Kenzior, O.; Folk, W.; Blasi, F.; Crippa, M.P. MAPK and JNK transduction pathways can phosphorylate Sp1 to activate the uPA minimal promoter element and endogenous gene transcription. Blood 2004, 104, 256–262. [Google Scholar] [CrossRef]
- Hammoud, L.; Burger, D.E.; Lu, X.; Feng, Q. Tissue inhibitor of metalloproteinase-3 inhibits neonatal mouse cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling. Am. J. Physiol. Cell Physiol. 2009, 296, C735–C745. [Google Scholar] [CrossRef]
- Reddy, V.S.; Prabhu, S.D.; Mummidi, S.; Valente, A.J.; Venkatesan, B.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF- B activation. AJP Heart Circ. Physiol. 2010, 299, H1242–H1254. [Google Scholar] [CrossRef]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trümper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Oishi, I.; Endo, M.; Nishita, M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: Their implications in developmental morphogenesis and human diseases. Dev. Dyn. 2010, 239, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.C.H.; Bao, H.L.; Cheh, P.L.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, K. Crosstalk of Sp1 and Stat3 signaling in pancreatic cancer pathogenesis. Cytokine Growth Factor Rev. 2012, 23, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An Epigenetic Switch Involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Cheneby, J.; Gheorghe, M.; Artufel, M.; Mathelier, A.; Ballester, B. ReMap 2018: An updated regulatory regions atlas from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic. Acids Res. 2017, 46, 267–275. [Google Scholar] [CrossRef]
- Bartis, D.; Csongei, V.; Weich, A.; Kiss, E.; Barko, S.; Kovacs, T.; Avdicevic, M.; D’Souza, V.K.; Rapp, J.; Kvell, K.; et al. Down-Regulation of Canonical and Up-Regulation of Non-Canonical Wnt Signalling in the Carcinogenic Process of Squamous Cell Lung Carcinoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Bayerlová, M.; Klemm, F.; Kramer, F.; Pukrop, T.; Beißbarth, T.; Bleckmann, A. Newly constructed network models of different WNT signaling cascades applied to breast cancer expression data. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M.; et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D. Histone variant H2A.Z can serve as a new target for breast cancer therapy. Curr. Med. Chem. 2010, 17, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef] [PubMed]
- Syring, I.; Klümper, N.; Offermann, A.; Braun, M.; Deng, M.; Boehm, D.; Queisser, A.; von Mässenhausen, A.; Brägelmann, J.; Vogel, W.; et al. Comprehensive analysis of the transcriptional profile of the Mediator complex across human cancer types. Oncotarget 2016, 7, 23043–23055. [Google Scholar] [CrossRef]
- Costa-Pereira, A.P.; Tininini, S.; Strobl, B.; Alonzi, T.; Schlaak, J.F.; Is’harc, H.; Gesualdo, I.; Newman, S.J.; Kerr, I.M.; Poli, V. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc. Natl. Acad. Sci. USA 2002, 99, 8043–8047. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genom. Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively Activated Stat3 Induces Tumorigenesis and Enhances Cell Motility of Prostate Epithelial Cells through Integrin 6. Mol. Cell. Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef] [PubMed]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed]
- Willert, J.; Epping, M.; Pollack, J.R.; Brown, P.O.; Nusse, R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2002, 2. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by WICKHAM, H. Biometrics 2011. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RhoU Primers |
| mRhoU-forward 5′-AGGGCAGGAGGAACTGGAGAGC-3′ |
| mRhoU-reverse, 5′-TACCCCTGGCCCCTGCTGTG-3′ |
| hRHOU-forward, 5′-ATAAAGGTTCACGGCATGCC-3′ |
| hRHOU-reverse, 5′-TAACTGCAGCTGATCGTGTG-3′ |
| β-globin Primers |
| β-glo-forward, 5′-CTCCCCCTCACTCTGTTCTG-3′ |
| β-glo-reverse, 5′-AGGAGGAGGGGAAGCTGATA-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteleone, E.; Orecchia, V.; Corrieri, P.; Schiavone, D.; Avalle, L.; Moiso, E.; Savino, A.; Molineris, I.; Provero, P.; Poli, V. SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer. Cancers 2019, 11, 101. https://doi.org/10.3390/cancers11010101
Monteleone E, Orecchia V, Corrieri P, Schiavone D, Avalle L, Moiso E, Savino A, Molineris I, Provero P, Poli V. SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer. Cancers. 2019; 11(1):101. https://doi.org/10.3390/cancers11010101
Chicago/Turabian StyleMonteleone, Emanuele, Valeria Orecchia, Paola Corrieri, Davide Schiavone, Lidia Avalle, Enrico Moiso, Aurora Savino, Ivan Molineris, Paolo Provero, and Valeria Poli. 2019. "SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer" Cancers 11, no. 1: 101. https://doi.org/10.3390/cancers11010101
APA StyleMonteleone, E., Orecchia, V., Corrieri, P., Schiavone, D., Avalle, L., Moiso, E., Savino, A., Molineris, I., Provero, P., & Poli, V. (2019). SP1 and STAT3 Functionally Synergize to Induce the RhoU Small GTPase and a Subclass of Non-canonical WNT Responsive Genes Correlating with Poor Prognosis in Breast Cancer. Cancers, 11(1), 101. https://doi.org/10.3390/cancers11010101