A Comprehensive Survey of Immune Cytolytic Activity-Associated Gene Co-Expression Networks across 17 Tumor and Normal Tissue Types

Abstract
1. Introduction
2. Result
2.1. Gene Co-Expression Network Construction for Tumor and Normal Tissues
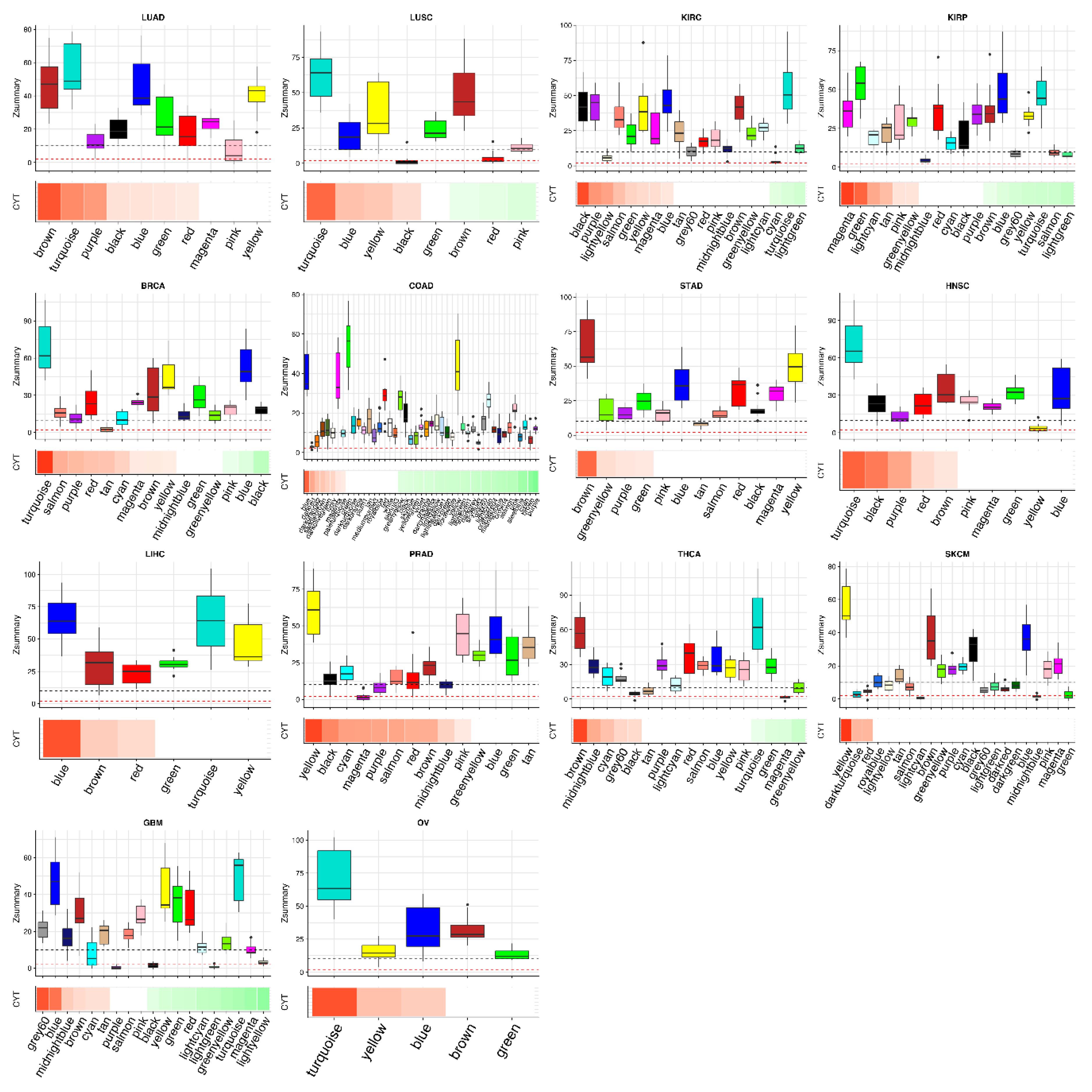
2.2. Tumor Networks Have More Diverse Cytolytic Immune Activity (CYT)-Associated Gene Signatures than Normal Networks
2.3. Some CYT-Associated Modules Are Specific to Tumor Types
2.4. Different Tumors Have a Common CYT-Associated Sub-Network
3. Discussion
4. Materials and Methods
4.1. Data Processing
4.2. Co-Expression Network Construction
4.3. Analysis of Somatic Mutation and Clinical Information
4.4. Module Preservation Statistics
4.5. Consensus Network Construction
4.6. Gene Ontology Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the curative potential of adoptive t-cell therapy for cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Prieto, P.A.; Yang, J.C.; Sherry, R.M.; Hughes, M.S.; Kammula, U.S.; White, D.E.; Levy, C.L.; Rosenberg, S.A.; Phan, G.Q. Ctla-4 blockade with ipilimumab: Long-term follow-up of 177 patients with metastatic melanoma. Clin. Cancer Res. 2012, 18, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, A.M.; Calabro, L.; Danielli, R.; Fonsatti, E.; Bertocci, E.; Pesce, I.; Fazio, C.; Cutaia, O.; Giannarelli, D.; Miracco, C.; et al. Long-term survival and immunological parameters in metastatic melanoma patients who responded to ipilimumab 10 mg/kg within an expanded access programme. Cancer Immunol. Immunother. 2013, 62, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-pd-l1 antibody mpdl3280a in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to ctla-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tian, Y.; Zhang, Z. Network biology in medicine and beyond. Circ. Cardiovasc. Genet. 2014, 7, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Furlong, L.I. Human diseases through the lens of network biology. Trends Genet. 2013, 29, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.J.; Borenstein, E.; Petrov, D.A. Broker genes in human disease. Genome Biol. Evol. 2010, 2, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Wgcna: An r package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Fast r functions for robust correlations and hierarchical clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Hsu, A.K.; Sajdak, J.; Qin, J.; Pavlidis, P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004, 14, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Sharma, N.K.; Zhang, B. Integrative network analysis reveals different pathophysiological mechanisms of insulin resistance among caucasians and african americans. BMC Med. Genom. 2015, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, H.; Thayanithy, V.; Subramanian, S.; Oberg, A.L.; Cunningham, J.M.; Cerhan, J.R.; Steer, C.J.; Thibodeau, S.N. Gene networks and micrornas implicated in aggressive prostate cancer. Cancer Res. 2009, 69, 9490–9497. [Google Scholar] [CrossRef] [PubMed]
- Ivliev, A.E.; AC’t Hoent, P.; Sergeeva, M.G. Coexpression network analysis identifies transcriptional modules related to proastrocytic differentiation and sprouty signaling in glioma. Cancer Res. 2010, 70, 10060–10070. [Google Scholar] [CrossRef] [PubMed]
- Schlager, J.J.; Powis, G. Cytosolic nad(p)h:(quinone-acceptor)oxidoreductase in human normal and tumor tissue: Effects of cigarette smoking and alcohol. Int. J. Cancer 1990, 45, 403–409. [Google Scholar] [CrossRef] [PubMed]
- MatÉs, J.M.; Pérez-Gómez, C.; De Castro, I.N. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Schmitz, G.; Muller, G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 1991, 32, 1539–1570. [Google Scholar] [PubMed]
- Wright, J.R. Pulmonary surfactant: A front line of lung host defense. J. Clin. Investig. 2003, 111, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.H., Jr. Membranous nephropathy and malignancy. Semin. Nephrol. 2010, 30, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging roles of epigenetic regulator sin3 in cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Li, C.R.; Su, J.J.; Wang, W.Y.; Lee, M.T.; Wang, T.Y.; Jiang, K.Y.; Li, C.F.; Hsu, J.M.; Chen, C.K.; Chen, M.; et al. Molecular profiling of prostatic acinar morphogenesis identifies pdcd4 and klf6 as tissue architecture-specific prognostic markers in prostate cancer. Am. J. Pathol. 2013, 182, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. Signaling at neuro/immune synapses. J. Clin. Investig. 2012, 122, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- De la Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying cancer and normal rna sequencing data from different sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. The genotype-tissue expression (gtex) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qiu, P.; Ji, Y. Tcga-assembler: Open-source software for retrieving and processing tcga data. Nat. Methods 2014, 11, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The dynamic tree cut package for r. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53. [Google Scholar] [CrossRef]
- Horvath, S. Weighted Network Analysis: Applications in Genomics and Systems Biology; Springer: New York, NY, USA, 2011. [Google Scholar]
- Wang, J.; Zhou, X.; Zhu, J.; Gu, Y.; Zhao, W.; Zou, J.; Guo, Z. Go-function: Deriving biologically relevant functions from statistically significant functions. Brief. Bioinform. 2012, 13, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Yekutieli, D.; Benjamini, Y. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LUAD | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| brown | 0.824296 | 2.22 × 10−145 | 261 | GO:0019221: cytokine-mediated signaling pathway | 45 | <2.2 × 10−16 |
| turquoise | 0.59894496 | 1.25 × 10−51 | 398 | GO:0006952: defense response | 141 | <2.2 × 10−16 |
| purple | 0.522830484 | 7.96 × 10−37 | 25 | GO:0002250: adaptive immune response | 7 | 0.017494691 |
| black | 0.156820899 | 0.001366798 | 53 | GO:0001944: vasculature development | 20 | 1.69 × 10−11 |
| blue | 0.152476114 | 0.001569748 | 331 | GO:0007059: chromosome segregation | 93 | <2.2 × 10−16 |
| green | 0.1433875 | 0.002702892 | 103 | GO:0005615: extracellular space | 54 | <2.2 × 10−16 |
| red | 0.113753405 | 0.018649506 | 74 | GO:0031012: extracellular matrix | 16 | 5.74 × 10−6 |
| LUSC | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| turquoise | 0.747554212 | 3.95 × 10−98 | 1622 | GO:0006952: defense response | 432 | <2.2 × 10−16 |
| blue | 0.329301026 | 3.60 × 10−13 | 999 | GO:0001568: blood vessel development | 100 | <2.2 × 10−16 |
| yellow | 0.2925106 | 1.40 × 10−10 | 379 | GO:0005615: extracellular space | 106 | <2.2 × 10−16 |
| black | 0.189279714 | 6.43 × 10−5 | 41 | GO:0042599: lamellar body | 3 | 0.01760483 |
| brown | −0.10539746 | 0.023468284 | 796 | GO:0006397: mRNA processing | 59 | 2.36 × 10−10 |
| red | −0.136069487 | 0.003582747 | 45 | GO:0016491: oxidoreductase activity | 21 | <2.2 × 10−16 |
| pink | −0.18133647 | 0.000111756 | 23 | GO:0005654: nucleoplasm | 16 | 0.011973696 |
| Lung | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment adjust p |
| salmon | 0.685802999 | 5.03 × 10−16 | 76 | GO:0002250: adaptive immune response | 23 | <2.2 × 10−16 |
| royalblue | 0.252541858 | 0.074263653 | 29 | GO:0030198: extracellular matrix organization | 14 | 0.000755567 |
| black | 0.231568485 | 0.099027542 | 324 | GO:0000139: Golgi membrane | 31 | 2.50 × 10−2 |
| KIRC | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| black | 0.853237771 | 7.92 × 10−175 | 442 | GO:0006952: defense response | 147 | <2.2 × 10−16 |
| purple | 0.540851383 | 1.38 × 10−40 | 371 | GO:0006952: defense response | 123 | <2.2 × 10−16 |
| light yellow | 0.483655412 | 3.41 × 10−31 | 24 | GO:0008064: regulation of actin polymerization or depolymerization | 6 | 0.006282833 |
| salmon | 0.334088519 | 4.48 × 10−14 | 122 | GO:0007059: chromosome segregation | 55 | <2.2 × 10−16 |
| yellow | 0.195625539 | 2.79 × 10−5 | 895 | GO:0005739: mitochondrion | 265 | <2.2 × 10−16 |
| magenta | 0.181397917 | 9.45 × 10−5 | 378 | GO:0005634: nucleus | 202 | 7.71 × 10−5 |
| blue | 0.132458919 | 0.005354675 | 1718 | GO:0005929: cilium | 70 | 0.003733141 |
| cyan | −0.149121774 | 0.001600027 | 106 | GO:0046395: carboxylic acid catabolic process | 16 | 8.63 × 10−8 |
| turquoise | −0.190383188 | 4.25 × 10−5 | 2322 | GO:0005654: nucleoplasm | 630 | <2.2 × 10−16 |
| KIRP | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| magenta | 0.890605949 | 7.90 × 10−121 | 261 | GO:0042110: T cell activation | 57 | <2.2 × 10−16 |
| green | 0.765114172 | 3.91 × 10−61 | 810 | GO:0006952: defense response | 254 | <2.2 × 10−16 |
| lightcyan | 0.455276412 | 3.39 × 10−15 | 43 | GO:0072359: circulatory system development | 20 | 1.52 × 10−9 |
| tan | 0.351260084 | 6.85 × 10−9 | 107 | GO:0005578: proteinaceous extracellular matrix | 21 | 2.31 × 10−11 |
| pink | 0.150757415 | 0.019363345 | 311 | GO:0005654: nucleoplasm | 94 | 0.00567888 |
| green yellow | 0.1482446 | 0.020133545 | 113 | GO:0003735: structural constituent of ribosome | 64 | <2.2 × 10−16 |
| brown | −0.113504399 | 0.081563375 | 1623 | GO:0005929: cilium | 73 | 0.000130924 |
| blue | −0.17038931 | 0.007845967 | 2117 | GO:0005794: Golgi apparatus | 285 | 4.72 × 10−13 |
| yellow | −0.235236938 | 0.000162479 | 1124 | GO:0051252: regulation of RNA metabolic process | 391 | <2.2 × 10−16 |
| turquoise | −0.242691756 | 0.000120124 | 2258 | GO:0006355: regulation of transcription, DNA-templated | 651 | <2.2 × 10−16 |
| Kidney | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| red | 0.901777428 | 2.39 × 10−47 | 762 | GO:0006952: defense response | 237 | <2.2 × 10−16 |
| green yellow | 0.642030403 | 5.42 × 10−13 | 264 | GO:0071944: cell periphery | 119 | 1.20 × 10−7 |
| dark green | 0.43921949 | 1.18 × 10−5 | 30 | GO:0006412: translation | 20 | < 2.2 × 10−16 |
| light green | 0.408015885 | 5.90 × 10−5 | 66 | GO:0003735: structural constituent of ribosome | 44 | <2.2 × 10−16 |
| salmon | 0.36223561 | 0.000495518 | 179 | GO:0072006: nephron development | 12 | 9.11 × 10−5 |
| lighty ellow | −0.201536545 | 0.099115723 | 58 | GO:0006355: regulation of transcription, DNA-templated | 29 | 0.003488448 |
| light cyan | −0.323038323 | 0.002327917 | 122 | GO:0006119: oxidative phosphorylation | 35 | <2.2 × 10−16 |
| dark red | −0.514005641 | 1.07 × 10−7 | 30 | GO:0005740: mitochondrial envelope | 11 | 8.30 × 10−5 |
| turquoise | −0.557270749 | 3.68 × 10−9 | 1462 | GO:0000786: nucleosome | 17 | 0.0025 |
| BRCA | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| turquoise | 0.911502399 | 0 | 796 | GO:0006952: defense response | 292 | <2.2 × 10−16 |
| salmon | 0.420106646 | 1.23 × 10−46 | 33 | GO:0051607: defense response to virus | 20 | <2.2 × 10−16 |
| red | 0.325051705 | 4.20 × 10−27 | 210 | GO:0001944: vasculature development | 36 | <2.2 × 10−16 |
| tan | 0.316862948 | 8.28 × 10−26 | 34 | GO:0005811: lipid particle | 6 | 9.47 × 10−5 |
| cyan | 0.242642809 | 2.68 × 10−15 | 32 | GO:0031012: extracellular matrix | 9 | 0.004274631 |
| magenta | 0.119753573 | 0.000159247 | 55 | GO:0003735: structural constituent of ribosome | 50 | <2.2 × 10−16 |
| brown | 0.117448707 | 0.000197181 | 302 | GO:0005615: extracellular space | 92 | <2.2 × 10−16 |
| yellow | 0.102251834 | 0.001113399 | 287 | GO:0007059: chromosome segregation | 89 | <2.2 × 10−16 |
| blue | −0.165492877 | 1.17 × 10−7 | 523 | GO:0010468: regulation of gene expression | 226 | <2.2 × 10−16 |
| Breast | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p |
| magenta | 0.651782549 | 1.06 × 10−14 | 78 | GO:0006955: immune response | 50 | <2.2 × 10−16 |
| tan | 0.47330672 | 6.74 × 10−7 | 28 | GO:0001944: vasculature development | 9 | 0.037762588 |
| green yellow | 0.359311022 | 0.000433761 | 33 | GO:0005615: extracellular space | 18 | 5.37 × 10−9 |
| red | −0.199648593 | 0.078699329 | 255 | GO:0016580: Sin3 complex | 5 | 0.022387309 |
| turquoise | −0.215023696 | 0.062018398 | 1820 | GO:0005634: nucleus | 1104 | <2.2 × 10−16 |
| purple | −0.249189975 | 0.027500087 | 66 | GO:0016569: regulation of transcription, DNA-templated | 35 | 0.0001246457 |
| Module | CYT Correlation | CYT Adjust p | Size | Representative GO | Intersect Number | Enrichment Adjust p | Median Z-Summary |
|---|---|---|---|---|---|---|---|
| LUSC:black | 0.189279714 | 0.0000643 | 41 | GO:0042599: lamellar body | 3 | 0.01760483 | 0.212845087 |
| LUSC:red | −0.136069487 | 0.003582747 | 45 | GO:0016491: oxidoreductase activity | 21 | <2.2 × 10−16 | 1.532464475 |
| Prostate adenocarcinoma (PRAD):magenta | 0.492176 | 1.28 × 10−30 | 427 | GO:0005615: extracellular space | 49 | 0.000711 | 1.263953 |
| Glioblastoma multiforme (GBM):black | −0.142172924 | 0.096014779 | 133 | GO:0007268: chemical synaptic transmission | 40 | <2.2 × 10−16 | 1.641635813 |
| Module | Size | Representative GO ID | Representative GO | Intersect Number | Enrichment Adjust p |
|---|---|---|---|---|---|
| turquoise | 745 | GO:0006355 | regulation of transcription, DNA-templated | 276 | <2.2 × 10−16 |
| blue | 523 | GO:0006952 | defense response | 212 | <2.2 × 10−16 |
| yellow | 113 | GO:0033108 | mitochondrial respiratory chain complex assembly | 19 | <2.2 × 10−16 |
| green | 112 | GO:0007059 | chromosome segregation | 49 | <2.2 × 10−16 |
| red | 89 | GO:0010468 | regulation of gene expression | 48 | 4.33 × 10−10 |
| black | 72 | GO:0008380 | RNA splicing | 14 | 1.79 × 10−6 |
| pink | 70 | GO:0003735 | structural constituent of ribosome | 62 | <2.2 × 10−16 |
| magenta | 68 | GO:0031981 | nuclear lumen | 49 | 5.51 × 10−12 |
| green yellow | 60 | GO:0071944 | cell periphery | 39 | 1.92 × 10−6 |
| tan | 58 | GO:0072359 | circulatory system development | 21 | 1.95 × 10−7 |
| salmon | 50 | GO:0005578 | proteinaceous extracellular matrix | 26 | <2.2 × 10−16 |
| cyan | 33 | GO:0006119 | oxidative phosphorylation | 8 | 3.35 × 10−6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, T.; Wan, J.; Han, Y.; Liu, H.; Gao, F.; Pan, Y.; Song, Q.; Wei, Z. A Comprehensive Survey of Immune Cytolytic Activity-Associated Gene Co-Expression Networks across 17 Tumor and Normal Tissue Types. Cancers 2018, 10, 307. https://doi.org/10.3390/cancers10090307
Tian T, Wan J, Han Y, Liu H, Gao F, Pan Y, Song Q, Wei Z. A Comprehensive Survey of Immune Cytolytic Activity-Associated Gene Co-Expression Networks across 17 Tumor and Normal Tissue Types. Cancers. 2018; 10(9):307. https://doi.org/10.3390/cancers10090307
Chicago/Turabian StyleTian, Tian, Ji Wan, Yan Han, Haoran Liu, Feng Gao, Youdong Pan, Qi Song, and Zhi Wei. 2018. "A Comprehensive Survey of Immune Cytolytic Activity-Associated Gene Co-Expression Networks across 17 Tumor and Normal Tissue Types" Cancers 10, no. 9: 307. https://doi.org/10.3390/cancers10090307
APA StyleTian, T., Wan, J., Han, Y., Liu, H., Gao, F., Pan, Y., Song, Q., & Wei, Z. (2018). A Comprehensive Survey of Immune Cytolytic Activity-Associated Gene Co-Expression Networks across 17 Tumor and Normal Tissue Types. Cancers, 10(9), 307. https://doi.org/10.3390/cancers10090307