HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell


 , ,
, , 
Abstract
1. Introduction
2. Results
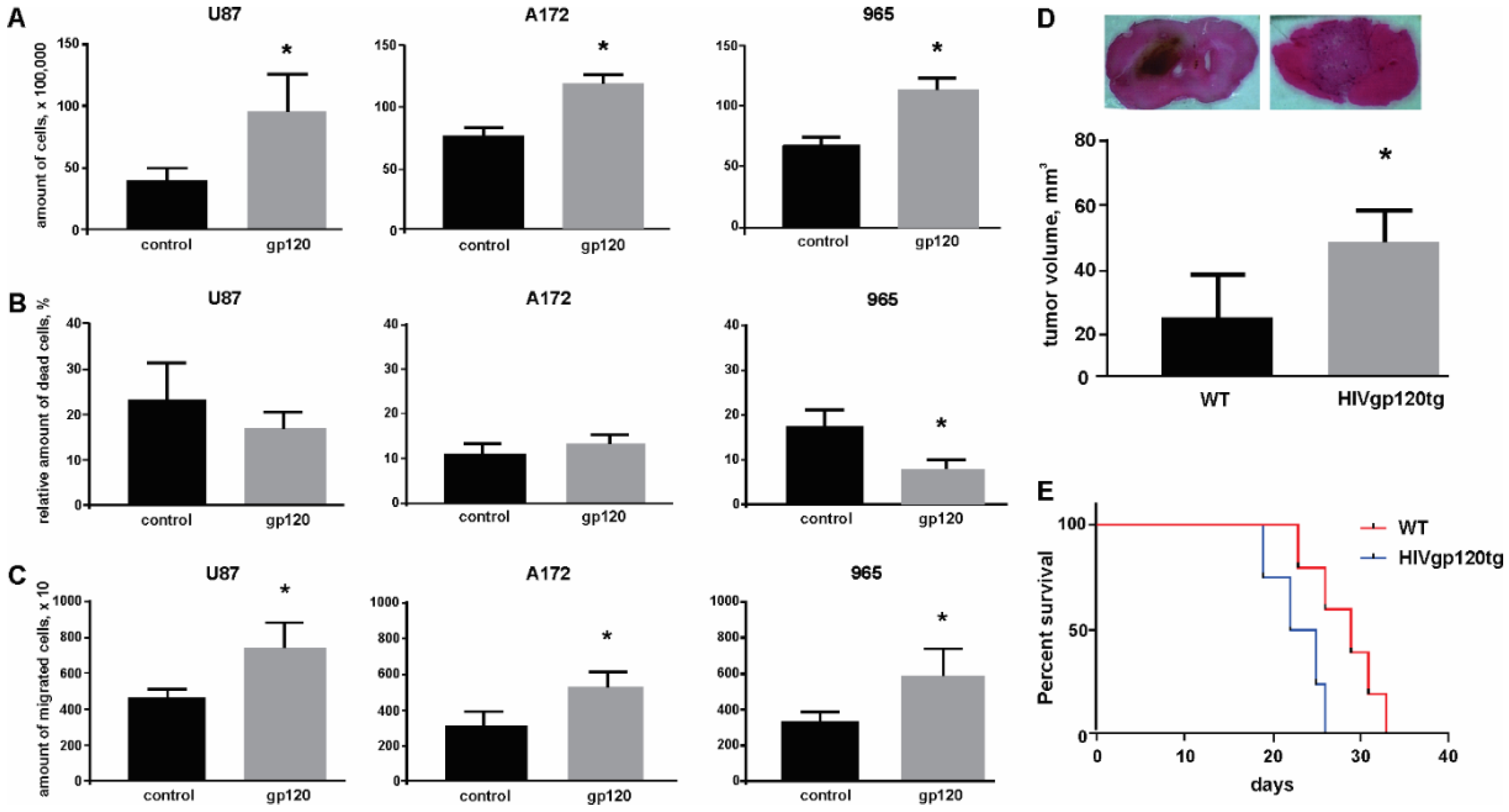
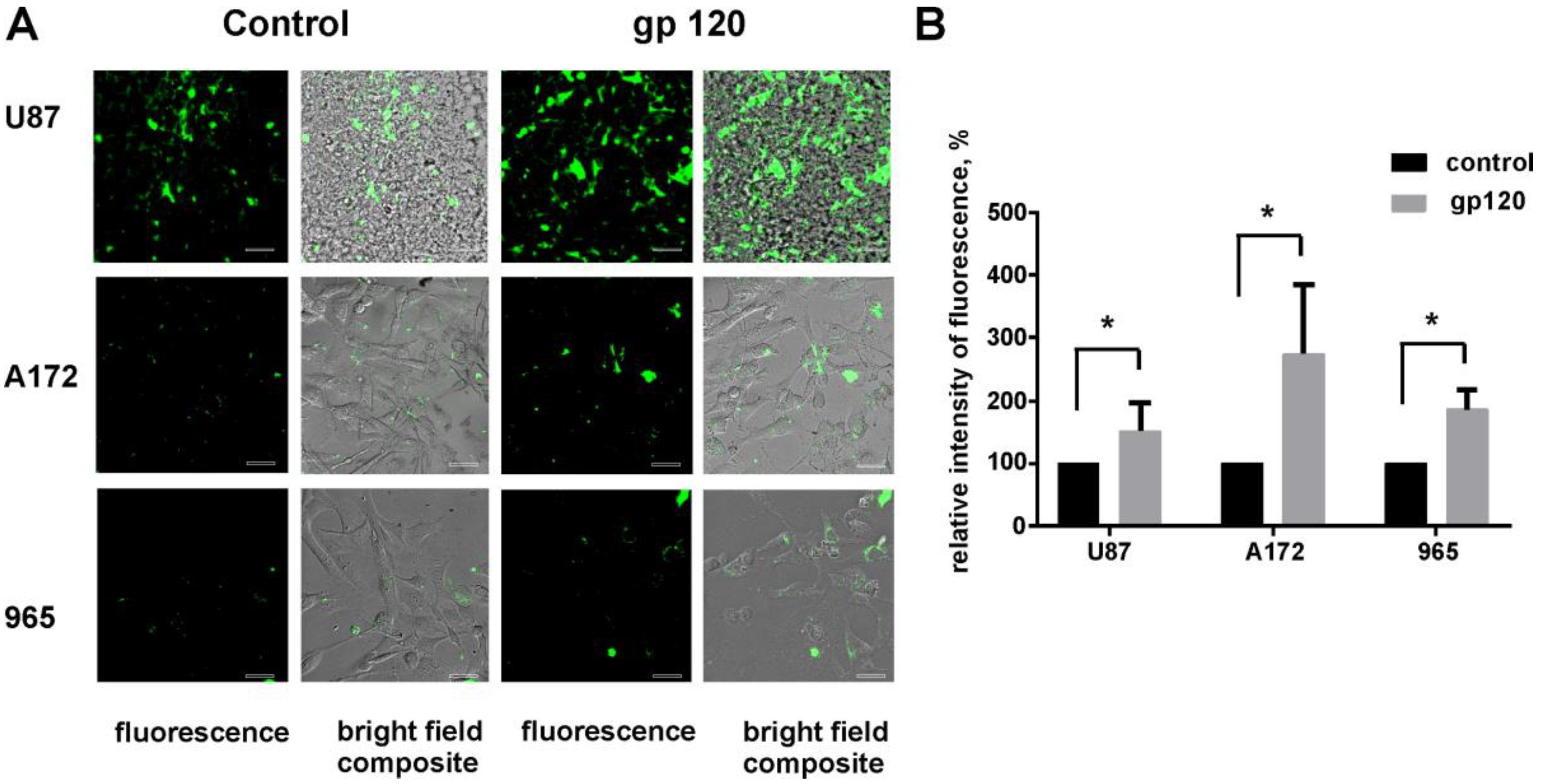
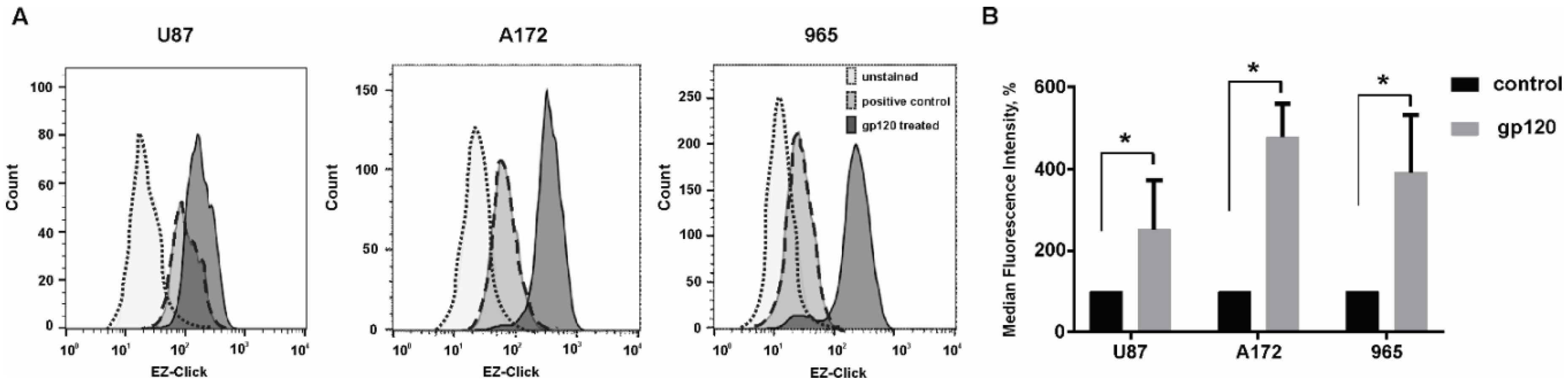
2.1. Gp120 Stimulates Proliferation and Migration in Glioma Cells
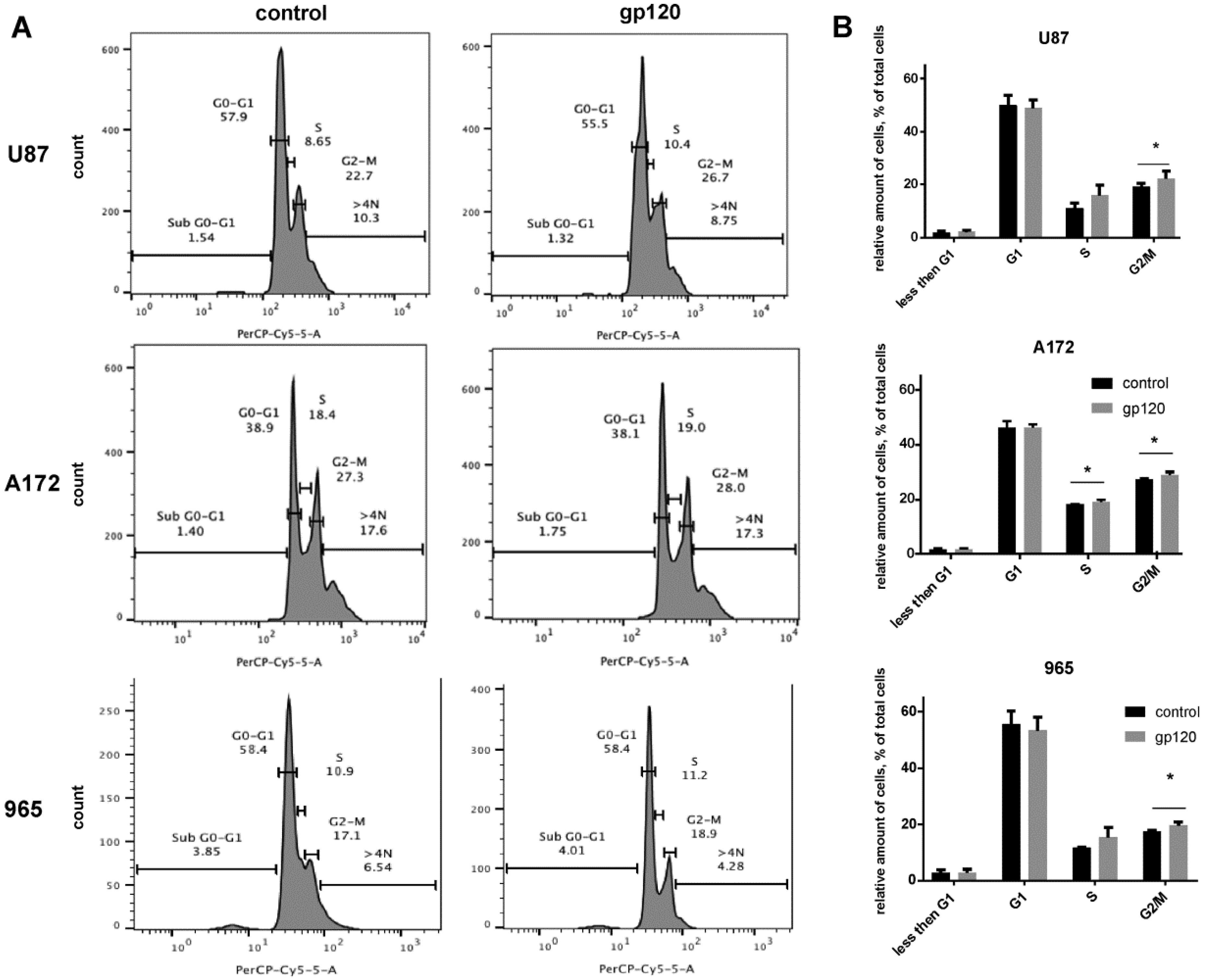
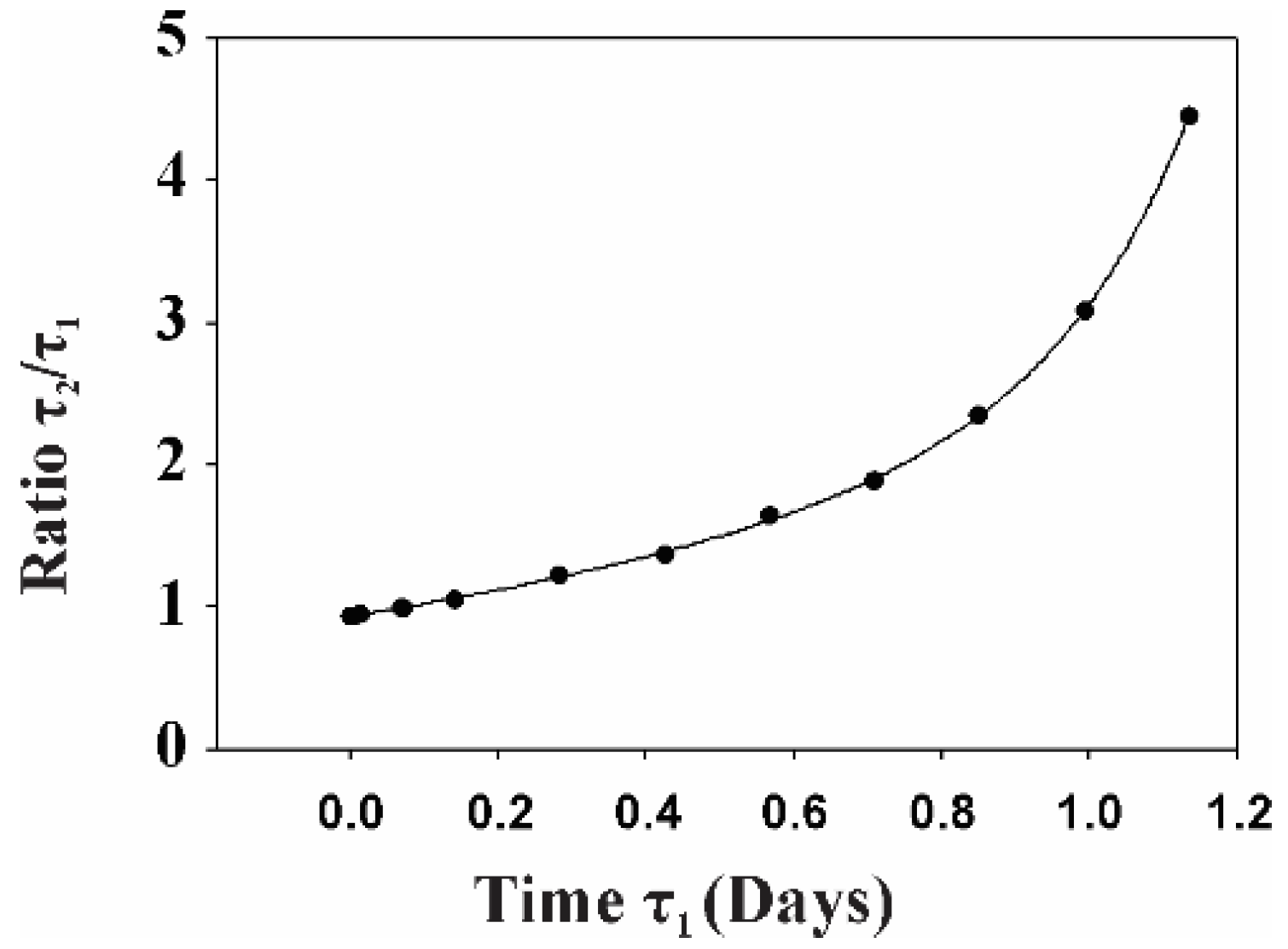
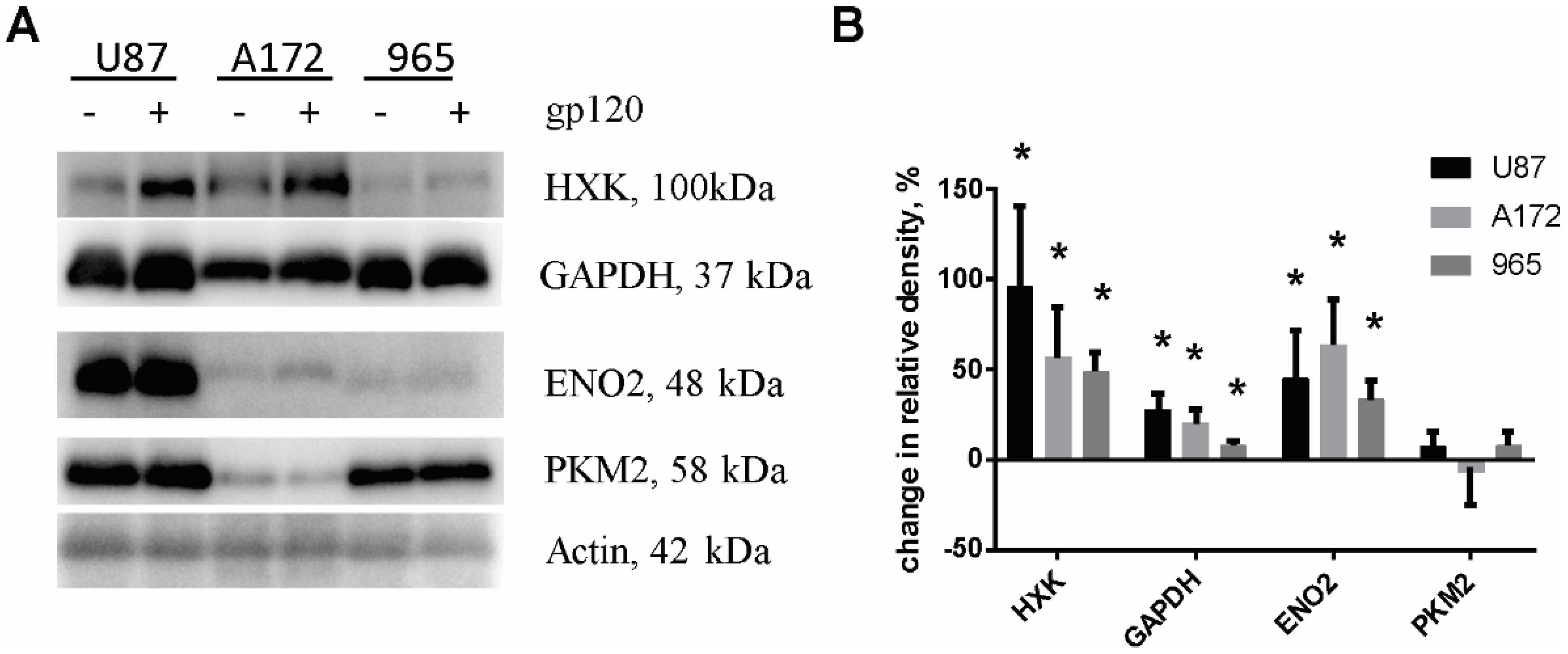
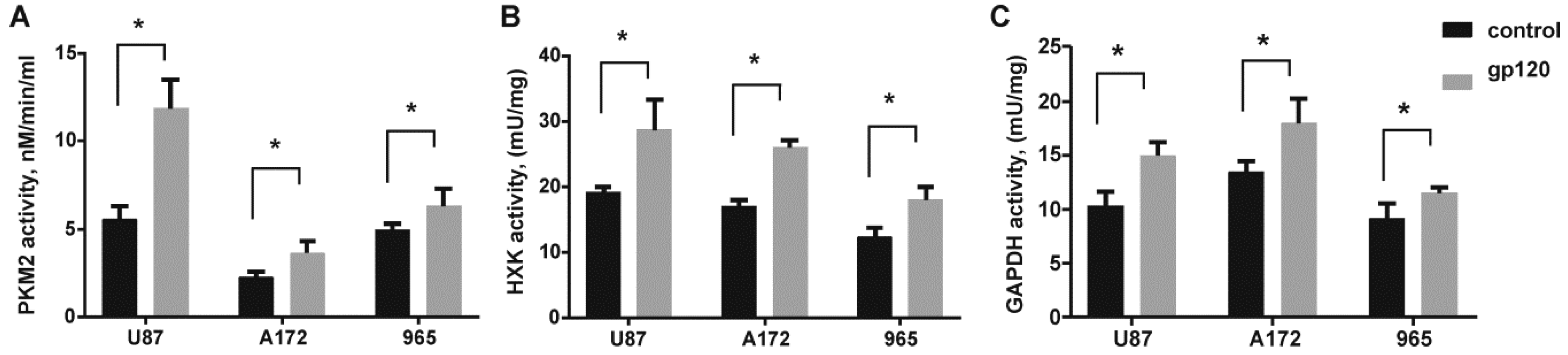
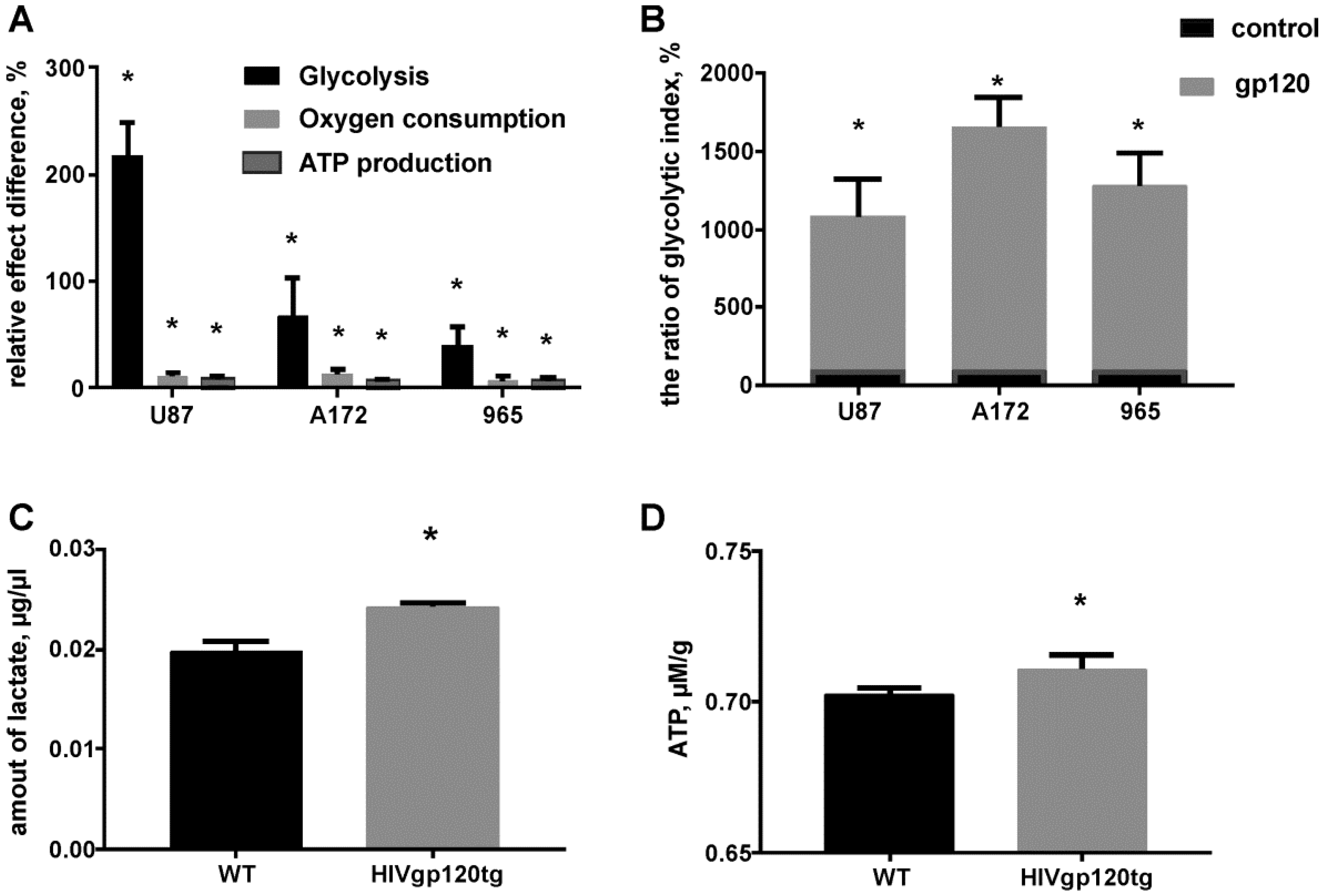
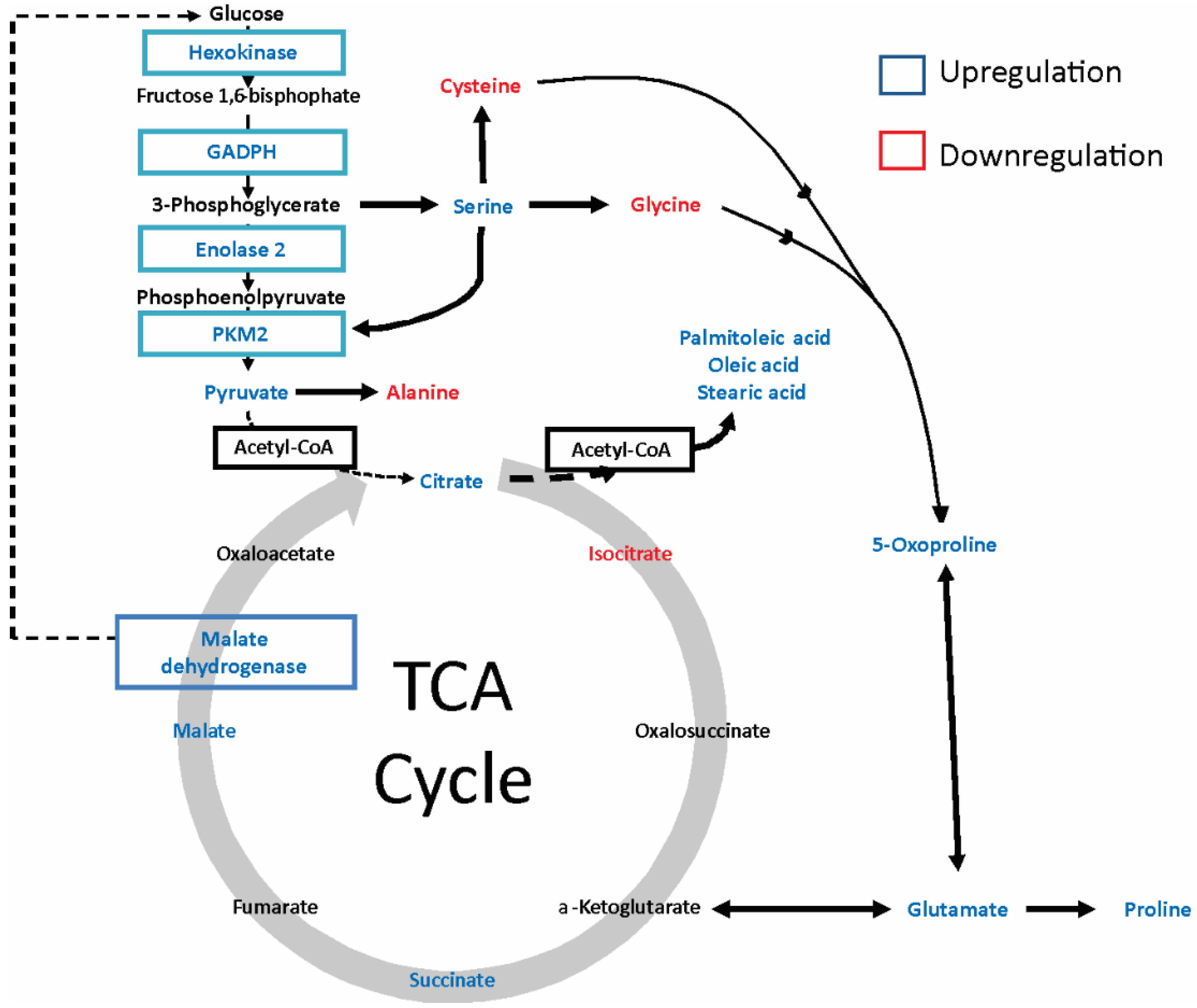
2.2. Gp120 Causes Upregulation in the Expression of Glycolytic Enzymes and Increases the Ratio of Glycolytic Index in Glioma Cells
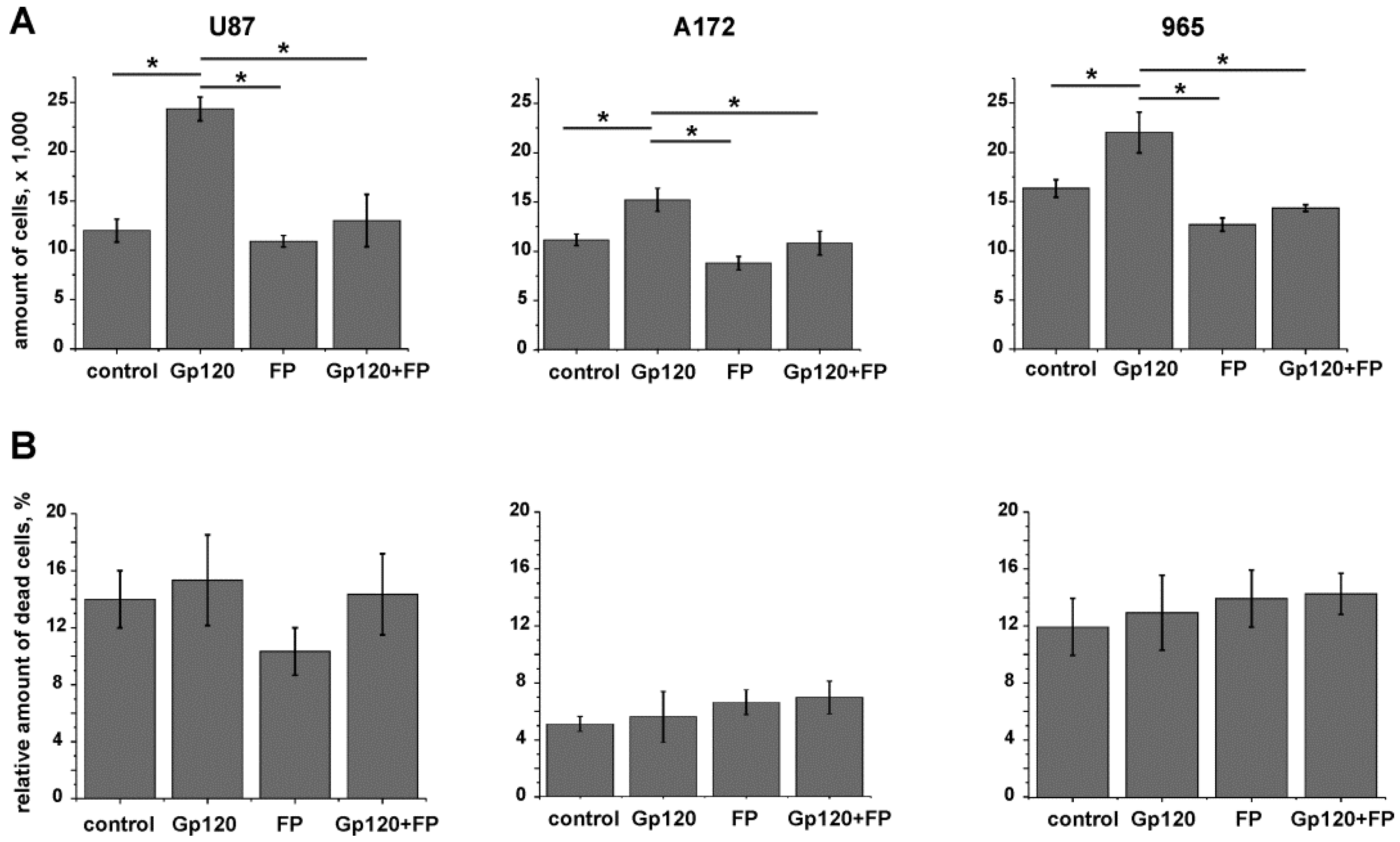
2.3. Inhibition of Glycolysis Eliminates the Stimulatory Effect of gp120 on Glioma Cell Growth
2.4. gp120 Causes Stimulation of Protein and Lipid Synthesis and a Reduction of Protein Degradation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. Intracranial Implantation of Glioma Cells
4.4. Tumor Size Evaluation
4.5. Survival Analysis
4.6. In Vitro Viability Assay
4.7. Migration Assay
4.8. Cell Cycle Assays
4.9. Western Blot Analysis
4.10. Pyruvate Kinase, Hexokinase and Glyceraldehyde 3-Phosphate Dehydrogenase Activity Assays
4.11. Glucose Uptake Assays
4.12. Glycolysis and Extracellular Oxygen Consumption Assays
4.13. ATP and Lactate Assays
4.14. Metabolomics Analysis
4.15. Protein Synthesis Assay
4.16. Chemicals and Reagents
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frisch, M.; Biggar, R.J.; Engels, E.A.; Goedert, J.J. Association of cancer with AIDS-related immunosuppression in adults. JAMA 2001, 285, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Short, S.C. Management of glioblastoma multiforme in HIV patients: A case series and review of published studies. Clin. Oncol. (R. Coll. Radiol.) 2009, 21, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, N.S.; Ahmad, F.U.; Blieden, C.; Benveniste, R.J. Brainstem anaplastic glioma in patients with AIDS: A case report and review of the literature. BMJ Case Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, M.; Lacoste, D.; Brossard, G.; Morlat, P.; Dupon, M.; Salm, L.; Dabis, F.; Groupe d’Epidémiologie Clinique du SIDA en Aquitaine. Non-acquired immune deficiency syndrome-defining malignancies in a hospital-based cohort of human immunodeficiency virus-infected patients: Bordeaux, France, 1985–1991. J. Natl. Cancer Inst. 1992, 84, 1593–1595. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, D.T.; Raizer, J.J.; Rosenblum, M.K.; Bilsky, M.H.; Hariharan, S.; Abrey, L.E. Primary intracranial neoplasms in patients with HIV. Neurology 1999, 52, 1648–1651. [Google Scholar] [CrossRef] [PubMed]
- Moulignier, A.; Mikol, J.; Pialoux, G.; Eliaszewicz, M.; Thurel, C.; Thiebaut, J.B. Cerebral glial tumors and human immunodeficiency virus-1 infection. More than a coincidental association. Cancer 1994, 74, 686–692. [Google Scholar] [CrossRef]
- Tacconi, L.; Stapleton, S.; Signorelli, F.; Thomas, D.G. Acquired immune deficiency syndrome (AIDS) and cerebral astrocytoma. Clin. Neurol. Neurosurg. 1996, 98, 149–151. [Google Scholar] [CrossRef]
- Choy, W.; Lagman, C.; Lee, S.J.; Bui, T.T.; Safaee, M.; Yang, I. Impact of Human Immunodeficiency Virus in the Pathogenesis and Outcome of Patients with Glioblastoma Multiforme. Brain Tumor Res. Treat 2016, 4, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kasantikul, V.; Kaoroptham, S.; Hanvanich, M. Acquired immunodeficiency syndrome associated with cerebral astrocytoma. Clin. Neuropathol. 1992, 11, 25–27. [Google Scholar] [PubMed]
- Acevedo, N.; Pillai, C.; Welch, M. Diagnosis and management of high-grade glioma in patients with HIV. Neuro. Oncol. 2015, 17 (Suppl. 5). [Google Scholar] [CrossRef]
- Wiemels, J.L.; Wilson, D.; Patil, C.; Patoka, J.; McCoy, L.; Rice, T.; Schwartzbaum, J.; Heimberger, A.; Sampson, J.H.; Chang, S.; et al. IgE, allergy and risk of glioma: Update from the San Francisco Bay Area Adult Glioma Study in the temozolomide era. Int. J. Cancer 2009, 125, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Salvati, M.; Frati, A.; Caroli, E.; Russo, N.; Polli, F.M.; Domenicucci, M.; Delfini, R. Glioblastoma in kidney transplant recipients. Report of five cases. J. Neurooncol. 2003, 63, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Gliomas in patients with acquired immune deficiency syndrome. Cancer 1994, 74, 1912–1914. [Google Scholar] [CrossRef]
- Wolff, R.; Zimmermann, M.; Marquardt, G.; Lanfermann, H.; Nafe, R.; Seifert, V. Glioblastoma multiforme of the brain stem in a patient with acquired immunodeficiency syndrome. Acta Neurochir. (Wien) 2002, 144. [Google Scholar] [CrossRef] [PubMed]
- Simard, E.P.; Pfeiffer, R.M.; Engels, E.A. Cumulative incidence of cancer among individuals with acquired immunodeficiency syndrome in the United States. Cancer 2011, 117, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Simard, E.P.; Pfeiffer, R.M.; Engels, E.A. Spectrum of cancer risk late after AIDS onset in the United States. Arch. Intern. Med. 2010, 170, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.J.; Lau, B.; Achenbach, C.J.; Jing, Y.; Althoff, K.N.; D’Souza, G.; Engels, E.A.; Hessol, N.A.; Brooks, J.T.; Burchell, A.N.; et al. North American AIDS Cohort Collaboration on Research and Design of the International Epidemiologic Databases to Evaluate AIDS. Cumulative Incidence of Cancer among Persons with HIV in North America: A Cohort Study. Ann. Intern. Med. 2015, 163, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Hoque, S.A.; Tanaka, A.; Islam, S.; Ahsan, G.U.; Jinno-Oue, A.; Hoshino, H. Suppression of HIV-1 Infectivity by Human Glioma Cells. AIDS Res Hum Retroviruses 2016, 32, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Haedicke, J.; Brown, C.; Naghavi, M.H. The brain-specific factor FEZ1 is a determinant of neuronal susceptibility to HIV-1 infection. Proc. Natl. Acad. Sci. USA 2009, 106, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J. Neurovirol. 2004, 10 (Suppl. 1), 21–24. [Google Scholar] [CrossRef]
- Wesselingh, S.L.; Thompson, K.A. Immunopathogenesis of HIV-associated dementia. Curr. Opin. Neurol. 2001, 14, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Ostalecki, C.; Wittki, S.; Lee, J.H.; Geist, M.M.; Tibroni, N.; Harrer, T.; Schuler, G.; Fackler, O.T.; Baur, A.S. HIV Nef- and Notch1-dependent Endocytosis of ADAM17 Induces Vesicular TNF Secretion in Chronic HIV Infection. EBioMedicine 2016, 13, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Similarity of neuronal cell injury and death in AIDS dementia and focal cerebral ischemia: Potential treatment with NMDA open-channel blockers and nitric oxide-related species. Brain Pathol. 1996, 6, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Power, C.; Gill, M.J.; Johnson, R.T. Progress in clinical neurosciences: The neuropathogenesis of HIV infection: Host-virus interaction and the impact of therapy. Can. J. Neurol. Sci. 2002, 29, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 12 (Suppl. 1), 878–892. [Google Scholar] [CrossRef] [PubMed]
- Calinescu, A.A.; Yadav, V.N.; Carballo, E.; Kadiyala, P.; Tran, D.; Zamler, D.; Doherty, R.; Srikanth, M.; Lowenstein, P.R.; Castro, M.G. Survival and proliferation of neural progenitor derived glioblastomas under hypoxic stress is controlled by a CXCL12/CXCR4 autocrine positive feedback mechanism. Clin. Cancer Res. 2017, 23, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.J. CXCR4 and Glioblastoma. Anticancer Agents Med. Chem. 2016, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Luo, D.; Liu, C.; Harrison, J.K. CCL5, CCR1 and CCR5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J. Neuroimmunol. 2012, 246, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kouno, J.; Nagai, H.; Nagahata, T.; Onda, M.; Yamaguchi, H.; Adachi, K.; Takahashi, H.; Teramoto, A.; Emi, M. Up-regulation of CC chemokine, CCL3L1 and receptors, CCR3, CCR5 in human glioblastoma that promotes cell growth. J. Neurooncol. 2004, 70, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell Neurosci. 2014, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- López, S.N.; Rodríguez-Valentín, M.; Rivera, M.; Rodríguez, M.; Babu, M.; Cubano, L.A.; Xiong, H.; Wang, G.; Kucheryavykh, L.; Boukli, N.M. HIV-1 Gp120 clade B/C induces a GRP78 driven cytoprotective mechanism in astrocytoma. Oncotarget 2017, 8, 68415–68438. [Google Scholar] [CrossRef] [PubMed]
- Rychert, J.; Strick, D.; Bazner, S.; Robinson, J.; Rosenberg, E. Detection of HIV gp120 in Plasma During Early HIV Infection Is Associated with Increased Proinflammatory and Immunoregulatory Cytokines. AIDS Res. Hum. Retroviruses 2010, 26, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Thaney, V.E.; O’Neill, A.M.; Hoefer, M.M.; Maung, R.; Sanchez, A.B.; Kaul, M. IFNβ Protects Neurons from Damage in a Murine Model of HIV-1 Associated Brain Injury. Sci. Rep. 2017, 7, 46514. [Google Scholar] [CrossRef] [PubMed]
- Maung, R.; Hoefer, M.M.; Sanchez, A.B.; Sejbuk, N.E.; Medders, K.E.; Desai, M.K.; Catalan, I.C.; Dowling, C.C.; de Rozieres, C.M.; Garden, G.A.; et al. CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J. Immunol. 2014, 193, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Toggas, S.M.; Masliah, E.; Rockenstein, E.M.; Rall, G.F.; Abraham, C.R.; Mucke, L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 1994, 367, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Asquith, B.; Debacq, C.; Florins, A.; Gillet, N.; Sanchez-Alcaraz, T.; Mosley, A.; Willems, L. Quantifying lymphocyte kinetics in vivo using carboxyfluorein diacetate succinimidyl ester. Proc. Biol. Sci. 2006, 273, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Metges, C.C.; Yu, Y.M.; Cai, W.; Lu, X.M.; Wong, S.; Regan, M.M.; Ajami, A.; Young, V.R. Oxoproline kinetics and oxoproline urinary excretion during glycine- or sulfur amino acid-free diets in humans. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E868–E876. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.A.; Persaud, C.; Hall, M.; Smith, S.; Evans, N.; Rutter, N. Urinary excretion of 5-L-oxoproline (pyroglutamic acid) during early life in term and preterm infants. Arch. Dis. Child. 1997, 76, F152–F157. [Google Scholar] [CrossRef]
- Lubert, S. Biochemistry, 4th ed.; Freeman and Company: New York, NY, USA, 1995; pp. 559–565, 614–623. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Gatenby and Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Barré-Sinoussi, F.; Ross, A.L.; Delfraissy, J.F. Past present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A. HIV entry and tropism: The chemokine receptor connection. AIDS 1997, 11 (Suppl. A), S3–S6. [Google Scholar]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Chevigne, A.; Delhalle, S.; Counson, M.; Beaupain, N.; Rybicki, A.; Verschueren, C.; Staub, T.; Schmit, J.C.; Seguin-Devaux, C.; Deroo, S. Isolation of an HIV-1 neutralizing peptide mimicking the CXCR4 and CCR5 surface from the heavy-chain complementary determining region 3 repertoire of a viremic controller. AIDS 2016, 30, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Barbero, S.; Bonavia, R.; Bajetto, A.; Porcile, C.; Pirani, P.; Ravetti, J.L.; Zona, G.L.; Spaziante, R.; Florio, T.; Schettini, G. Stromal cell-derived factor 1alpha stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res. 2003, 63, 1969–1974. [Google Scholar] [PubMed]
- Gao, D.; Rahbar, R.; Fish, E.N. CCL5 activation of CCR5 regulates cell metabolism to enhance proliferation of breast cancer cells. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Burke, J.D.; Gao, D.F.; Fish, E.N. The Chemokine CCL5 Regulates Glucose Uptake and AMP Kinase Signaling in Activated T Cells to Facilitate Chemotaxis. J. Biol. Chem. 2012, 287, 29406–29416. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Cecchetti, S.; Ricci, A.; Pacella, A.; Cigliana, G.; Bozzuto, G.; Podo, F.; Iorio, E.; Carpinelli, G. Phosphatidylcholine-specific phospholipase C inhibition down-regulates CXCR4 expression and interferes with proliferation, invasion and glycolysis in glioma cells. PLoS ONE 2017, 12, E0176108. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Clippinger, A.J.; Pierciey, FJ. Jr.; Alwine, J.C. Viruses and metabolism: Alterations of glucose and glutamine metabolism mediated by human cytomegalovirus. Adv. Virus Res. 2011, 80, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T-cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T lymphocyte metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Fukuda, T.; Krstic-Demonacos, M.; Demonacos, C.; Okumura, K.; Isogai, H.; Hayashi, M.; Saito, K.; Isogai, E. miR-663a regulates growth of colon cancer cells, after administration of antimicrobial peptides, by targeting CXCR4-p21 pathway. BMC Cancer 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Wu, H.H.; Liu, S.C.; Wang, P.C.; Ou, W.C.; Chou, W.Y.; Shen, Y.S.; Tang, C.H. CCL5 and CCR5 interaction promotes cell motility in human osteosarcoma. PLoS ONE 2012, 7, e35101. [Google Scholar] [CrossRef]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell. Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.; Gilman, V.; Khanzenzon, N.; Lesche, R.; Li, A.; Qiao, R.; Liu, X.; Wu, H. Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol. Cell. Biol. 2002, 22, 3842–3851. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Kawakami, K.; Nakada, M.; Mai, W.; Shakoori, A.; Fujisawa, H.; Hayashi, Y.; Hamada, J.; Minamoto, T. Potential therapeutic effect of glycogen synthase kinase 3beta inhibition against human glioblastoma. Clin. Cancer Res. 2009, 15, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Panigrahi, M.; Challa, S.; Mahadevan, A.; Babu, P.P. Activation of Wnt/beta-catenin/Tcf signaling pathway in human astrocytomas. Neurochem. Int. 2009, 55, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Schiapparelli, P.; Nguyen, H.N.; Igarashi, A.; Zhang, Q.; Abbadi, S.; Amzel, L.M.; Sesaki, H.; Quinones-Hinojosa, A.; Iijima, M. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene 2017, 36, 3673–3685. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Ohbayashi, K.; Suzuki, T.; Sasaki, T. Cell cycle arrest and astrocytic differentiation resulting from PTEN expression in glioma cells. J. Neurosurg. 1999, 91, 822–830. [Google Scholar] [CrossRef] [PubMed]
- De Saldanha da Gama Fischer, J.; Liao, L.; Carvalho, P.C.; Barbosa, V.C.; Domont, G.B.; da Gloria da Costa Carvalho, M.; Yates, J.R. Dynamic Proteomic Overview of Glioblastoma Cells (A172) Exposed to Perillyl Alcohol. J. Proteomics 2010, 73, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Steták, A.; Veress, R.; Ovádi, J.; Csermely, P.; Kéri, G.; Ullrich, A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007, 67, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.-W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, Ra73. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Shinagawa, T.; Ishii, S. Modulation of M2-type pyruvate kinase activity by the cytoplasmic PML tumor suppressor protein. Genes Cells 2008, 13, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Barger, J.F.; Plas, D.R. Balancing biosynthesis and bioenergetics: Metabolic programs in oncogenesis. Endocr. Relat. Cancer 2010, 17, R287–R304. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.B.; Elhassan, G.O.; Ibrahim, M.E.; David Polo Orozco, J.; Cardone, R.A.; Reshkin, S.J.; Harguindey, S. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Mesecar, A.; Heath, P.J.; Shi, W.; Nowak, T.; Stoddard, B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998, 6, 195–210. [Google Scholar] [CrossRef]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Käkönen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melinoemail, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1A5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [PubMed]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. 2015ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Pecqueur, C.; Oliver, L.; Oizel, K.; Lalier, L.; Vallette, F.M. Targeting Metabolism to Induce Cell Death in Cancer Cells and Cancer Stem Cells. Int. J. Cell. Biol. 2013, 2013, 805975. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Vina, J.R. How Glutamate Is Managed by the Blood-Brain Barrier. Biology 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Gopal, E.; Babu, E.; Srinivas, S.R.; Kubo, Y.; Umapathy, N.S.; Thakkar, S.V.; Ganapathy, V.; Prasad, P.D. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim. Biophys. Acta 2010, 1798, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [PubMed]
- Reversade, B.; Escande-Beillard, N.; Dimopoulou, A.; Fischer, B.; Chng, S.C.; Li, Y.; Tham, P.Y.; Kayserili, H.; Al-Gazali, L.; Shahwan, M.; et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 2009, 41, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhoise, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- George, C. Prendergast Cancer: Why tumours eat tryptophan. Nature 2011, 478, 192–194. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Schiapparelli, P.; Ap Rhys, C.; Guerrero-Cazares, H.; Smith, C.; Kim, D.H.; Kone, L.; Farber, H.; Lee, D.Y.; An, S.S.; et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Schiapparelli, P.; Guerrero-Cazares, H.; Magaña-Maldonado, R.; Hamilla, S.M.; Ganaha, S.; Goulin Lippi Fernandes, E.; Huang, C.H.; Aranda-Espinoza, H.; Devreotes, P.; Quinones-Hinojosa, A. NKCC1 Regulates Migration Ability of Glioblastoma Cells by Modulation of Actin Dynamics and Interacting with Cofilin. EBioMedicine 2017, 21, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Rolón-Reyes, K.; Kucheryavykh, Y.V.; Cubano, L.A.; Inyushin, M.; Skatchkov, S.N.; Eaton, M.J.; Harrison, J.K.; Kucheryavykh, L.Y. Microglia Activate Migration of Glioma Cells through a Pyk2 Intracellular Pathway. PLoS ONE 2015, 10, e0131059. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Nurnberger, N.; Kaspar, H.; Gruber, M.A.; Almstetter, M.F.; Oefner, P.J. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: Optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 2011, 399, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Babushok, V.I.; Linstrom, P.J.; Reed, J.J.; Zenkevich, I.G.; Brown, R.L.; Mallard, W.G.; Stein, S.E. Development of a database of gas chromatographic retention properties of organic compounds. J. Chromatogr. A 2007, 1157, 414–421. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID HMDB | Compound ID | Retention Time (min) | Fold Change | p Value (t Test) | Characteristic Fragment Ions | Up/Down Regulation |
|---|---|---|---|---|---|---|
| MDB00190 | L-lactic acid | 10.27 | 1.612 | 2.50E-01 | 73,147,261 | + |
| HMDB00161 | L-alanine | 10.99 | 0.223 | 1.90E-02 | 73,158,232 | − |
| HMDB00123 | glycine | 11.30 | 0.281 | 3.37E-02 | 73,147,218 | − |
| HMDB00294 | UREA | 12.60 | 0.165 | 9.25E-05 | 104,147,231 | − |
| HMDB00883 | L-valine | 12.72 | 0.357 | 4.53E-02 | 73,186,260 | − |
| HMDB00687 | L-leucine | 13.29 | 0.236 | 1.32E-02 | 73,200,274 | − |
| HMDB00172 | L-isoleucine | 13.74 | 0.013 | 1.29E-02 | 73,200,274 | − |
| HMDB00162 | L-proline | 14.25 | 23.786 | 2.70E-02 | 73,184,258 | + |
| HMDB00267 | 5-oxoproline | 16.61 | 2.243 | 1.41E-02 | 73,272,300 | + |
| HMDB00696 | L-methionine | 16.83 | 2.094 | 2.42E-02 | 73,218,292 | + |
| HMDB00187 | L-serine | 17.11 | 3.14 | 2.59E-03 | 73,288,390 | + |
| HMDB00812 | N-acetyl-L-aspartic acid | 17.30 | 0.728 | 7.33E-01 | 73,346 | − |
| HMDB00167 | L-Threonine | 17.51 | 0.993 | 6.35E-01 | 73,303,404 | − |
| HMDB00806 | Myristic acid | 18.18 | 4.479 | 2.25E-01 | 75,285 | + |
| HMDB00159 | L-phenylalanine | 18.32 | 0.899 | 9.85E-01 | 73,234,302 | − |
| HMDB00254 | Succinic acid | 14.1 | 1.723 | 2.92E-01 | 73,147,289 | + |
| HMDB00156 | Malic acid | 18.53 | 1.73 | 0.28091 | 73,115,419 | + |
| HMDB00191 | L-aspartic acid | 19.03 | 0.783 | 2.63E-01 | 73,302,418 | − |
| HMDB00574 | L-cysteine | 19.62 | 0.633 | 1.84E-01 | 73,378,406 | − |
| HMDB03229 | Palmitoleic acid | 20.16 | 3.205 | 1.85E-02 | 75,129,311 | + |
| HMDB00123 | L-glutamic acid | 20.33 | 2.501 | 4.10E-02 | 73,272,432 | + |
| HMDB00220 | Palmitic acid | 20.43 | 1.093 | 8.04E-01 | 75,129,313 | + |
| HMDB00641 | L-glutamine | 21.91 | 1.05 | 6.64E-01 | 73,147,431 | + |
| HMDB00207 | Oleic acid | 22.27 | 4.44 | 4.41E-02 | 75,129,339 | + |
| HMDB00827 | Stearic acid | 22.50 | 3.63 | 3.28E-03 | 75,129,341 | + |
| HMDB00094 | Citric acid | 23.84 | 1.506 | 5.22E-01 | 73,459,591 | + |
| HMDB00158 | L-tyrosine | 24.05 | 0.846 | 9.93E-01 | 73,302 | − |
| HMDB00929 | L-tryptophan | 24.49 | 15.672 | 1.81E-03 | 73,302,375 | + |
| HMDB01874 | Isocitric acid | 24.60 | 0.312 | 7.72E-01 | 73,345 | − |
| HMDB00067 | Cholesterol | 37.20 | 1.010 | 4.59E-01 | 75,443 | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentín-Guillama, G.; López, S.; Kucheryavykh, Y.V.; Chorna, N.E.; Pérez, J.; Ortiz-Rivera, J.; Inyushin, M.; Makarov, V.; Valentín-Acevedo, A.; Quinones-Hinojosa, A.; et al. HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers 2018, 10, 301. https://doi.org/10.3390/cancers10090301
Valentín-Guillama G, López S, Kucheryavykh YV, Chorna NE, Pérez J, Ortiz-Rivera J, Inyushin M, Makarov V, Valentín-Acevedo A, Quinones-Hinojosa A, et al. HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers. 2018; 10(9):301. https://doi.org/10.3390/cancers10090301
Chicago/Turabian StyleValentín-Guillama, Gabriel, Sheila López, Yuriy V. Kucheryavykh, Nataliya E. Chorna, Jose Pérez, Jescelica Ortiz-Rivera, Michael Inyushin, Vladimir Makarov, Aníbal Valentín-Acevedo, Alfredo Quinones-Hinojosa, and et al. 2018. "HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell" Cancers 10, no. 9: 301. https://doi.org/10.3390/cancers10090301
APA StyleValentín-Guillama, G., López, S., Kucheryavykh, Y. V., Chorna, N. E., Pérez, J., Ortiz-Rivera, J., Inyushin, M., Makarov, V., Valentín-Acevedo, A., Quinones-Hinojosa, A., Boukli, N., & Kucheryavykh, L. Y. (2018). HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers, 10(9), 301. https://doi.org/10.3390/cancers10090301