Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled


Abstract
1. Introduction
1.1. Tumor Microenvironment (TME) Associated with Ovarian Neoplasms
1.2. Basic Epigenetic Mechanisms at a Glance
- DNA methylation—addition of methyl groups to DNA CpG sites without altering DNA nucleotide sequence. Methylation occurs by means of enzymes called DNA methyltransferases (DNMTs), which place methyl groups on symmetric cytosine residues in double-stranded CpG sites [46,47]. Hypermethylation of CpG islands (nucleotide sequences enriched for CpG sites) in the promoter regions of tumor suppressor genes (TSGs) and growth regulatory genes prompts gene silencing [46,47] as attached methyl groups physically block binding of transcription factors to the gene promoters. Alternatively, dense DNA methylation interferes with the proper nucleosome positioning [48]. Within the DNMT family (including three active enzymes, DNMT1, DNMT3a, and DNMT3b), DNMT1 exhibits high preference for hemimethylated DNA (in which one of two complimentary DNA strands already possess attached methyl groups), and is therefore responsible for so called “maintenance methylation” [49,50]. DNMT3a and DNMT3b are primarily responsible for the “de novo” methylation of previously unmethylated CpG regions [51,52], but both of these methyltransferases have been shown to carry out maintenance methylation as well [53]. Importantly, in human neoplastic cells, it has been shown that DNMT1 provides both de novo and maintenance DNA methylation of TSGs [54,55,56]. The demethylating agents (or hypomethylating agents (HMAs) that inhibit these enzymes (azacitidine or AZA; decitabine or DAC; SGI-110 or guadecitabine) are discussed below).
- Histone modifications—various posttranslational modifications (PTMs) at histone protein N-terminal tails, which impair proper interactions between adjacent nucleosomes to affect the compact packing of the chromatin and impede the binding ability of other factors/enzymes that are involved in gene transcription [57,58]. The most common and well-characterized PTM, histone acetylation, is a dynamic, reversible process in which positively charged histone lysine residues are neutralized via the addition of acetyl groups by histone acetyltransferases (HATs), resulting in the attenuation of bonds between negatively charged DNA string and a histone complex. In the reverse reaction, deacetylation, enzymes histone deacetylases (HDACs) remove acetyl groups, and reinforce positive charge of the lysines, securing compact wrapping of DNA around histones [59,60]. Similarly, histone (de)phosphorylation utilizes protein kinases and phosphatases to attach or remove negatively charged phosphate groups, respectively, influencing chemical attraction between DNA and histone tails (reviewed in [60,61]). Histone (de)methylation—addition/removal of methyl groups by histone-specific methyltransferases and demethylases—can either activate or silence gene transcription. Remarkably, the functional consequences of each histone (de)methylation event depend on the histone, amino acid and residue methylated, degree of modification (mono-, di- or tri-methylation), and attraction of additional function-specific protein cofactors to the site, as well as existence of other methyl or acetyl groups in close proximity (reviewed in [62]). Comprehensive analyses of currently known histone PTMs, including those less common (ubiquitylation, sumoylation, deamination, etc.), their functional outcomes and complex interplay between the DNA methylation and histone modifications have been recently published [63,64].
- Chromatin remodeling—rearrangement of chromatin organization through complete or partial nucleosome repositioning and altering gene access for transcription. Chromatin remodeling can occur via nucleosome sliding (movement of the core histone octamer nexus across DNA segment with no evident disintegration of the octamer itself), nucleosome ejection (nucleosome segregation from the chromatin chain), or histone eviction (removal of histone H2A/H2B dimers from the DNA-associated nucleosome, sometimes with an alternative histone replacement) [65,66,67]. These processes are mediated by a number of ATP-dependent chromatin remodelers with high binding affinity to modified core histone tails, as well as transcriptional enzymes, which are extensively described in [65,66,67]. In particular, ARID1A and SMARCA4 are prominent chromatin remodeler examples in ovarian cancer. ARID1A is frequently mutated in ovarian clear cell (~50%) and low grade ovarian endometrioid (30%) carcinomas [68,69]. Most interestingly, tumors with ARID1A mutations acquire sensitivity to pan-HDAC inhibitors, thus making ARID1A-bearing cancers attractive for HDAC-based therapy [70]. SMARCA4 is frequently (over 90%) mutated in ovarian small cell carcinomas of the hypercalcemic type [71,72], however, to our knowledge, the first case of a germline SMARCA4 mutation in a patient with HGSOC was recently reported [73]. Further investigation on the role and clinical applicability of SMARCA4 and ARID1A in HGSOC is warranted [74,75].Altogether, the three epigenetic mechanisms that are described above work closely to mediate DNA (un)coiling around the core histones and ensure dynamic chromatin reassembly between heterochromatin (condensed or closed, silent) and euchromatin (loose or open, transcription-permissive) states.
- Non-coding RNA interference—a group of epigenetic regulatory mechanisms that involves microRNAs (miRNAs; miR) and long non-coding RNAs (lncRNAs). MiRNAs are short (~22 nucleotides) non-messenger RNAs that act primarily at a posttranscriptional level by base pairing with their complimentary mRNA targets to alter mRNA translation into protein [76]. Remarkably, one miRNA may complement a variety of mRNAs, whereas the same mRNA transcript might be a target of multiple miRNAs. Additionally, miRNAs may act as mRNA destabilizers causing poly-A-tail shortening [77] or interfere at the gene transcriptional level by means of PTMs (e.g., initiation of histone H3 lysine9 methylation with RNA interference machinery, followed by DNA methylation and gene transcription repression) and heterochromatic silencing [78]. LncRNAs are long (>200 for up to a hundred thousand nucleotides) non-messenger RNA that execute epigenetic regulation via several mechanisms: engage in post-translational histone modifications through association with chromatin-modifying proteins as an obligatory active player in the complex or as a scaffold that brings different protein complexes in close vicinity for proper functioning; serve as endogenous competitors to mRNA by base pairing with miRNAs and uncovering mRNAs for effective protein translation; or, serve as precursor RNAs for miRNAs (all mechanisms are detailed in [79,80,81]).
2. Epigenetic Crosstalk between EOC Cells and TME Cellular Components
2.1. Cancer-Associated Fibroblasts (CAFs)
2.2. Cancer-Associated Adipocytes (CAAs)
2.3. Mesenchymal Stem Cells (MSCs)
2.4. Tumor-Associated Endothelial Cells (TECs)
2.5. Pericytes
2.6. Tumor-Associated Macrophages (TAMs)
2.7. Tumor-Infiltrating Lymphocytes (TILs)
2.8. Plasmacytoid Dendritic Cells (PDCs)
2.9. Mesothelial Cells (MCs)
3. Ovarian TME: Potential Epigenomic-Based Therapeutic Strategies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D. Rethinking Ovarian Cancer: Recommendations for Improving Outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2018; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Ahmedin, J. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Jacques, F.; Isabelle, S.; Rajesh, D.; Sultan, E.; Colin, M.; Marise, R.; Parkin, D.M.; David, F.; Freddie, B. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2011; National Cancer Institute: Bethesda, MD, USA, 2013. Available online: http://Seer.Cancer.Gov/Csr/1975_2011/ (accessed on 20 April 2014).
- Marcus, C.S.; Maxwell, G.L.; Darcy, K.M.; Hamilton, C.A.; McGuire, W.P. Current Approaches and Challenges in Managing and Monitoring Treatment Response in Ovarian Cancer. J. Cancer 2014, 5, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A. Rethinking Ovarian Cancer II: Reducing Mortality from High-Grade Serous Ovarian Cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.T.; Choi, K.; Kang, S.K.; Leung, P.C.K. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. The Origin of Ovarian Cancers—Hypotheses and Controversies. Front. Biosci. 2013, 5, 709–719. [Google Scholar] [CrossRef]
- Bowen, N.J.; Walker, L.D.; Matyunina, L.V.; Logani, S.; Totten, K.A.; Benigno, B.B.; McDonald, J.F. Gene Expression Profiling Supports the Hypothesis that Human Ovarian Surface Epithelia are Multipotent and Capable of Serving as Ovarian Cancer Initiating Cells. BMC Med. Genom. 2009, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Chene, G.; Dauplat, J.; Radosevic-Robin, N.; Cayre, A.; Penault-Llorca, F. Tu-be Or Not Tu-be: That is the Question… About Serous Ovarian Carcinogenesis. Crit. Rev. Oncol. 2013, 88, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.; Cheng, C.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian Surface Epithelium at the Junction Area Contains a Cancer-Prone Stem Cell Niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Yemelyanova, A.; Zaino, R.J.; Kurman, R.J. The Fallopian Tube-Peritoneal Junction: A Potential Site of Carcinogenesis. Int. J. Gynecol. Pathol. 2011, 30, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Earp, M.A.; Cunningham, J.M. DNA Methylation Changes in Epithelial Ovarian Cancer Histotypes. Genomics 2015, 106, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Barton, C.A.; Hacker, N.F.; Clark, S.J.; O’Brien, P.M. DNA Methylation Changes in Ovarian Cancer: Implications for Early Diagnosis, Prognosis and Treatment. Gynecol. Oncol. 2008, 109, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Montavon, C.; Gloss, B.S.; Warton, K.; Barton, C.A.; Statham, A.L.; Scurry, J.P.; Tabor, B.; Nguyen, T.V.; Qu, W.; Samimi, G. Prognostic and Diagnostic Significance of DNA Methylation Patterns in High Grade Serous Ovarian Cancer. Gynecol. Oncol. 2012, 124, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Jen, J.; Harris, C.C. Clinical Outcomes of TP53 Mutations in Cancers. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Pu, T.; Xiong, L.; Liu, Q.; Zhang, M.; Cai, Q.; Liu, H.; Sood, A.K.; Li, G.; Kang, Y.; Xu, C. Delineation of Retroperitoneal Metastatic Lymph Nodes in Ovarian Cancer with Near-infrared Fluorescence Imaging. Oncol. Lett. 2017, 14, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.; Choi, H.J.; Bischoff, F.Z. Hematogenous Metastasis of Ovarian Cancer: Rethinking Mode of Spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular Spheroids in Ovarian Cancer Metastases: Biology and Pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic Plasticity of Neoplastic Ovarian Epithelium: Unique Cadherin Profiles in Tumor Progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B. Ovarian Carcinoma and Serous Effusions. Changing Views Regarding Tumor Progression and Review of Current Literature1. Anal. Cell. Pathol. 2001, 23, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P.N. Ovarian Carcinoma Ascites Spheroids Adhere to Extracellular Matrix Components and Mesothelial Cell Monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Chang, S.; Hsiao, C.; Chien, T.; Shih, D.T. Isolation and Characterization of Stromal Progenitor Cells from Ascites of Patients with Epithelial Ovarian Adenocarcinoma. J. Biomed. Sci. 2012, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K. Getting to Know Ovarian Cancer Ascites: Opportunities for Targeted Therapy-Based Translational Research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose Tissue and Adipocytes Support Tumorigenesis and Metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.; Galandi, D.; Blum, H.E. Malignant Ascites: Systematic Review and Guideline for Treatment. Eur. J. Cancer 2006, 42, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Ross, L.; Holash, J.; Nakanishi, M.; Hu, L.; Hofmann, J.I.; Yancopoulos, G.D.; Jaffe, R.B. Vascular Endothelial Growth Factor-Trap Decreases Tumor Burden, Inhibits Ascites, and Causes Dramatic Vascular Remodeling in an Ovarian Cancer Model. Clin. Cancer Res. 2003, 9, 5721–5728. [Google Scholar] [PubMed]
- Fang, X.; Schummer, M.; Mao, M.; Yu, S.; Tabassam, F.H.; Swaby, R.; Hasegawa, Y.; Tanyi, J.L.; LaPushin, R.; Eder, A. Lysophosphatidic Acid is a Bioactive Mediator in Ovarian Cancer. Biochim. Biophys. Acta 2002, 1582, 257–264. [Google Scholar] [CrossRef]
- Garrison, R.N.; Galloway, R.H.; Heuser, L.S. Mechanisms of Malignant Ascites Production. J. Surg. Res. 1987, 42, 126–132. [Google Scholar] [CrossRef]
- Gotlieb, W.H.; Abrams, J.S.; Watson, J.M.; Velu, T.J.; Berek, J.S.; Martínez-Maza, O. Presence of Interleukin 10 (IL-10) in the Ascites of Patients with Ovarian and Other Intra-Abdominal Cancers. Cytokine 1992, 4, 385–390. [Google Scholar] [CrossRef]
- Hu, L.; Hofmann, J.; Zaloudek, C.; Ferrara, N.; Hamilton, T.; Jaffe, R.B. Vascular Endothelial Growth Factor Immunoneutralization Plus Paclitaxel Markedly Reduces Tumor Burden and Ascites in Athymic Mouse Model of Ovarian Cancer. Am. J. Pathol. 2002, 161, 1917–1924. [Google Scholar] [CrossRef]
- Mesiano, S.; Ferrara, N.; Jaffe, R.B. Role of Vascular Endothelial Growth Factor in Ovarian Cancer: Inhibition of Ascites Formation by Immunoneutralization. Am. J. Pathol. 1998, 153, 1249–1256. [Google Scholar] [CrossRef]
- Moradi, M.M.; Carson, L.F.; Twiggs, L.B.; Weinberg, J.B.; Haney, A.; Ramakrishnan, S. Serum and Ascitic Fluid Levels of Interleukin-1, Interleukin-6, and Tumor Necrosis Factor-alpha in Patients with Ovarian Epithelial Cancer. Cancer 1993, 72, 2433–2440. [Google Scholar] [CrossRef]
- Pedersen, N.; Schmitt, M.; Ronne, E.; Nicoletti, M.I.; Hoyer-Hansen, G.; Conese, M.; Giavazzi, R.; Dano, K.; Kuhn, W.; Janicke, F. A Ligand-Free, Soluble Urokinase Receptor is Present in the Ascitic Fluid from Patients with Ovarian Cancer. J. Clin. Investig. 1993, 92, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Cannon, M.J.; Pecorelli, S.; Parham, G.P. Secretion of Vascular Endothelial Growth Factor in Ovarian Cancer. Eur. J. Gynaecol. Oncol. 1999, 20, 177–181. [Google Scholar] [PubMed]
- Topalak, O.; Saygili, U.; Soyturk, M.; Karaca, N.; Batur, Y.; Uslu, T.; Erten, O. Serum, Pleural Effusion, and Ascites CA-125 Levels in Ovarian Cancer and Nonovarian Benign and Malignant Diseases: A Comparative Study. Gynecol. Oncol. 2002, 85, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, O.; Hafter, R.; Coppenrath, E.; Pflanz, M.A.; Schmitt, M.; Babic, R.; Linke, R.; Gossner, W.; Graeff, H. Fibrin-Fibronectin Compounds in Human Ovarian Tumor Ascites and their Possible Relation to the Tumor Stroma. Cancer Res. 1988, 48, 3507–3514. [Google Scholar] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic Acid as a Potential Biomarker for Ovarian and Other Gynecologic Cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Yabushita, H.; Shimazu, M.; Noguchi, M.; Kishida, T.; Narumiya, H.; Sawaguchi, K.; Noguchi, M. Vascular Endothelial Growth Factor Activating Matrix Metalloproteinase in Ascitic Fluid during Peritoneal Dissemination of Ovarian Cancer. Oncol. Rep. 2003, 10, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Carduner, L.; Leroy-Dudal, J.; Picot, C.R.; Gallet, O.; Carreiras, F.; Kellouche, S. Ascites-induced shift along epithelial-mesenchymal spectrum in ovarian cancer cells: Enhancement of their invasive behavior partly dependant on αv integrins. Clin. Exp. Metastasis 2014, 31, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C. Embryology, Epigenetics and Biogenetics. Nature 1956, 177, 1241. [Google Scholar] [CrossRef]
- Esteller, M. Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Huff, J.; Zilberman, D. Dnmt1-Independent CG Methylation Contributes to Nucleosome Positioning in Diverse Eukaryotes. Cell 2014, 156, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and Sequencing of a cDNA Encoding DNA Methyltransferase of Mouse Cells: The Carboxyl-Terminal Domain of the Mammalian Enzymes is Related to Bacterial Restriction Methyltransferases. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(Cytosine-C5)-Methyltransferase Methylates DNA Processively with High Preference for Hemimethylated Target Sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F.; Ramsahoye, B.H.; Kashevsky, H.; Tudor, M.; Mastrangelo, M.; Orr-Weaver, T.L.; Jaenisch, R. Mammalian (Cytosine-5) Methyltransferases Cause Genomic DNA Methylation and Lethality in Drosophila. Nat. Genet. 1999, 23, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA Methylation De Novo. Science 1999, 286, 2287–2288. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Jair, K.; Yen, R.C.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG Methylation is Maintained in Human Cancer Cells Lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Jair, K.W.; Bachman, K.E.; Suzuki, H.; Ting, A.H.; Rhee, I.; Yen, R.W.; Baylin, S.B.; Schuebel, K.E. De Novo CpG Island Methylation in Human Cancer Cells. Cancer Res. 2006, 66, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.H.; Jair, K.W.; Schuebel, K.E.; Baylin, S.B. Differential Requirement for DNA Methyltransferase 1 in Maintaining Human Cancer Cell Gene Promoter Hypermethylation. Cancer Res. 2006, 66, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is Required to Maintain CpG Methylation and Aberrant Gene Silencing in Human Cancer Cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å. Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in Cancer: Targeting Chromatin Modifications. Mol. Cancer. Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Seto, E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of Nucleosome Dynamics by Histone Modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Oki, M.; Aihara, H.; Ito, T. Role of histone phosphorylation in chromatin dynamics and its implications in diseases. In Chromatin and Disease; Springer: Berlin, Germany, 2007; pp. 323–340. [Google Scholar]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA Methylation and Histone Modification: Patterns and Paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hamiche, A.; Sandaltzopoulos, R.; Gdula, D.A.; Wu, C. ATP-Dependent Histone Octamer Sliding Mediated by the Chromatin Remodeling Complex NURF. Cell 1999, 97, 833–842. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. Chromatin remodeling complexes. In Fundamentals of Chromatin; Springer: Berlin, Germany, 2014; pp. 69–146. [Google Scholar]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih, I.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 Mutations in Small Cell Carcinoma of the Ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B. Germline and Somatic SMARCA4 Mutations Characterize Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Muppala, R.; Donenberg, T.; Huang, M.S.; Schlumbrecht, M.P. SMARCA4 Germline Gene Mutation in a Patient with Epithelial Ovarian: A Case Report. Gynecol. Oncol. Rep. 2017, 22, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Fukumoto, T.; Magno, E. SWI/SNF Complexes in Ovarian Cancer: Mechanistic Insights and Therapeutic Implications. Mol. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.; Conejo-Garcia, J.R. Synthetic Lethality by Targeting EZH2 Methyltransferase Activity in ARID1A-Mutated Cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA Destabilization is the Dominant Effect of Mammalian microRNAs by the Time Substantial Repression Ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of Heterochromatic Silencing and Histone H3 Lysine-9 Methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Ollikainen, N.; Guttman, M. Long Non-Coding RNAs: Spatial Amplifiers that Control Nuclear Structure and Gene Expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and Cross-Talk between Non-Coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Jones, P. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The Epigenetics of Cancer Etiology. Semin. Cancer Biol. 2004, 14, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Schneider, R. Targeting Histone Modifications—Epigenetics in Cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sawan, C.; Herceg, Z. Histone modifications and cancer. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2010; pp. 57–85. [Google Scholar]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Fang, F.; Matei, D.E.; Huang, T.H.; Nephew, K.P. Minireview: Epigenetic Changes in Ovarian Cancer. Endocrinology 2009, 150, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Matei, D.E.; Huang, T.H.; Nephew, K.P. Role of Epigenomics in Ovarian and Endometrial Cancers. Epigenomics 2010, 2, 419–447. [Google Scholar] [CrossRef] [PubMed]
- Natanzon, Y.; Goode, E.L.; Cunningham, J.M. Epigenetics in Ovarian Cancer. Semin. Cancer Biol. 2018, 51, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Volinia, S.; Bonome, T.; Calin, G.A.; Greshock, J.; Yang, N.; Liu, C.; Giannakakis, A.; Alexiou, P.; Hasegawa, K. Genomic and Epigenetic Alterations Deregulate microRNA Expression in Human Epithelial Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 7004–7009. [Google Scholar] [CrossRef] [PubMed]
- Kinose, Y.; Sawada, K.; Nakamura, K.; Kimura, T. The Role of microRNAs in Ovarian Cancer. Biomed. Res. Int. 2014, 2014, 249393. [Google Scholar] [CrossRef] [PubMed]
- Meryet-Figuiere, M.; Lambert, B.; Gauduchon, P.; Vigneron, N.; Brotin, E.; Poulain, L.; Denoyelle, C. An Overview of Long Non-Coding RNAs in Ovarian Cancers. Oncotarget 2016, 7, 44719–44734. [Google Scholar] [CrossRef] [PubMed]
- Worku, T.; Bhattarai, D.; Ayers, D.; Wang, K.; Wang, C.; Rehman, Z.U.; Talpur, H.S.; Yang, L. Long Non-Coding RNAs: The New Horizon of Gene Regulation in Ovarian Cancer. Cell. Physiol. Biochem. 2017, 44, 948–966. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, R.; Hyde, C.A.; Zhong, X.Y. Epigenetics of Ovarian Cancer: From the Lab to the Clinic. Gynecol. Oncol. 2010, 118, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Huang, T.H.; Brown, R.; Nephew, K.P. The Epigenetics of Ovarian Cancer Drug Resistance and Resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ashworth, A. Marked for Death: Targeting Epigenetic Changes in Cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell. Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New Horizons in Tumor Microenvironment Biology: Challenges and Opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of Tumor Microenvironment in Tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. Immune-Suppressing Cellular Elements of the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2017, 1, 241–255. [Google Scholar] [CrossRef]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lin, J. Seed-in-Soil: Pancreatic Cancer Influenced by Tumor Microenvironment. Cancers 2017, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A. Molecular Characterization of the Tumor Microenvironment in Breast Cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Omura, N.; Hong, S.; Griffith, M.; Goggins, M. Pancreatic Cancer Associated Fibroblasts Display Normal Allelotypes. Cancer Biol. Ther. 2008, 7, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Hu, M.; Sridhar, A.; Opeskin, K.; Fox, S.; Shipitsin, M.; Trivett, M.; Thompson, E.R.; Ramakrishna, M.; Gorringe, K.L. No Evidence of Clonal Somatic Genetic Alterations in Cancer-Associated Fibroblasts from Human Breast and Ovarian Carcinomas. Nat. Genet. 2008, 40, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.A.; Gillespie, J.W.; Grover, A.; Tangrea, M.A.; Chuaqui, R.F.; Emmert-Buck, M.R.; Tangrea, J.A.; Libutti, S.K.; Linehan, W.M.; Woodson, K.G. Gene Promoter Methylation in Prostate Tumor–Associated Stromal Cells. J. Natl. Cancer Inst. 2006, 98, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; van den Brûle, F.; Velculescu, V.; Polyak, K. Distinct Epigenetic Changes in the Stromal Cells of Breast Cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Mathot, P.; Grandin, M.; Devailly, G.; Souaze, F.; Cahais, V.; Moran, S.; Campone, M.; Herceg, Z.; Esteller, M.; Juin, P. DNA Methylation Signal Has a Major Role in the Response of Human Breast Cancer Cells to the Microenvironment. Oncogenesis 2017, 6, e390. [Google Scholar] [CrossRef] [PubMed]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M. DNA Methylation Variations are Required for Epithelial-to-Mesenchymal Transition Induced by Cancer-Associated Fibroblasts in Prostate Cancer Cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Serrano, C.H.; Benamar, S.; Croce, O. Epigenetic Switch Drives the Conversion of Fibroblasts into Proinvasive Cancer-Associated Fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Busch, J.I.; Tang, B.; Flanders, K.C.; Oshima, A.; Sutton, E.; Karpova, T.S.; Roberts, A.B.; Wakefield, L.M.; Niederhuber, J.E. Transient Tumor-Fibroblast Interactions Increase Tumor Cell Malignancy by a TGF-B Mediated Mechanism in a Mouse Xenograft Model of Breast Cancer. PLoS ONE 2010, 5, e9832. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-B Induces Global Changes in DNA Methylation during the Epithelial-to-Mesenchymal Transition in Ovarian Cancer Cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-Beta in CAF-Mediated Tumor Growth and Metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-Beta Modulates Ovarian Cancer Invasion by Upregulating CAF-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-Wide Mapping of Polycomb Target Genes Unravels their Roles in Cell Fate Transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.; Hsu, C.; Peng, K.; Chen, C.; Kuo, W.; Eva, Y.L.; Shew, J.; Chang, K.; Juan, L.; Lee, W. Breast Cancer Cells Induce Stromal Fibroblasts to Secrete ADAMTS1 for Cancer Invasion through an Epigenetic Change. PLoS ONE 2012, 7, e35128. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, R. Role of EZH2 in Epithelial Ovarian Cancer: From Biological Insights to Therapeutic Target. Front. Oncol. 2013, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA Expression Analysis of Cancer-Associated Fibroblasts and Normal Fibroblasts in Breast Cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, S.; Le, S.; Lu, R.; Rader, J.S.; Meyers, C.; Zheng, Z. Aberrant Expression of Oncogenic and Tumor-Suppressive microRNAs in Cervical Cancer is Required for Cancer Cell Growth. PLoS ONE 2008, 3, e2557. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Sakamoto, N.; Oue, N.; Yashiro, M.; Sentani, K.; Yanagihara, K.; Hirakawa, K.; Yasui, W. MicroRNA-143 Regulates Collagen Type III Expression in Stromal Fibroblasts of Scirrhous Type Gastric Cancer. Cancer Sci. 2014, 105, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Sacca, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D. Control of Tumor and Microenvironment Cross-Talk by miR-15a and miR-16 in Prostate Cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Yamamichi, N.; Shimomura, R.; Inada, K.; Sakurai, K.; Haraguchi, T.; Ozaki, Y.; Fujita, S.; Mizutani, T.; Furukawa, C.; Fujishiro, M.; et al. Locked Nucleic Acid in Situ Hybridization Analysis of miR-21 Expression during Colorectal Cancer Development. Clin. Cancer Res. 2009, 15, 4009–4016. [Google Scholar]
- Schepeler, T.; Reinert, J.T.; Ostenfeld, M.S.; Christensen, L.L.; Silahtaroglu, A.N.; Dyrskjot, L.; Wiuf, C.; Sorensen, F.J.; Kruhoffer, M.; Laurberg, S.; et al. Diagnostic and Prognostic microRNAs in Stage II Colon Cancer. Cancer Res. 2008, 68, 6416–6424. [Google Scholar] [CrossRef] [PubMed]
- Enkelmann, A.; Heinzelmann, J.; von Eggeling, F.; Walter, M.; Berndt, A.; Wunderlich, H.; Junker, K. Specific Protein and miRNA Patterns Characterise Tumour-Associated Fibroblasts in Bladder Cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs Reprogram Normal Fibroblasts into Cancer-Associated Fibroblasts in Ovarian Cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ji, G.; Le, X.; Wang, C.; Xu, L.; Feng, M.; Zhang, Y.; Yang, H.; Xuan, Y.; Yang, Y.; et al. Long Noncoding RNA LINC00092 Acts in Cancer-Associated Fibroblasts to Drive Glycolysis and Progression of Ovarian Cancer. Cancer Res. 2017, 77, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Vafaee, F.; Colvin, E.K.; Mok, S.C.; Howell, V.M.; Samimi, G. Functional Prediction of Long Non-Coding RNAs in Ovarian Cancer-Associated Fibroblasts Indicate a Potential Role in Metastasis. Sci. Rep. 2017, 7, 10374. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Rio, M. The role of cancer-associated adipocytes (CAA) in the dynamic interaction between the tumor and the host. In Tumor-Associated Fibroblasts and Their Matrix; Springer: Berlin, Germany, 2011; pp. 111–123. [Google Scholar]
- Hoy, A.J.; Balaban, S.; Saunders, D.N. Adipocyte–Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Venkateswaran, V. Periprostatic Adipose Tissue and Prostate Cancer Progression: New Insights into the Tumor Microenvironment. Clin. Genitourin. Cancer 2014, 12, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-Associated Adipocytes Promotes Breast Tumor Radioresistance. Biochem. Biophys. Res. Commun. 2011, 411, 102–106. [Google Scholar]
- Tabuso, M.; Homer-Vanniasinkam, S.; Adya, R.; Arasaradnam, R.P. Role of Tissue Microenvironment Resident Adipocytes in Colon Cancer. World J. Gastroenterol. 2017, 23, 5829–5835. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.C.; Wang, J.; Kong, J.; Qi, Y.; Quigg, R.J.; Li, X. miR-17-92 Cluster Accelerates Adipocyte Differentiation by Negatively Regulating Tumor-Suppressor Rb2/p130. Proc. Natl. Acad. Sci. USA 2008, 105, 2889–2894. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, B.S.; Ryu, H.S.; Lee, H.; Lee, M.; Park, I.A.; Kim, J.; Han, W.; Noh, D.; Moon, H. Transition into Inflammatory Cancer-Associated Adipocytes in Breast Cancer Microenvironment Requires microRNA Regulatory Mechanism. PLoS ONE 2017, 12, e0174126. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal Transfer of Stroma-Derived miR21 Confers Paclitaxel Resistance in Ovarian Cancer Cells through Targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, A.R.; Helland, Å.; Gromov, P.; Wielenga, V.T.; Talman, M.M.; Brunner, N.; Sandhu, V.; Børresen-Dale, A.; Gromova, I.; Haakensen, V.D. Profiling of microRNAs in Tumor Interstitial Fluid of Breast Tumors—A Novel Resource to Identify Biomarkers for Prognostic Classification and Detection of Cancer. Mol. Oncol. 2017, 11, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.; Monteiro, C.; Catalán, V.; Hu, P.; Cunha, V.; Rodríguez, A.; Gómez-Ambrosi, J.; Fraga, A.; Príncipe, P.; Lobato, C. Obesity and Prostate Cancer: Gene Expression Signature of Human Periprostatic Adipose Tissue. BMC Med. 2012, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Monteiro, C.; Matos, A.; You, J.; Fraga, A.; Pereira, C.; Catalán, V.; Rodríguez, A.; Gómez-Ambrosi, J.; Frühbeck, G. Epigenome-Wide DNA Methylation Profiling of Periprostatic Adipose Tissue in Prostate Cancer Patients with Excess Adiposity—A Pilot Study. Clin. Epigenet. 2018, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An Analysis of DNA Methylation in Human Adipose Tissue Reveals Differential Modification of Obesity Genes before and After Gastric Bypass and Weight Loss. Genome Biol. 2015, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A. Obesity-Induced DNA Hypermethylation of the Adiponectin Gene Mediates Insulin Resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Pulliam, N.; Ozes, A.; Buechlein, A.; Ding, N.; Rusch, D.; Keer, H.; O’Hagan, H.; Stack, M.S.; Nephew, K.P. Epigenetic Targeting of Adipocytes Inhibits High-Grade Serous Ovarian Cancer Cell Migration and Invasion. Mol. Cancer. Res. 2018, 16, 1226–1240. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z. Progress and Prospects of Long Noncoding RNAs in Lipid Homeostasis. Mol. Metab. 2016, 5, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Liu, L.; Li, H.; Sun, Y.; Luo, H.; Li, T.; Wang, S.; Dalton, S.; Zhao, R.C.; Chen, R. Long Noncoding RNA ADINR Regulates Adipogenesis by Transcriptionally Activating C/EBPα. Stem Cell Rep. 2015, 5, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jin, C.; Zheng, Y.; Li, X.; Zhang, S.; Zhang, Y.; Jia, L.; Li, W. Knockdown of lncRNA MIR31HG Inhibits Adipocyte Differentiation of Human Adipose-Derived Stem Cells Via Histone Modification of FABP4. Sci. Rep. 2017, 7, 8080. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long Noncoding RNAs Regulate Adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Nikpayam, E.; Tasharrofi, B.; Sarrafzadeh, S.; Ghafouri-Fard, S. The Role of Long Non-Coding RNAs in Ovarian Cancer. Iran. Biomed. J. 2017, 21, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.S.; Meryet-Figuiere, M.; Sabzalipoor, H.; Kashani, H.H.; Nikzad, H.; Asemi, Z. Dysregulated Expression of Long Noncoding RNAs in Gynecologic Cancers. Mol. Cancer 2017, 16, 107. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal Stem Cells Reside in Virtually all Post-Natal Organs and Tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human Ovarian Carcinoma-Associated Mesenchymal Stem Cells Regulate Cancer Stem Cells and Tumorigenesis Via Altered BMP Production. J. Clin. Investig. 2011, 121, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Choi, Y.J.; McLean, K.; Allen, B.L.; di Magliano, M.P.; Buckanovich, R.J. Human Carcinoma-Associated Mesenchymal Stem Cells Promote Ovarian Cancer Chemotherapy Resistance Via a BMP4/HH Signaling Loop. Oncotarget 2016, 7, 6916–6932. [Google Scholar] [CrossRef] [PubMed]
- Touboul, C.; Lis, R.; Al Farsi, H.; Raynaud, C.M.; Warfa, M.; Althawadi, H.; Mery, E.; Mirshahi, M.; Rafii, A. Mesenchymal Stem Cells Enhance Ovarian Cancer Cell Infiltration through IL6 Secretion in an Amniochorionic Membrane Based 3D Model. J. Transl. Med. 2013, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Lis, R.; Touboul, C.; Mirshahi, P.; Ali, F.; Mathew, S.; Nolan, D.J.; Maleki, M.; Abdalla, S.A.; Raynaud, C.M.; Querleu, D. Tumor Associated Mesenchymal Stem Cells Protects Ovarian Cancer Cells from Hyperthermia through CXCL12. Int. J. Cancer 2011, 128, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from Ovarian Cancer Cells Induce Adipose Tissue-Derived Mesenchymal Stem Cells to Acquire the Physical and Functional Characteristics of Tumor-Supporting Myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Bu, S.; Wang, Q.; Zhang, Q.; Sun, J.; He, B.; Xiang, C.; Liu, Z.; Lai, D. Human Endometrial Mesenchymal Stem Cells Exhibit Intrinsic Anti-Tumor Properties on Human Epithelial Ovarian Cancer Cells. Sci. Rep. 2016, 6, 37019. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chen, M.; Wang, L.; Ning, Y.; Liang, J.; Zhang, H.; Xu, C.; Chen, S.; Yao, L. Systemic Mesenchymal Stem Cells Reduce Growth Rate of Cisplatin-Resistant Ovarian Cancer. Int. J. Clin. Exp. Pathol. 2013, 6, 2506–2514. [Google Scholar] [PubMed]
- Zhang, Y.; Wang, J.; Ren, M.; Li, M.; Chen, D.; Chen, J.; Shi, F.; Wang, X.; Dou, J. Gene Therapy of Ovarian Cancer Using IL-21-Secreting Human Umbilical Cord Mesenchymal Stem Cells in Nude Mice. J. Ovar. Res. 2014, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, W.; Zhuang, R.; Song, T.; Li, P. The Effect of Endostatin Mediated by Human Mesenchymal Stem Cells on Ovarian Cancer Cells in Vitro. J. Cancer Res. Clin. Oncol. 2010, 136, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Wilson, S.M.; Spaeth, E.L.; Studeny, M.; Zompetta, C.; Samudio, I.; Roby, K.; Andreeff, M.; Marini, F.C. Tumor Stroma Engraftment of Gene-Modified Mesenchymal Stem Cells as Anti-Tumor Therapy Against Ovarian Cancer. Cytotherapy 2013, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wei, D.; Sun, L.; Wang, Y.; Wu, X.; Li, Y.; Fang, Z.; Shang, H.; Wei, Z. A Preliminary Study on the Construction of Double Suicide Gene Delivery Vectors by Mesenchymal Stem Cells and the in Vitro Inhibitory Effects on SKOV3 Cells. Oncol. Rep. 2014, 31, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Amara, I.; Touati, W.; Beaune, P.; de Waziers, I. Mesenchymal Stem Cells as Cellular Vehicles for Prodrug Gene Therapy Against Tumors. Biochimie 2014, 105, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Iliou, M.S.; Esteller, M. Epigenetic Alterations Involved in Cancer Stem Cell Reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Leu, Y.; Huang, T.H.; Hsiao, S. Epigenetic reprogramming of mesenchymal stem cells. In Epigenetic Alterations in Oncogenesis; Springer: Berlin, Germany, 2013; pp. 195–211. [Google Scholar]
- Teven, C.M.; Liu, X.; Hu, N.; Tang, N.; Kim, S.H.; Huang, E.; Yang, K.; Li, M.; Gao, J.L.; Liu, H.; et al. Epigenetic Regulation of Mesenchymal Stem Cells: A Focus on Osteogenic and Adipogenic Differentiation. Stem Cells Int. 2011, 2011, 201371. [Google Scholar] [CrossRef] [PubMed]
- Mortada, I.; Mortada, R. Epigenetic Changes in Mesenchymal Stem Cells Differentiation. Eur. J. Med. Genet. 2017, 61, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.B.; Benkusky, N.A.; Sen, B.; Rubin, J.; Pike, J.W. Epigenetic Plasticity Drives Adipogenic and Osteogenic Differentiation of Marrow-Derived Mesenchymal Stem Cells. J. Biol. Chem. 2016, 291, 17829–17847. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bruno, S.; Deregibus, M.C.; Tetta, C.; Camussi, G. MicroRNAs and mesenchymal stem cells. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2011; pp. 291–320. [Google Scholar]
- Reza, A.M.M.T.; Choi, Y.-J.; Yasuda, H.; Kim, J.-H. Human Adipose Mesenchymal Stem Cell-Derived Exosomal-miRNAs are Critical Factors for Inducing Anti-Proliferation Signalling to A2780 and SKOV-3 Ovarian Cancer Cells. Sci. Rep. 2016, 6, 38498. [Google Scholar] [CrossRef] [PubMed]
- Bellayr, I.H.; Kumar, A.; Puri, R.K. MicroRNA Expression in Bone Marrow-Derived Human Multipotent Stromal Cells. BMC Genom. 2017, 18, 605. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, L.; Chen, Y.; Shao, Q.; Qin, W. Bafilomycin A1 Inhibits the Growth and Metastatic Potential of the BEL-7402 Liver Cancer and HO-8910 Ovarian Cancer Cell Lines and Induces Alterations in their microRNA Expression. Exp. Ther. Med. 2015, 10, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Zang, D.; Wu, S.; Liu, A.; Zhu, J.; Wu, G.; Li, J.; Jiang, L. Upregulation of miR-572 Transcriptionally Suppresses SOCS1 and p21 and Contributes to Human Ovarian Cancer Progression. Oncotarget 2015, 6, 15180–15193. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Huang, Y.; Zhang, L.; Tian, G.; Liao, Q.; Chen, S. MiR-572 Prompted Cell Proliferation of Human Ovarian Cancer Cells by Suppressing PPP2R2C Expression. Biomed. Pharmacother. 2016, 77, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Lopatina, T.; Gai, C.; Deregibus, M.C.; Kholia, S.; Camussi, G. Cross Talk between Cancer and Mesenchymal Stem Cells through Extracellular Vesicles Carrying Nucleic Acids. Front. Oncol. 2016, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Shih, D.T.; Hsiao, C.; Huang, S.; Chang, S.; Cheng, W. Gene Methylation of Human Ovarian Carcinoma Stromal Progenitor Cells Promotes Tumorigenesis. J. Transl. Med. 2015, 13, 367. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular Differentiation, Cytidine Analogs and DNA Methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Akino, T.; Hida, K.; Hida, Y.; Tsuchiya, K.; Freedman, D.; Muraki, C.; Ohga, N.; Matsuda, K.; Akiyama, K.; Harabayashi, T.; et al. Cytogenetic Abnormalities of Tumor-Associated Endothelial Cells in Human Malignant Tumors. Am. J. Pathol. 2009, 175, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Konerding, M.; Malkusch, W.; Klapthor, B.; Van Ackern, C.; Fait, E.; Hill, S.; Parkins, C.; Chaplin, D.; Presta, M.; Denekamp, J. Evidence for Characteristic Vascular Patterns in Solid Tumours: Quantitative Studies using Corrosion Casts. Br. J. Cancer 1999, 80, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; Croix, B.S. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell 2007, 11, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Yu, J.; Old, M.; Teknos, T.N.; Kumar, P. Tumor-Associated Endothelial Cells Promote Tumor Metastasis by Chaperoning Circulating Tumor Cells and Protecting them from Anoikis. PLoS ONE 2015, 10, e0141602. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation Via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Ohga, N.; Akiyama, K.; Maishi, N.; Hida, Y. Heterogeneity of Tumor Endothelial Cells. Cancer Sci. 2013, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M. Heterogeneity of Tumor Endothelial Cells: Comparison between Tumor Endothelial Cells Isolated from High-and Low-Metastatic Tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N. Tumour Endothelial Cells in High Metastatic Tumours Promote Metastasis Via Epigenetic Dysregulation of Biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Jair, K.W.; Vire, E.; Eguchi, S.; Hoebers, N.T.; Fraga, M.F.; Esteller, M.; Fuks, F.; Baylin, S.B.; van Engeland, M.; et al. Angiostatic Activity of DNA Methyltransferase Inhibitors. Mol. Cancer Ther. 2006, 5, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.; Lee, S.; Moon, E.; Kim, H.; Lee, S.; Chung, H.Y. Histone Deacetylases Induce Angiogenesis by Negative Regulation of Tumor Suppressor Genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Castermans, K.; Vire, E.; Dings, R.P.; Hoebers, N.T.; Mayo, K.H.; Oude Egbrink, M.G.; Molema, G.; Fuks, F.; van Engeland, M.; et al. Epigenetic Regulation of Tumor Endothelial Cell Anergy: Silencing of Intercellular Adhesion Molecule-1 by Histone Modifications. Cancer Res. 2006, 66, 10770–10777. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Melotte, V.; Vire, E.; Langenkamp, E.; Molema, G.; Fuks, F.; Herman, J.G.; Van Criekinge, W.; Griffioen, A.W.; van Engeland, M. Identification of Epigenetically Silenced Genes in Tumor Endothelial Cells. Cancer Res. 2007, 67, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Bonome, T.; Li, Y.; Kamat, A.A.; Han, L.Y.; Schmandt, R.; Coleman, R.L.; Gershenson, D.M.; Jaffe, R.B.; Birrer, M.J.; et al. Gene Alterations Identified by Expression Profiling in Tumor-Associated Endothelial Cells from Invasive Ovarian Carcinoma. Cancer Res. 2007, 67, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Varambally, S.; Arend, R.C. Histone Methyltransferase EZH2: A Therapeutic Target for Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A. Regulation of Tumor Angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, J.; Wang, H. MicroRNA-298 Inhibits Malignant Phenotypes of Epithelial Ovarian Cancer by Regulating the Expression of EZH2. Oncol. Lett. 2016, 12, 3926–3932. [Google Scholar] [CrossRef] [PubMed]
- Özeş, A.R.; Wang, Y.; Zong, X.; Fang, F.; Pilrose, J.; Nephew, K.P. Therapeutic Targeting using Tumor Specific Peptides Inhibits Long Non-Coding RNA HOTAIR Activity in Ovarian and Breast Cancer. Sci. Rep. 2017, 7, 894. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Farrington-Rock, C.; Crofts, N.J.; Doherty, M.J.; Ashton, B.A.; Griffin-Jones, C.; Canfield, A.E. Chondrogenic and Adipogenic Potential of Microvascular Pericytes. Circulation 2004, 110, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Washington, R.; Wagnerova, J.; Dore-Duffy, P. CNS Microvascular Pericytes Express Macrophage-Like Function, Cell Surface Integrin αM, and Macrophage Marker ED-2. Microvasc. Res. 1996, 52, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Okamoto, O.K. Combined Effects of Pericytes in the Tumor Microenvironment. Stem Cells Int. 2015, 2015, 868475. [Google Scholar] [CrossRef] [PubMed]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an Overlooked Player in Vascular Pathobiology. Pharmacol. Ther. 2017, 171, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Chong, L.; George, J.; Schluter, H.; Monchgesang, S.; Mills, S.; Li, J.; Parish, C.; Bowtell, D.; Kaur, P.; et al. Pericytes Promote Malignant Ovarian Cancer Progression in Mice and Predict Poor Prognosis in Serous Ovarian Cancer Patients. Clin. Cancer Res. 2016, 22, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shahzad, M.M.; Moreno-Smith, M.; Lin, Y.; Jennings, N.B.; Allen, J.K.; Landen, C.N.; Mangala, L.S.; Armaiz-Pena, G.N.; Schmandt, R. Targeting Pericytes with a PDGF-B Aptamer in Human Ovarian Carcinoma Models. Cancer Biol. Ther. 2010, 9, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Perotti, A.; Sessa, C.; Mancuso, A.; Noberasco, C.; Cresta, S.; Locatelli, A.; Carcangiu, M.L.; Passera, K.; Braghetti, A.; Scaramuzza, D.; et al. Clinical and Pharmacological Phase I Evaluation of Exherin (ADH-1), a Selective Anti-N-Cadherin Peptide in Patients with N-Cadherin-Expressing Solid Tumours. Ann. Oncol. 2009, 20, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Xiang, W.; Zhang, Q.; Cen, Y.; Su, Q.; Zhang, F.; Lu, Y.; Zhao, H.; Fu, P. Human Glioblastoma-Derived Mesenchymal Stem Cell to Pericytes Transition and Angiogenic Capacity in Glioblastoma Microenvironment. Cell Physiol. Biochem. 2018, 46, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Truettner, J.S.; Katyshev, V.; Esen-Bilgin, N.; Dietrich, W.D.; Dore-Duffy, P. Hypoxia Alters MicroRNA Expression in Rat Cortical Pericytes. Microrna 2013, 2, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y. Inflammation-Induced Endothelial Cell-Derived Extracellular Vesicles Modulate the Cellular Status of Pericytes. Sci. Rep. 2015, 5, 8505. [Google Scholar] [CrossRef] [PubMed]
- Kratzsch, T.; Kuhn, S.A.; Joedicke, A.; Hanisch, U.K.; Vajkoczy, P.; Hoffmann, J.; Fichtner, I. Treatment with 5-Azacitidine Delay Growth of Glioblastoma Xenografts: A Potential New Treatment Approach for Glioblastomas. J. Cancer Res. Clin. Oncol. 2018, 144, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Olin, E.; Buskirk, H.; Reineke, L. Cytotoxicity and Mode of Action of 5-Azacytidine on L1210 Leukemia. Cancer Res. 1970, 30, 2760–2769. [Google Scholar] [PubMed]
- Karén, J.; Rodriguez, A.; Friman, T.; Dencker, L.; Sundberg, C.; Scholz, B. Effects of the Histone Deacetylase Inhibitor Valproic Acid on Human Pericytes in Vitro. PLoS ONE 2011, 6, e24954. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Turkowski, K.; Mora, J.; Brune, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting Tumor-Associated Macrophages to Become Tumoricidal Effectors as a Novel Strategy for Cancer Therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian Cancer Cells Polarize Macrophages Toward a Tumor-Associated Phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Komohara, Y.; Takaishi, K.; Katabuchi, H.; Takeya, M. Detection of M2 Macrophages and Colony-stimulating Factor 1 Expression in Serous and Mucinous Ovarian Epithelial Tumors. Pathol. Int. 2009, 59, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K. Tumor-Associated Macrophages Contribute to Tumor Progression in Ovarian Cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M. A Transcriptome-Based Global Map of Signaling Pathways in the Ovarian Cancer Microenvironment Associated with Clinical Outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Finkernagel, F.; Reinartz, S.; Konzer, A.; Adhikary, T.; Nist, A.; Stiewe, T.; Wagner, U.; Looso, M.; Graumann, J.; et al. Proteotranscriptomics Reveal Signaling Networks in the Ovarian Cancer Microenvironment. Mol. Cell Proteom. 2018, 17, 270–289. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Chakraborty, S.; Mazumdar, M.; Ghosh, S.; Mukherjee, S.; Manna, A.; Mohanty, S.; Nakka, K.K.; Joshi, S.; De, A.; et al. Inhibition of Epithelial to Mesenchymal Transition by E-Cadherin Up-Regulation Via Repression of Slug Transcription and Inhibition of E-Cadherin Degradation: Dual Role of Scaffold/Matrix Attachment Region-Binding Protein 1 (SMAR1) in Breast Cancer Cells. J. Biol. Chem. 2014, 289, 25431–25444. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial Ovarian Cancer-Secreted Exosomal miR-222-3p Induces Polarization of Tumor-Associated Macrophages. Oncotarget 2016, 7, 43076–43087. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes Derived from Hypoxic Epithelial Ovarian Cancer Deliver microRNA-940 to Induce Macrophage M2 Polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wu, X.; Ying, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wu, Y.; Wang, X. Suppression of Endothelial Cell Migration by Tumor Associated Macrophage-Derived Exosomes is Reversed by Epithelial Ovarian Cancer Exosomal lncRNA. Cancer Cell Int. 2017, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, D.; Wu, A.; Qiu, X.; Di, W.; Huang, L.; Qiu, L. TWEAK-Stimulated Macrophages Inhibit Metastasis of Epithelial Ovarian Cancer Via Exosomal Shuttling of microRNA. Cancer Lett. 2017, 393, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, C.; Yang, C.; Tsai, I.; Hou, Y.; Chen, C.; Shan, Y. Tumor-Associated Macrophages Promote Epigenetic Silencing of Gelsolin through DNA Methyltransferase 1 in Gastric Cancer Cells. Cancer Immunol. Res. 2017, 5, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Jiang, C.; Liu, X.; Xu, S.; Zhang, Z.; Chen, H.; Zhang, Y.; Liu, Y.; Ma, D. Hypermethylation of IFN-Γ in Oral Cancer Tissues. Clin. Oral Investig. 2017, 21, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xiao, X.; Huang, T.; Du, C.; Wang, S.; Mo, Y.; Ma, N.; Murata, M.; Li, B.; Wen, W.; et al. Epigenetic Inactivation of Follistatin-Like 1 Mediates Tumor Immune Evasion in Nasopharyngeal Carcinoma. Oncotarget 2016, 7, 16433–16444. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Tsuchida, R.; Muramatsu, M.; Shimamura, T.; Wang, F.; Suehiro, J.; Kanki, Y.; Wada, Y.; Yuasa, Y.; Aburatani, H.; et al. Inhibition of Histone Demethylase JMJD1A Improves Anti-Angiogenic Therapy and Reduces Tumor-Associated Macrophages. Cancer Res. 2013, 73, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F., 4th; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic Regulation of the Alternatively Activated Macrophage Phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.; Klaske, A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.J.; Daemen, T.; Nijman, H.W. Prognostic Significance of Tumor-Infiltrating T-Lymphocytes in Primary and Metastatic Lesions of Advanced Stage Ovarian Cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef] [PubMed]
- James, F.R.; Jiminez-Linan, M.; Alsop, J.; Mack, M.; Song, H.; Brenton, J.D.; Pharoah, P.D.; Ali, H.R. Association between Tumour Infiltrating Lymphocytes, Histotype and Clinical Outcome in Epithelial Ovarian Cancer. BMC Cancer 2017, 17, 657. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.C.; Bean, S.M.; Whitaker, R.S.; Kondoh, E.; Baba, T.; Fujii, S.; Marks, J.R.; Dressman, H.K.; Murphy, S.K.; Berchuck, A. Ovarian Cancer Tumor Infiltrating T-Regulatory (T(Reg)) Cells are Associated with a Metastatic Phenotype. Gynecol. Oncol. 2010, 116, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Tsiatas, M.L.; Gyftaki, R.; Liacos, C.; Politi, E.; Rodolakis, A.; Dimopoulos, M.A.; Bamias, A. Study of T Lymphocytes Infiltrating Peritoneal Metastases in Advanced Ovarian Cancer: Associations with Vascular Endothelial Growth Factor Levels and Prognosis in Patients Receiving Platinum-Based Chemotherapy. Int. J. Gynecol. Cancer 2009, 19, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Napoletano, C.; Bellati, F.; Landi, R.; Pauselli, S.; Marchetti, C.; Visconti, V.; Sale, P.; Liberati, M.; Rughetti, A.; Frati, L. Ovarian Cancer Cytoreduction Induces Changes in T Cell Population Subsets Reducing Immunosuppression. J. Cell. Mol. Med. 2010, 14, 2748–2759. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor Infiltrating Lymphocytes in Ovarian Cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Sehouli, J.; Loddenkemper, C.; Cornu, T.; Schwachula, T.; Hoffmüller, U.; Grützkau, A.; Lohneis, P.; Dickhaus, T.; Gröne, J.; Kruschewski, M. Epigenetic Quantification of Tumor-Infiltrating T-Lymphocytes. Epigenetics 2011, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune Regulation by Low Doses of the DNA Methyltransferase Inhibitor 5-Azacitidine in Common Human Epithelial Cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer. Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.L.; et al. Epigenetic Therapy Activates Type I Interferon Signaling in Murine Ovarian Cancer to Reduce Immunosuppression and Tumor Burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [PubMed]
- Adair, S.J.; Hogan, K.T. Treatment of Ovarian Cancer Cell Lines with 5-Aza-2′-Deoxycytidine Upregulates the Expression of Cancer-Testis Antigens and Class I Major Histocompatibility Complex-Encoded Molecules. Cancer Immunol. Immunother. 2009, 58, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A. Inhibiting DNA Methylation Causes an Interferon Response in Cancer Via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Parameswaran, J.; Shapira, I.; Lovecchio, J.; Menzin, A.; Whyte, J.; Dos Santos, L.; Liang, S.; Bhuiya, T.; Keogh, M.; et al. Two microRNA Signatures for Malignancy and Immune Infiltration Predict overall Survival in Advanced Epithelial Ovarian Cancer. J. Investig. Med. 2017, 65, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- McKenna, K.; Beignon, A.S.; Bhardwaj, N. Plasmacytoid Dendritic Cells: Linking Innate and Adaptive Immunity. J. Virol. 2005, 79, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Soncini, M.; Melocchi, L.; Sozzani, S.; Facchetti, F. Plasmacytoid Dendritic Cells and Cancer. J. Leukoc. Biol. 2011, 90, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 1268. [Google Scholar] [CrossRef] [PubMed]
- Wertel, I.; Polak, G.; Bednarek, W.; Barczyński, B.; Roliński, J.; Kotarski, J. Dendritic Cell Subsets in the Peritoneal Fluid and Peripheral Blood of Women Suffering from Ovarian Cancer. Cytom. Part B Clin. Cytom. 2008, 74, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic Cell Subsets Differentially Regulate Angiogenesis in Human Ovarian Cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef] [PubMed]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L. Tumor-Infiltrating Dendritic Cell Precursors Recruited by a B-Defensin Contribute to Vasculogenesis Under the Influence of Vegf-A. Nat. Med. 2004, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid Dendritic Cells Induce CD8+ Regulatory T Cells in Human Ovarian Carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Treilleux, I.; Goddard-Leon, S.; Combes, J.; Blay, J.; Ray-Coquard, I.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid Dendritic Cells Infiltrating Ovarian Cancer are Associated with Poor Prognosis. Oncoimmunology 2012, 1, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Huarte, E.; Cubillos-Ruiz, J.R.; Nesbeth, Y.C.; Scarlett, U.K.; Martinez, D.G.; Buckanovich, R.J.; Benencia, F.; Stan, R.V.; Keler, T.; Sarobe, P.; et al. Depletion of Dendritic Cells Delays Ovarian Cancer Progression by Boosting Antitumor Immunity. Cancer Res. 2008, 68, 7684–7691. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Meng, L.; Zhang, Y. Epigenetic Regulation of Dendritic Cell Development and Function. Cancer J. 2017, 23, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Amit, I. Plasticity in the Transcriptional and Epigenetic Circuits Regulating Dendritic Cell Lineage Specification and Function. Curr. Opin. Immunol. 2014, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.; Park, J.H.; Kim, Y. Transcriptional and Epigenetic Networks in the Development and Maturation of Dendritic Cells. Epigenomics 2013, 5, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Boardman, D.A.; Tung, S.L.; Lechler, R.; Lombardi, G. MicroRNAs Affect Dendritic Cell Function and Phenotype. Immunology 2015, 144, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Valadkhan, S.; Gunawardane, L.S. lncRNA-Mediated Regulation of the Interferon Response. Virus Res. 2016, 212, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Baird, J.R.; Tesone, A.J.; Rutkowski, M.R.; Scarlett, U.K.; Camposeco-Jacobs, A.L.; Anadon-Arnillas, J.; Harwood, N.M.; Korc, M.; Fiering, S.N.; et al. Reprogramming Tumor-Associated Dendritic Cells in Vivo using miRNA Mimetics Triggers Protective Immunity Against Ovarian Cancer. Cancer Res. 2012, 72, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; Kalinski, P. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Davidowitz, R.A.; Iwanicki, M.P.; Brugge, J.S. In Vitro Mesothelial Clearance Assay that Models the Early Steps of Ovarian Cancer Metastasis. J. Vis. Exp. 2012, 60, 3888. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Loughran, E.; Yang, J.; Lombard, R.; Alber, M.; Stack, M. Cadherin Composition and Multicellular Aggregate Invasion in Organotypic Models of Epithelial Ovarian Cancer Intraperitoneal Metastasis. Oncogene 2017, 19, 5840–5851. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.; Zhao, Z.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic Acid is Constitutively Produced by Human Peritoneal Mesothelial Cells and Enhances Adhesion, Migration, and Invasion of Ovarian Cancer Cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, S.; Amberger, A.; Pollheimer, J.; Gastl, G.; Offner, F.A.; Margreiter, R.; Zeimet, A.G. Ovarian Carcinoma Cells and IL-1beta-Activated Human Peritoneal Mesothelial Cells are Possible Sources of Vascular Endothelial Growth Factor in Inflammatory and Malignant Peritoneal Effusions. Gynecol. Oncol. 2005, 97, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Kassim, S.K.; El-Salahy, E.M.; Fayed, S.T.; Helal, S.A.; Helal, T.; Azzam, E.E.; Khalifa, A. Vascular Endothelial Growth Factor and Interleukin-8 are Associated with Poor Prognosis in Epithelial Ovarian Cancer Patients. Clin. Biochem. 2004, 37, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.M.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of Vascular Endothelial Growth Factor in Growth and Peritoneal Dissemination of Ovarian Cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, D.; Zhang, H.; Kirmani, K.; Zhao, Z.; Steinmetz, R.; Xu, Y. Lysophosphatidic Acid Stimulates Cell Migration, Invasion, and Colony Formation as Well as Tumorigenesis/Metastasis of Mouse Ovarian Cancer in Immunocompetent Mice. Mol. Cancer Ther. 2009, 8, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Zeimet, A.G.; Stadlmann, S.; Natoli, C.; Widschwendter, M.; Hermann, M.; Abendstein, B.; Daxenbichler, G.; Offner, F.A.; Iacobelli, S.; Marth, C. Peritoneal Mesothelial Cells as a Significant Source of Ascitic Immunostimulatory Protein 90K. Anticancer Res. 2000, 20, 4507–4511. [Google Scholar] [PubMed]
- Zeimet, A.G.; Natoli, C.; Herold, M.; Fuchs, D.; Windbichler, G.; Daxenbichler, G.; Iacobelli, S.; Dapunt, O.; Marth, C. Circulating Immunostimulatory Protein 90K and Soluble Interleukin-2-receptor in Human Ovarian Cancer. Int. J. Cancer 1996, 68, 34–38. [Google Scholar] [CrossRef]
- Iacobelli, S.; Sismondi, P.; Giai, M.; d’Egidio, M.; Tinari, N.; Amatetti, C.; Di Stefano, P.; Natoli, C. Prognostic Value of a Novel Circulating Serum 90K Antigen in Breast Cancer. Br. J. Cancer 1994, 69, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Fornarini, B.; D’Ambrosio, C.; Natoli, C.; Tinari, N.; Silingardi, V.; Iacobelli, S. Adhesion to 90K (Mac-2 BP) as a Mechanism for Lymphoma Drug Resistance in Vivo. Blood 2000, 96, 3282–3285. [Google Scholar] [PubMed]
- Matte, I.; Lane, D.; Bachvarov, D.; Rancourt, C.; Piché, A. Role of Malignant Ascites on Human Mesothelial Cells and their Gene Expression Profiles. BMC Cancer 2014, 14, 288. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Munck, J.; Tang, J.; Taverna, P.; Wang, Y.; Miller, D.F.; Pilrose, J.; Choy, G.; Azab, M.; Pawelczak, K.S.; et al. The Novel, Small-Molecule DNA Methylation Inhibitor SGI-110 as an Ovarian Cancer Chemosensitizer. Clin. Cancer Res. 2014, 20, 6504–6516. [Google Scholar]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Balch, C.; Schilder, J.; Breen, T.; Zhang, S.; Shen, C.; Li, L.; Kulesavage, C.; Snyder, A.J.; Nephew, K.P. A Phase 1 and Pharmacodynamic Study of Decitabine in Combination with Carboplatin in Patients with Recurrent, Platinum-resistant, Epithelial Ovarian Cancer. Cancer 2010, 116, 4043–4053. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic Targeting of Ovarian Cancer Stem Cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin. Cancer Res. 2018, 24, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A Phase II Study of Vorinostat in the Treatment of Persistent or Recurrent Epithelial Ovarian Or Primary Peritoneal Carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; LaRochelle, W.J.; Ara, G.; Wu, F.; Petersen, K.D.; Thougaard, A.; Sehested, M.; Lichenstein, H.S.; Jeffers, M. Activity of PXD101, a Histone Deacetylase Inhibitor, in Preclinical Ovarian Cancer Studies. Mol. Cancer. Ther. 2006, 5, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.T.; Balch, C.; Kulp, S.K.; Mand, M.R.; Nephew, K.P.; Chen, C.S. A Rationally Designed Histone Deacetylase Inhibitor with Distinct Antitumor Activity Against Ovarian Cancer. Neoplasia 2009, 11, 552–563, 3 p following 563. [Google Scholar] [CrossRef] [PubMed]
- Halsall, J.A.; Turan, N.; Wiersma, M.; Turner, B.M. Cells Adapt to the Epigenomic Disruption Caused by Histone Deacetylase Inhibitors through a Coordinated, Chromatin-Mediated Transcriptional Response. Epigenet. Chromatin 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Combination Therapy with Histone Deacetylase Inhibitors and Lithium Chloride: A Novel Treatment for Carcinoid Tumors. Ann. Surg. Oncol. 2009, 16, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Halsall, J.A.; Turner, B.M. Histone Deacetylase Inhibitors for Cancer Therapy: An Evolutionarily Ancient Resistance Response May Explain Their Limited Success. Bioessays 2016, 38, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Drak Alsibai, K.; Nicolas, A.; Bieche, I.; Morillon, A. Long Noncoding RNAs as New Architects in Cancer Epigenetics, Prognostic Biomarkers, and Potential Therapeutic Targets. BioMed Res. Int. 2015, 2015, 320214. [Google Scholar] [CrossRef] [PubMed]
- Labiche, A.; Heutte, N.; Herlin, P.; Chasle, J.; Gauduchon, P.; Elie, N. Stromal Compartment as a Survival Prognostic Factor in Advanced Ovarian Carcinoma. Int. J. Gynecol. Cancer 2010, 20, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J. Combined Inhibition of DNA Methylation and Histone Acetylation Enhances Gene Re-Expression and Drug Sensitivity in Vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Nolan, L.; Crabb, S.; Packham, G. Epigenetic Therapy: Histone Acetylation, DNA Methylation and Anti-Cancer Drug Discovery. Curr. Cancer Drug Targets 2009, 9, 963–981. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.; Leung, C.S.; Mok, S.C. CAF Reprogramming Inhibits Ovarian Cancer Progression. Cell Cycle 2014, 13, 3783–3784. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, S.; Zhang, K.; Xu, T. A Comprehensive Overview of Exosomes in Ovarian Cancer: Emerging Biomarkers and Therapeutic Strategies. J. Ovar. Res. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Mirandola, L.; Kast, W.M.; Jenkins, M.R.; Cobos, E.; Cannon, M.J. Understanding the Cross-Talk between Ovarian Tumors and Immune Cells: Mechanisms for Effective Immunotherapies. Int. Rev. Immunol. 2011, 30, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.; Teran-Cabanillas, E.; Cubillos-Ruiz, J.R. Dendritic Cell Rehab: New Strategies to Unleash Therapeutic Immunity in Ovarian Cancer. Cancer Immunol. Immunother. 2017, 66, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Drakes, M.L.; Stiff, P.J. Understanding Dendritic Cell Immunotherapy in Ovarian Cancer. Expert Rev. Anticancer Ther. 2016, 16, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wefers, C.; Lambert, L.J.; Torensma, R.; Hato, S.V. Cellular Immunotherapy in Ovarian Cancer: Targeting the Stem of Recurrence. Gynecol. Oncol. 2015, 137, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K. Immunotherapy in Ovarian Cancer. Ann. Oncol. 2017, 28, viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Munster, P.N. Epigenetic Modifiers in Immunotherapy: A Focus on Checkpoint Inhibitors. Immunotherapy 2016, 8, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.; Strissel, P.L.; Baylin, S.B.; Chiappinelli, K.B. Unraveling the Molecular Pathways of DNA-Methylation Inhibitors: Human Endogenous Retroviruses Induce the Innate Immune Response in Tumors. Oncoimmunology 2016, 5, e1122160. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Ye, Q.; Santoro, S.; Fang, C.; Best, A.; Powell, D.J., Jr. Chimeric NKG2D CAR-Expressing T Cell-Mediated Attack of Human Ovarian Cancer is Enhanced by Histone Deacetylase Inhibition. Hum. Gene Ther. 2013, 24, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wargo, J.A.; Robbins, P.F.; Li, Y.; Zhao, Y.; El-Gamil, M.; Caragacianu, D.; Zheng, Z.; Hong, J.A.; Downey, S.; Schrump, D.S. Recognition of NY-ESO-1 Tumor Cells by Engineered Lymphocytes is Enhanced by Improved Vector Design and Epigenetic Modulation of Tumor Antigen Expression. Cancer Immunol. Immunother. 2009, 58, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y. Cancer/Testis Antigens: An Expanding Family of Targets for Cancer Immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic Potentiation of NY-ESO-1 Vaccine Therapy in Human Ovarian Cancer. Cancer. Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]




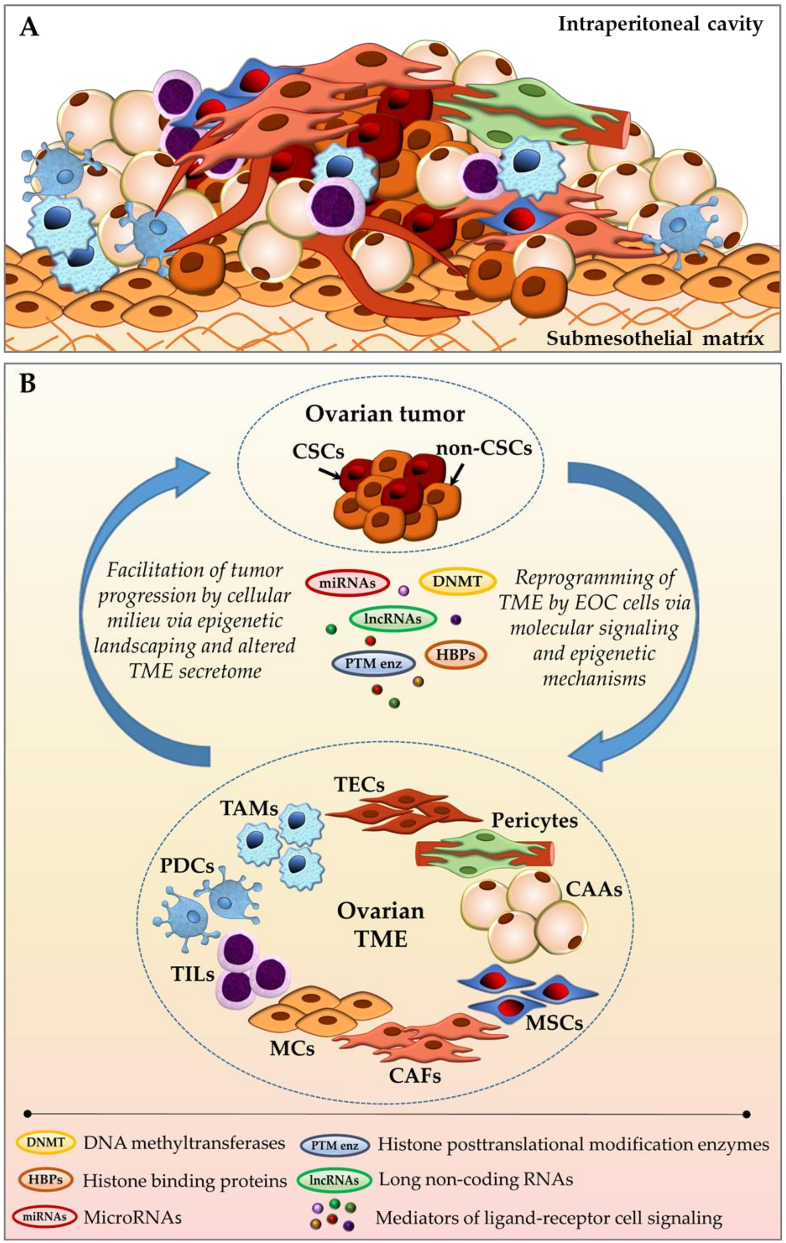{kind=link}
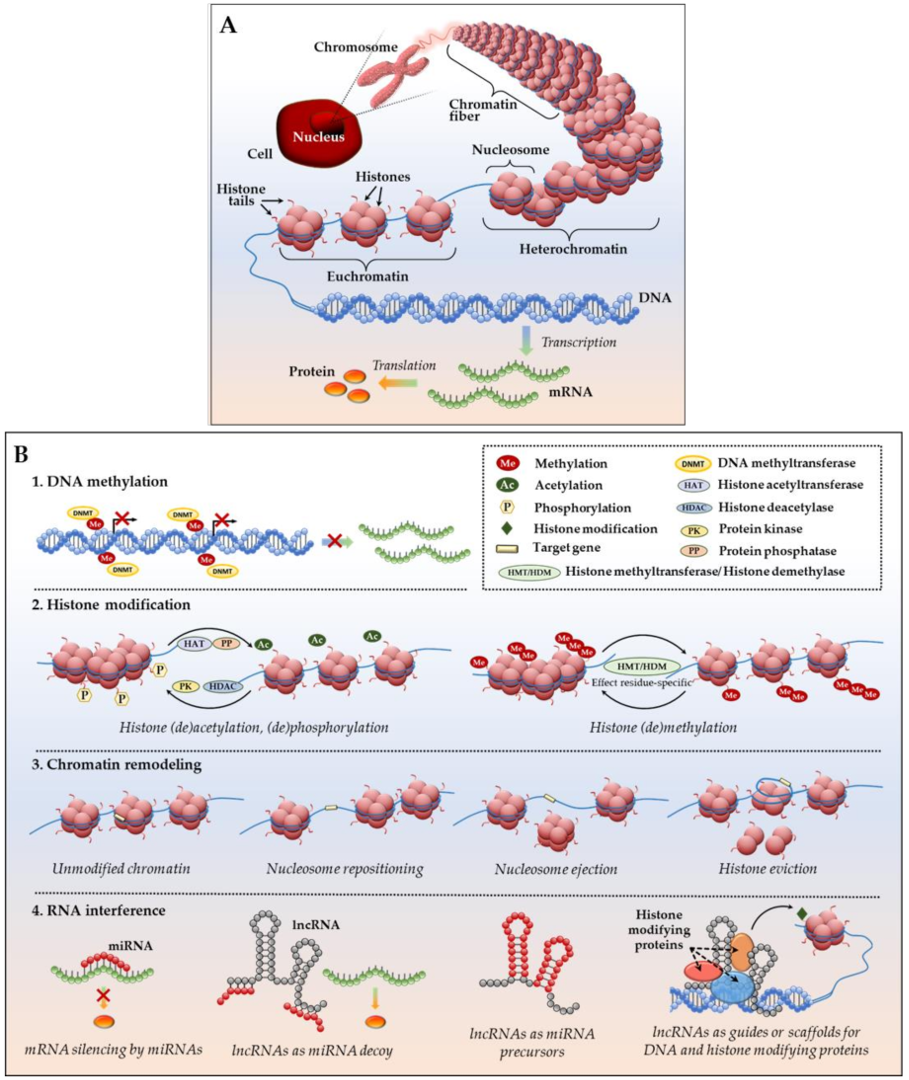{kind=link}
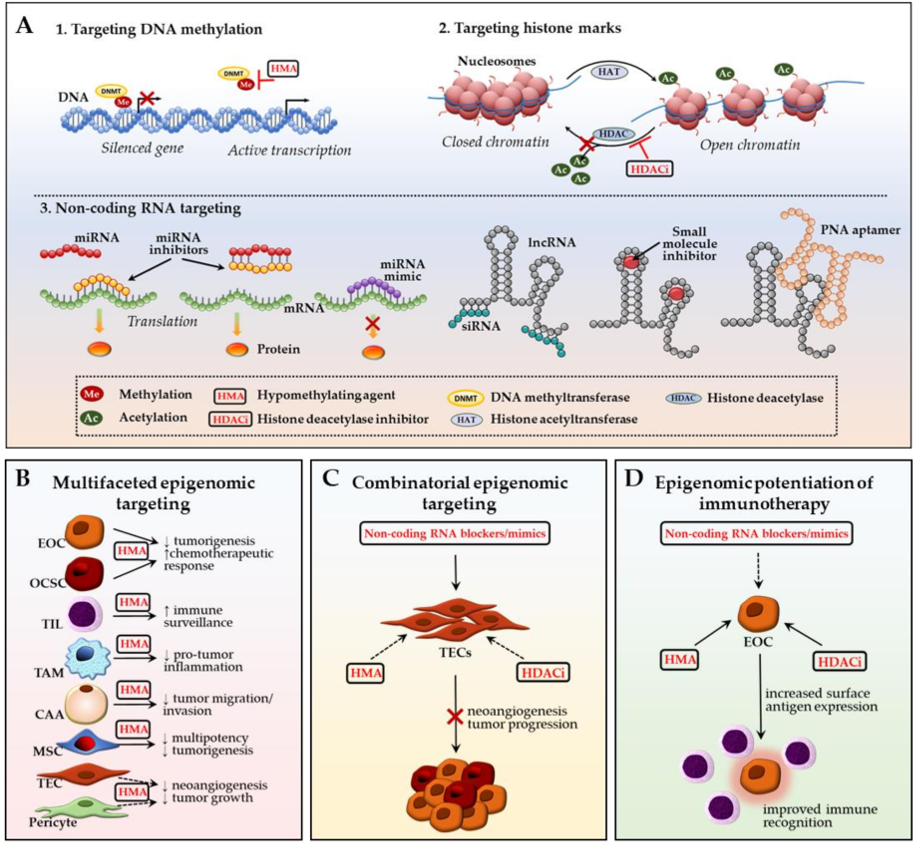{kind=link}
| Study Name | Phase | Status | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Decitabine, Vaccine Therapy, and Pegylated Liposomal Doxorubicin Hydrochloride in Treating Patients With Recurrent Ovarian Epithelial Cancer, Fallopian Tube Cancer, or Peritoneal Cancer | I | Completed Completion: June 2013 | NCT01673217 |
| Atezolizumab, Guadecitabine, and CDX-1401 Vaccine in Treating Patients With Recurrent Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | I/IIb | Ongoing Patient recruitment suspended Estimated completion: March 2020 | NCT03206047 |
| Guadecitabine and Pembrolizumab in Treating Patients With Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | II | Ongoing Recruiting patients Estimated completion: March 2020 | NCT02901899 |
| Genetically Modified T Cells and Decitabine in Treating Patients With Recurrent or Refractory Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | I | Ongoing Recruiting patients Estimated completion: August 2020 | NCT03017131 |
| Study of Azacitidine and Durvalumab in Advanced Solid Tumors (METADUR) | II | Ongoing Recruiting patients Estimated completion: January 2022 | NCT02811497 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klymenko, Y.; Nephew, K.P. Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers 2018, 10, 295. https://doi.org/10.3390/cancers10090295
Klymenko Y, Nephew KP. Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers. 2018; 10(9):295. https://doi.org/10.3390/cancers10090295
Chicago/Turabian StyleKlymenko, Yuliya, and Kenneth P. Nephew. 2018. "Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled" Cancers 10, no. 9: 295. https://doi.org/10.3390/cancers10090295
APA StyleKlymenko, Y., & Nephew, K. P. (2018). Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers, 10(9), 295. https://doi.org/10.3390/cancers10090295