The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Results
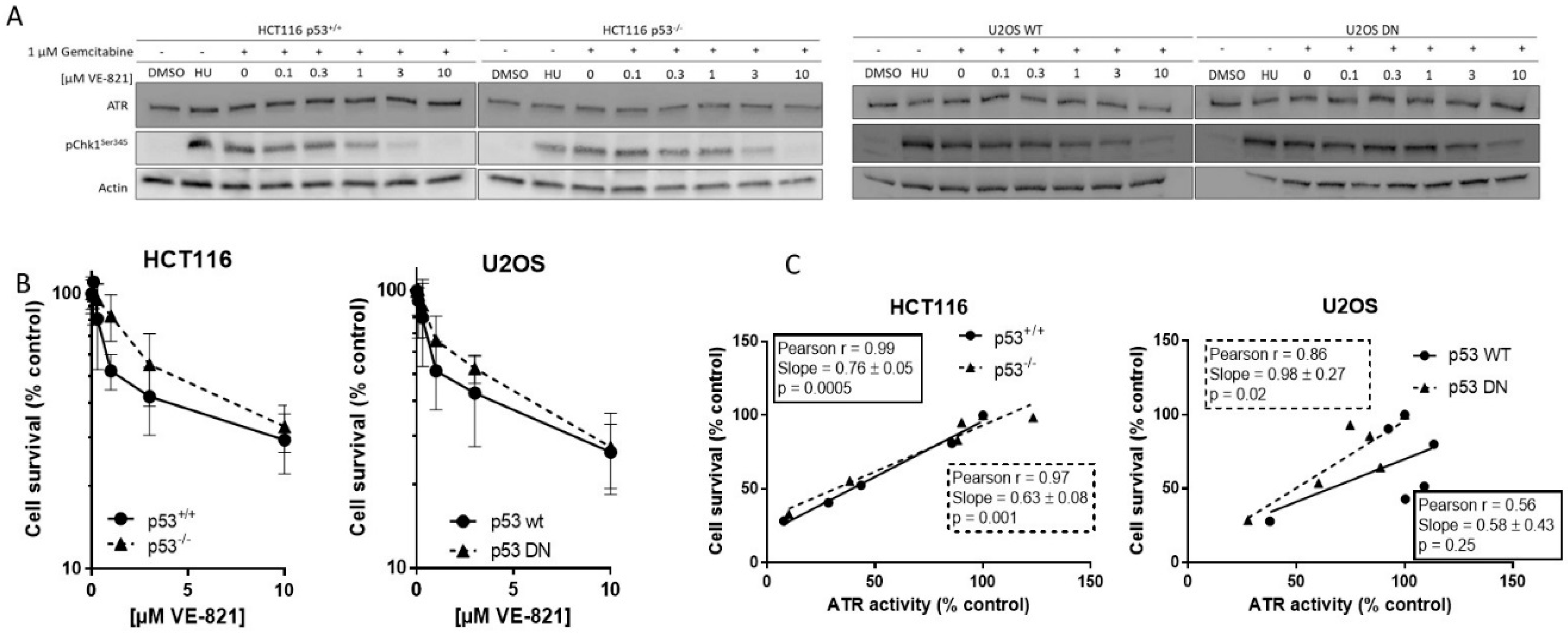
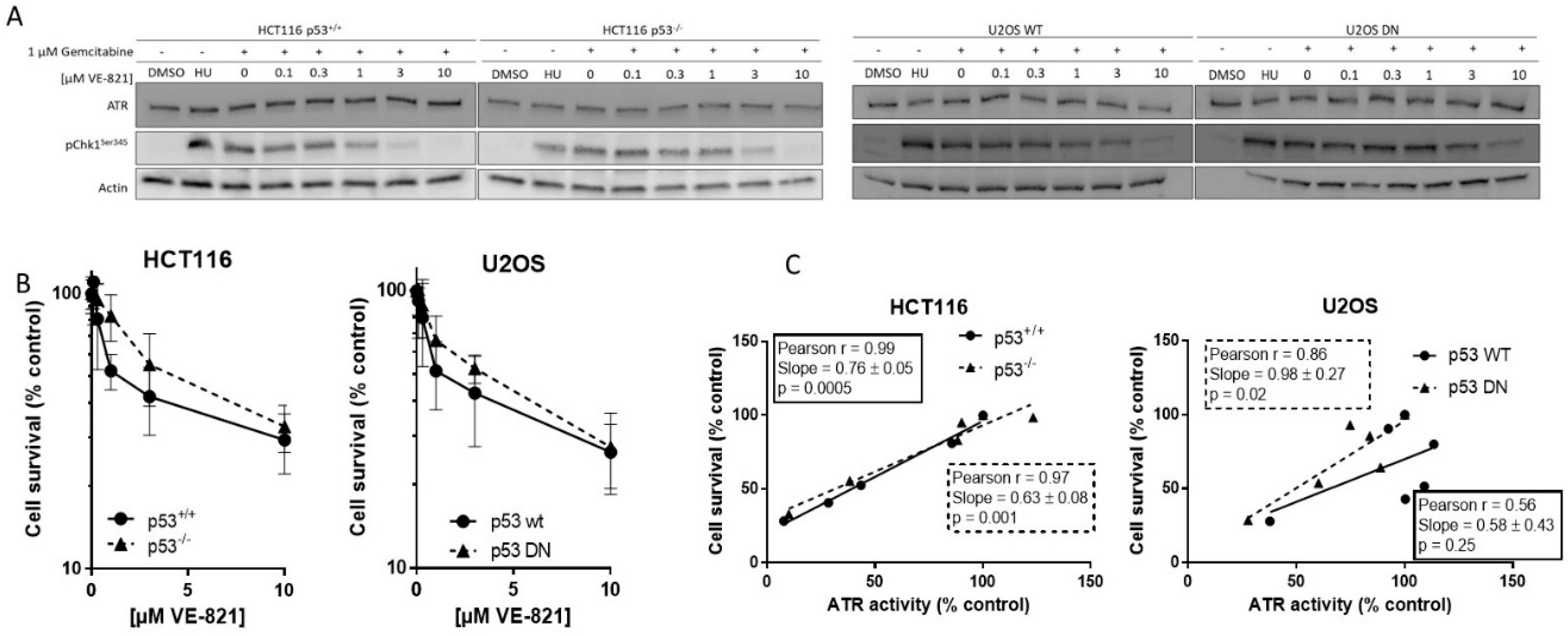
2.1. VE-821 Inhibits ATR Activity on All Cell Lines
2.2. Cytotoxicity of Single-Agent VE-821 Is Not Greater in p53 Mutant Cells and Cytotoxicity Is Directly Proportional to ATR Inhibition
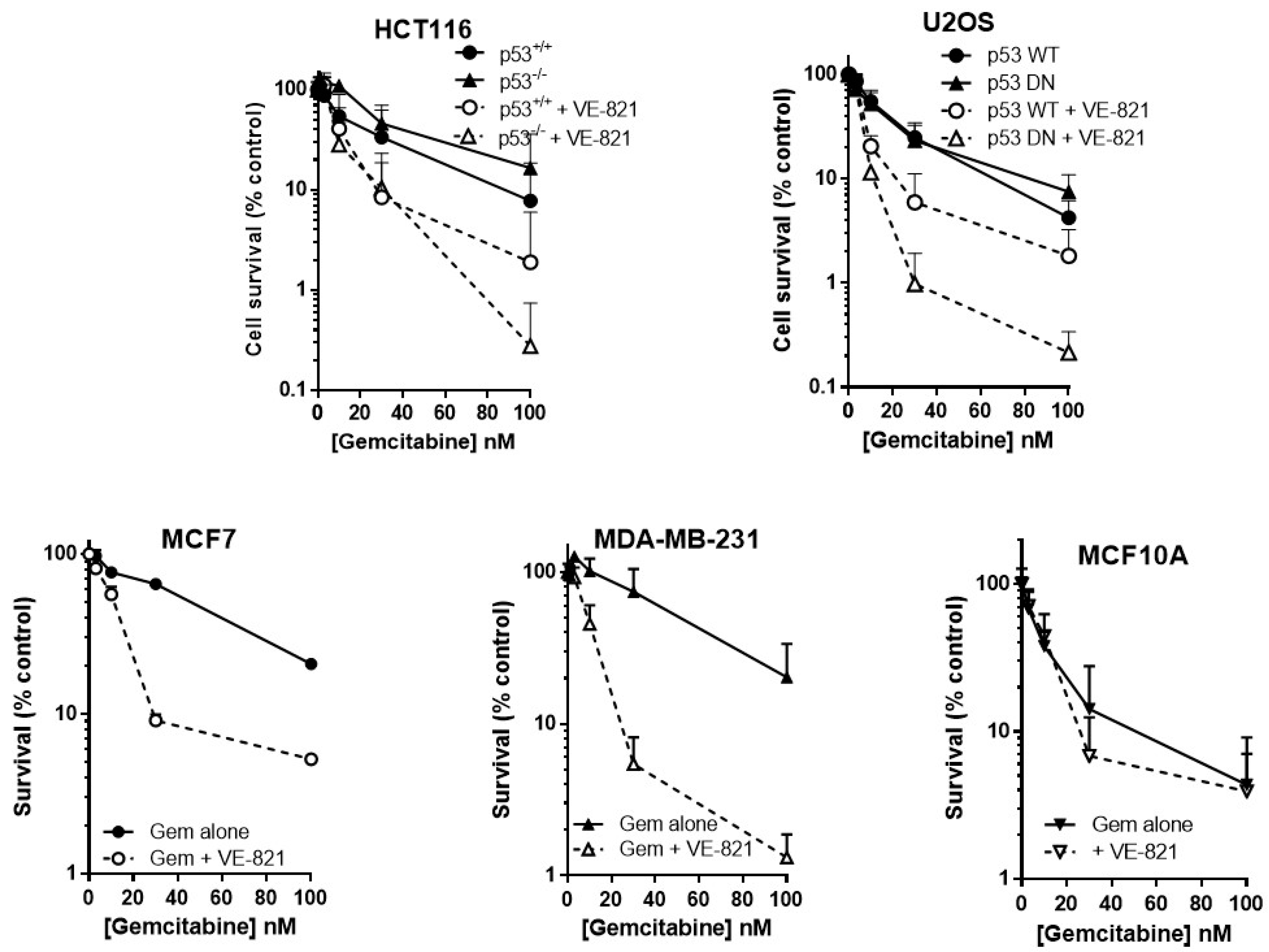
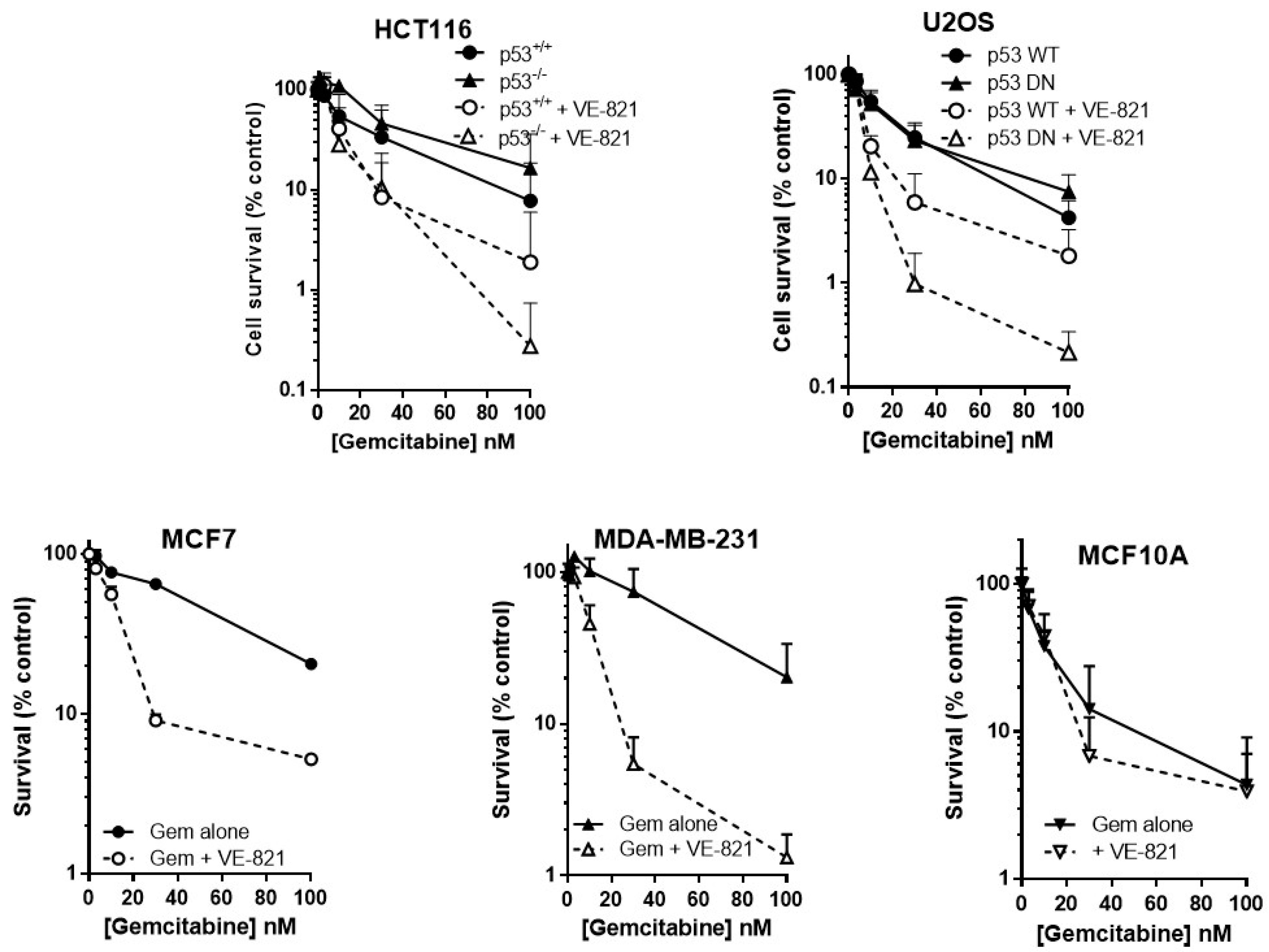
2.3. ATR Inhibition Potentiates Gemcitabine, in p53 Dysfunctional and wt Cells, but Not Nontumourigenic Immortalised Breast Cells
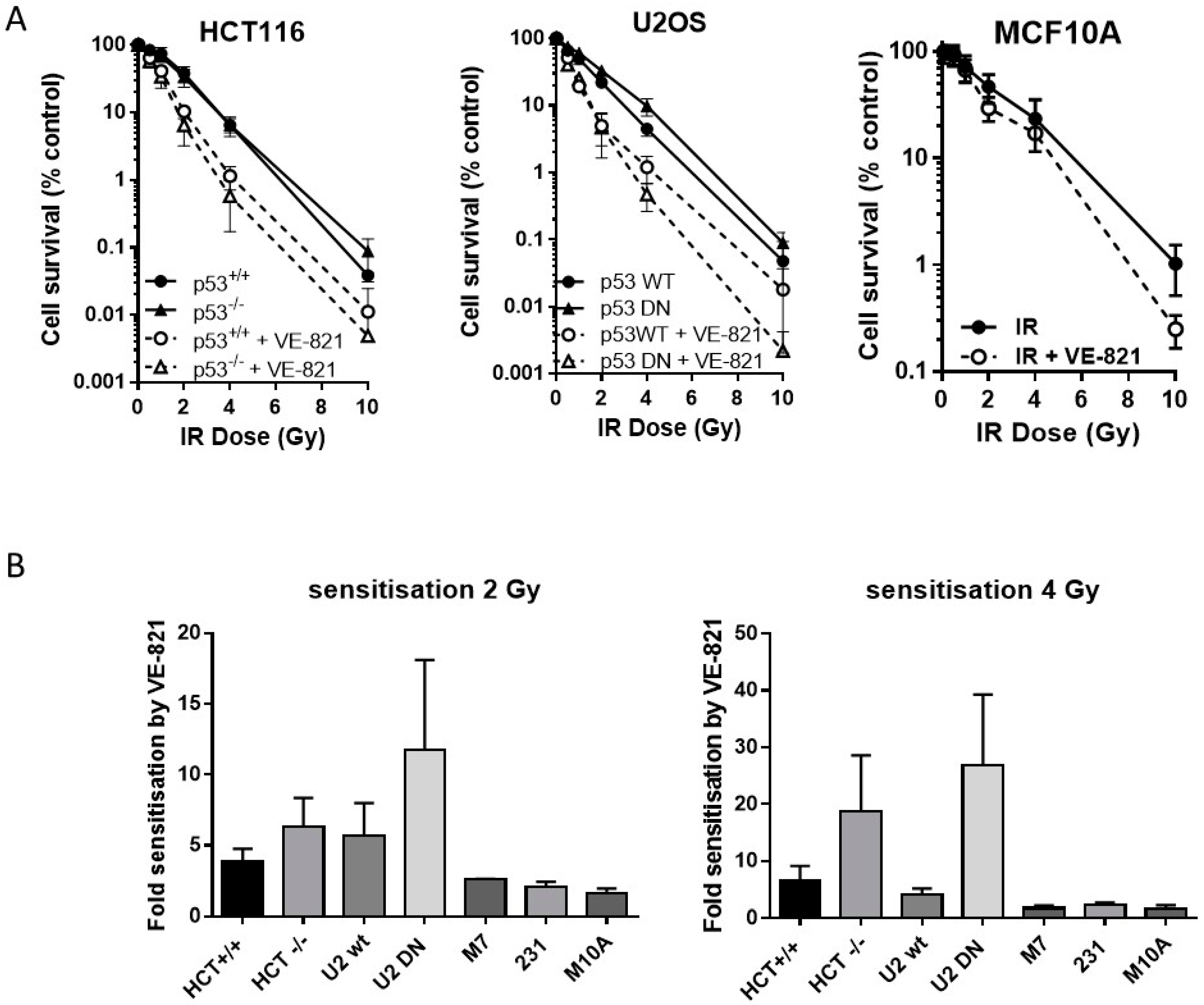
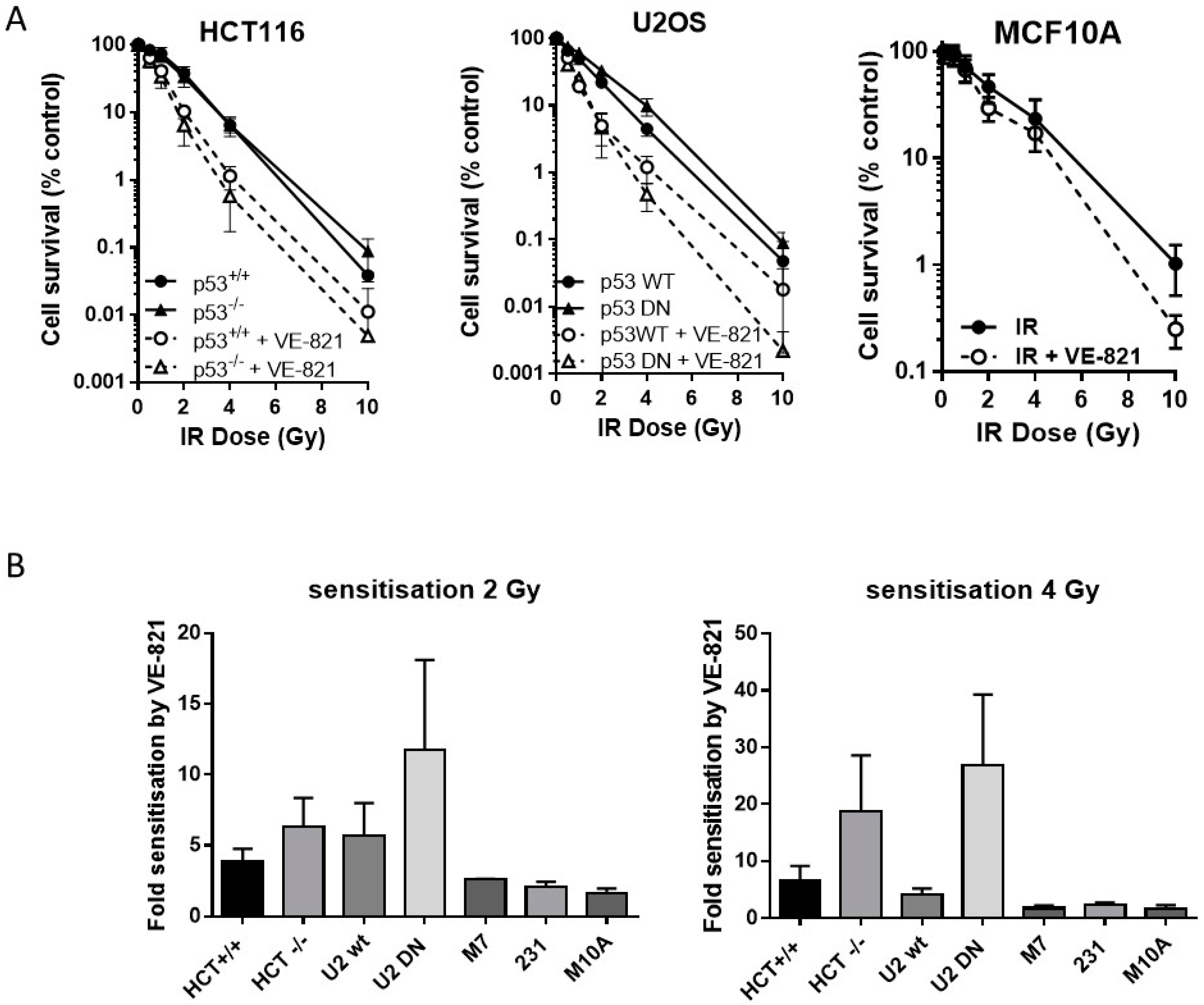
2.4. ATR Inhibition Increases Radiosensitivity, in p53 Dysfunctional and wt Cells, but Not Nontumourigenic Immortalised Breast Cells
3. Discussion
4. Methods
4.1. Chemicals and Reagents
4.2. Cell Lines
4.3. ATR Inhibition
4.4. Cytotoxicity
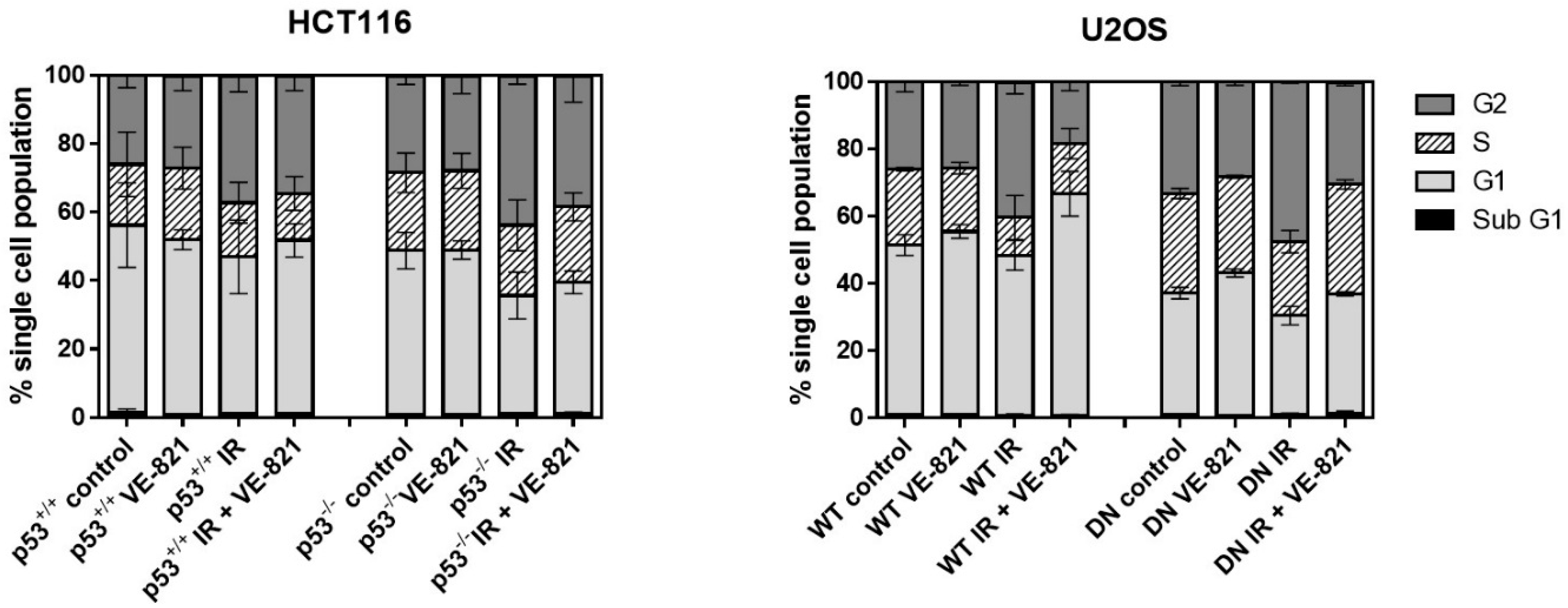
4.5. Cell Cycle Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation, from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Sangster-Guity, N.; Conrad, B.H.; Papadopoulos, N.; Bunz, F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011, 30, 2526–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses toDNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.; McCormick, A.; Edmondson, R.; May, F.E.B.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Middleton, F.K.; Falcon, S.; Reaper, P.M.; Pollard, J.R.; Curtin, N.J. Development of Pharmacodynamic Biomarkers for ATR inhibitors. Mol. Oncol. 2015, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.A.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-Chk1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Alsubhi, N.; Middleton, F.; Abdel-Fatah, T.M.A.; Stephens, P.; Doherty, R.; Arora, A.; Moseley, P.M.; Chan, S.Y.T.; Aleskandarany, M.A.; Green, A.R.; et al. CHK1 phosphorylated at serine 345 is a predictor of early local recurrence and radio-resistance in breast cancer. Mol. Oncol. 2016, 10, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- O’Carrigan, B.; de Miguel-Luken, M.J.; Papadatos-Pastos, D.; Brown, J.; Tunariu, N.; Lopez, R.P.; Ganegoda, M.; Riisnaes, R.; Figueiredo, I.; Carreira, S.; et al. Phase I trial of first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combinations (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J. Clin. Oncol. 2016, 34, 2504. [Google Scholar]
- Plummer, E.R.; Dean, E.; Evans, J.; Greystoke, A.; Herbschleb, K.; Ranson MBrown, J.; Zhang, Y.; Karan, S.; Pollard, J.; et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). J. Clin. Oncol. 2016, 34, 2513. [Google Scholar]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, H.E.; Patel, R.; McLaughlin, M.; Schick, U.; Zaidi, S.; Nutting, C.M.; Bhide, S.; Harrington, K.J. CHK1 Inhibition Radiosensitizes Head and Neck Cancers to Paclitaxel-Based Chemoradiotherapy. Mol. Cancer Ther. 2016, 15, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Haibe-Kains, B.; El-Hachem, N.; Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2013, 504, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, F.K. Exploiting ATR as a Selective Cancer Therapy. Master’s Thesis, Newcastle University, Newcastle upon Tyne, UK, 2011. [Google Scholar]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Gazin, C.; Park, S.M.; Zhu, L.J.; Debily, M.A.; Kittler, E.L.; Zapp, M.L.; Lapointe, D.; Gobeil, S.; Virbasius, C.M.; et al. A synthetic interaction screen identifies factors selectively required for proliferation and TERT transcription in p53-deficient human cancer cells. PLoS Genet. 2012, 8, e1003151. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Levesque, A.A.; Fanous, A.A.; Poh, A.; Eastman, A. Defective p53 signaling in p53 wild-type tumors attenuates p21waf1 induction and cyclin B repression rendering them sensitive to Chk1 inhibitors that abrogate DNA damage-induced S and G2 arrest. Mol. Cancer Ther. 2008, 7, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Li, Q.; Zhou, J.; Liu, Z.J.; Su, N.; Wilson, J.; Lu, Z.M.; Deng, D. Homeostatic maintenance of allele-specific p16 methylation in cancer cells accompanied by dynamic focal methylation and hydroxymethylation. PLoS ONE 2014, 9, e97785. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Park, M.J.; Kimura, K.; Shimizu, K.; Lee, S.H.; Yokota, J. Alterations in the INK4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet Cytogenet. 2003, 141, 5–13. [Google Scholar] [CrossRef]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Leteur, C.; Yang, C.; Zhang, P.; Castedo, M.; Pierré AGolsteyn, R.M.; Bourhis, J.; Kroemer, G.; Deutsch, E. Radiosensitization by Chir-124, a selective Chk1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle 2009, 8, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nghiem, P.; Park, P.K.; Kim, Y.S.; Desai, B.N.; Schreiber, S.L. ATR is not required for p53 activation but synergizes with p53 in the replication checkpoint. J. Biol. Chem. 2002, 8, 4428–4434. [Google Scholar] [CrossRef] [PubMed]
- Shaitelman, S.F.; Schlembach, P.J.; Arzu, I.; Ballo, M.; Bloom, E.S.; Buchholz, D.; Chronowski, G.M.; Dvorak, T.; Grade, E.; Hoffman, K.E.; et al. Acute and Short-term Toxic Effects of Conventionally Fractionated vs Hypofractionated Whole-Breast Irradiation: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Jagsi, R.; Griffith, K.A.; Boike, T.P.; Walker, E.; Nurushev, T.; Grills, I.S.; Moran, J.M.; Feng, M.; Hayman, J.; Pierce, L.J. Differences in the Acute Toxic Effects of Breast Radiotherapy by Fractionation Schedule: Comparative Analysis of Physician-Assessed and Patient-Reported Outcomes in a Large Multicenter Cohort. JAMA Oncol. 2015, 1, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Teckie, S.; Lok, B.H.; Rao, S.; Gutiontov, S.I.; Yamada, Y.; Berry, S.L.; Zelefsky, M.J.; Lee, N.Y. High-dose hypofractionated radiotherapy is effective and safe for tumors in the head-and-neck. Oral Oncol. 2016, 60, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, H.; Nakamura, S.; Suzuki, G.; Yoshida, K.; Yoshioka, Y.; Koizumi, M.; Ogawa, K. Hypofractionated Radiotherapy for Localized Prostate Cancer: A Challenging Accelerated Hypofractionated Radiotherapy. Anticancer Res. 2015, 35, 5167–5177. [Google Scholar] [PubMed]
- Plunkett, W.; Huang, P.; Searcy, C.E.; Gandhi, V. Gemcitabine: Preclinical Pharmacology and Mechanisms of Action. Semin. Oncol. 1996, 23, 3–15. [Google Scholar] [PubMed]
- Karnitz, L.M.; Flatten, K.S.; Wagner, J.M.; Loegering, D.; Hackbarth, J.S.; Arlander, S.J.; Vroman, B.T.; Thomas, M.B.; Baek, Y.U.; Hopkins, K.M.; et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol. Pharmacol. 2005, 68, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Zenvirt, S.; Kravchenko-Balasha, N.; Levitzki, A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene 2010, 29, 6149–6159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Trasoff, P.; Satterlee, W.; Raber, M.N.; Plunkett, W. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, R.; Kantarjian, H.; Du, M.; Fuacher, K.; Tarasoff, P.; Plunkett, W. Gemcitabine in leukemia: A phase I clinical, plasma, and cellular pharmacology study. J. Clin. Oncol. 1992, 10, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; McKenna, G.W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Price, A.; Yellowlees, A.; Keerie, C.; Russell, S.; Faivre-Finn, C.; Gilligan, D.; Snee, M.; Skailes, G.; Hatton, M.; Erridge, S.; et al. Radical radiotherapy with or without gemcitabine in patients with early stage medically inoperable non-small cell lung cancer. Lung Cancer 2012, 77, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Hudson, E.; Hurt, C.; Mort, D.; Brewster, A.E.; Iqbal, N.; Joseph, G.; Crosby, T.D.; Mukherjee, S.L. Induction chemotherapy followed by chemoradiation in locally advanced pancreatic cancer: An effective and well-tolerated treatment. Clin. Oncol. R. Coll. Radiol. 2010, 22, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Fried, M. P53-dependent apoptosis or growth arrest induced by different forms of radiation in U2OS cells: P21WAF1/CIP1 repression in UV induced apoptosis. Oncogene 1999, 18, 5403–5412. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middleton, F.K.; Pollard, J.R.; Curtin, N.J. The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers 2018, 10, 275. https://doi.org/10.3390/cancers10080275
Middleton FK, Pollard JR, Curtin NJ. The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers. 2018; 10(8):275. https://doi.org/10.3390/cancers10080275
Chicago/Turabian StyleMiddleton, Fiona K., John R. Pollard, and Nicola J. Curtin. 2018. "The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation" Cancers 10, no. 8: 275. https://doi.org/10.3390/cancers10080275
APA StyleMiddleton, F. K., Pollard, J. R., & Curtin, N. J. (2018). The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers, 10(8), 275. https://doi.org/10.3390/cancers10080275