Targeting the Microenvironment in High Grade Serous Ovarian Cancer



Abstract
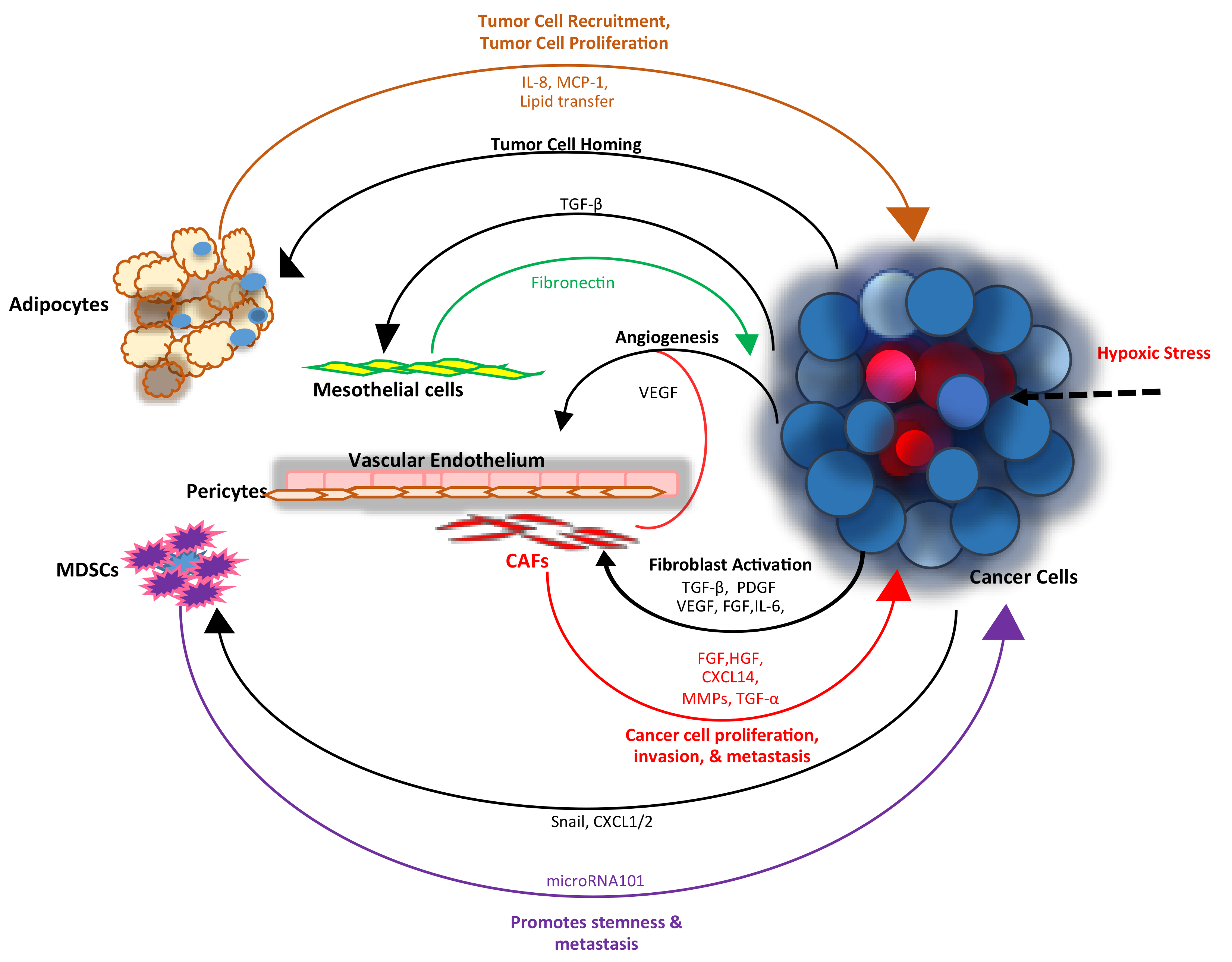
1. Introduction
2. Fibroblasts
3. Therapies Targeting Fibroblasts
4. Angiogenesis
5. Anti-Angiogenic Therapy (AAT)
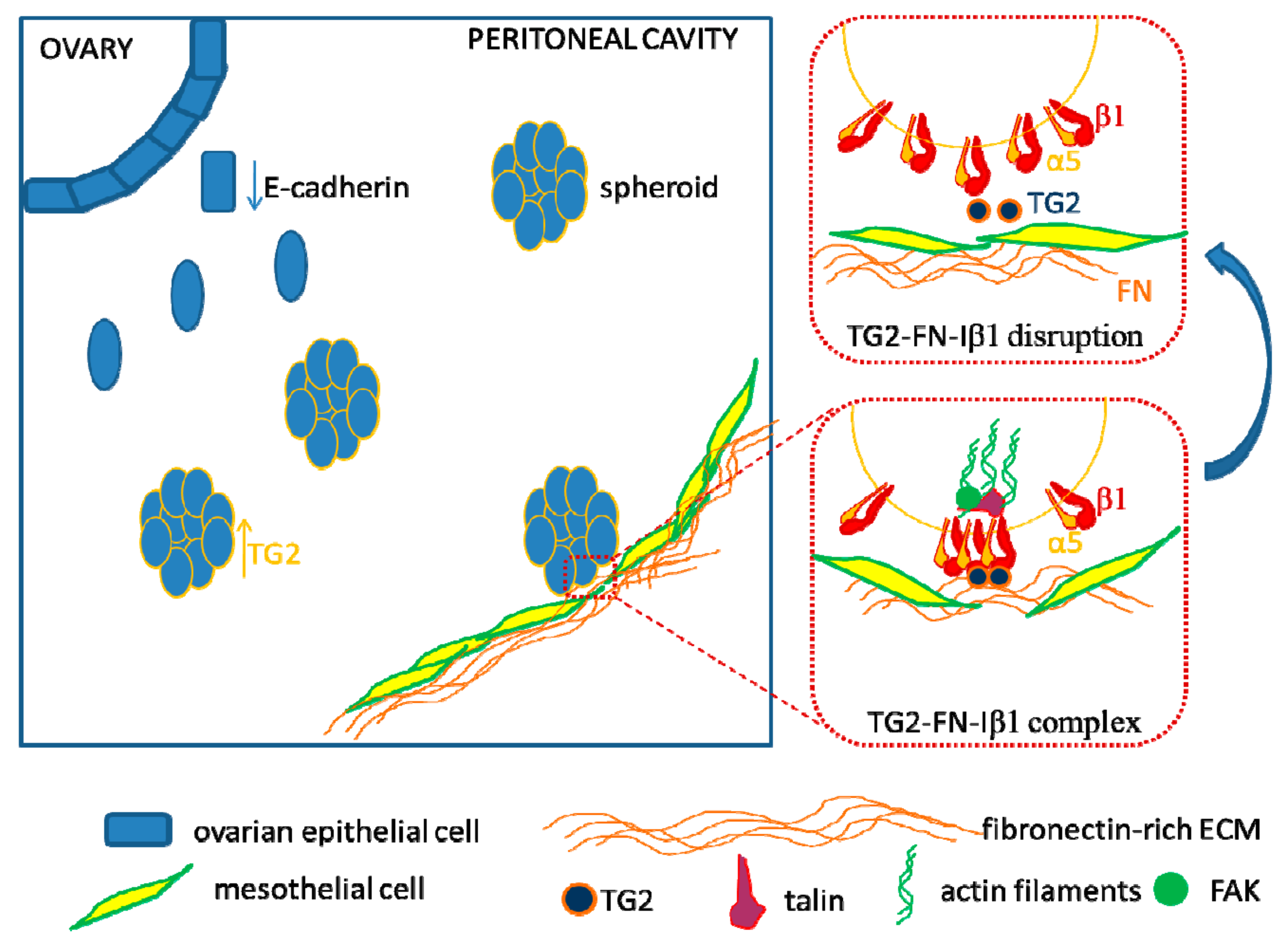
6. Interactions with the Mesothelial Matrix
7. Targeting Ovarian Cancer Cell Adhesion to the Peritoneal Matrix
8. Tumor Immune Response in Ovarian Cancer
9. Immune Checkpoint Inhibitors
10. Targeting Tumor Associated Macrophages (TAMs) and Myeloid-Derived Suppressor Cells (MDSCs)
11. Conclusions
Funding
Conflicts of Interest
References
- Freedman, R.S.; Deavers, M.; Liu, J.; Wang, E. Peritoneal inflammation—A microenvironment for epithelial ovarian cancer (eoc). J. Transl. Med. 2004, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Socha, M.J.; Olearczyk, J.J.; Elmarakby, A.A.; Imig, J.D.; Motamed, K. Normalization of the ovarian cancer microenvironment by sparc. Mol. Cancer Res. 2007, 5, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Hurteau, J.; Rodriguez, G.C.; Whitaker, R.S.; Shah, S.; Mills, G.; Bast, R.C.; Berchuck, A. Transforming growth factor-beta inhibits proliferation of human ovarian cancer cells obtained from ascites. Cancer 1994, 74, 93–99. [Google Scholar] [CrossRef]
- Gopinathan, G.; Milagre, C.; Pearce, O.M.; Reynolds, L.E.; Hodivala-Dilke, K.; Leinster, D.A.; Zhong, H.; Hollingsworth, R.E.; Thompson, R.; Whiteford, J.R.; et al. Interleukin-6 stimulates defective angiogenesis. Cancer Res. 2015, 75, 3098–3107. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D.; Croft, C.B. Ultrastructural features of wound healing in mouse skin. J. Anat. 1969, 105, 189–190. [Google Scholar] [PubMed]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [PubMed]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.B.; Cliff, W.J.; Gabbiani, G.; Irle, C.; Montandon, D.; Statkov, P.R.; Majno, G. Myofibroblasts in human granulation tissue. Hum. Pathol. 1974, 5, 55–67. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. Tgf-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4t1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Francesco, M.D.; Ascenzo, S.; Palmerini, M.G.; Macchiarelli, G.; Carta, G.; Dolo, V. Ovarian cancer-derived extracellular vesicles affect normal human fibroblast behavior. Cancer Biol. Ther. 2018, 19, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Qu, X.; Yang, Q.; Wei, M.; Kong, B. Clic4 mediates tgf-beta1-induced fibroblast-to-myofibroblast transdifferentiation in ovarian cancer. Oncol. Rep. 2009, 22, 541–548. [Google Scholar] [PubMed]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal mir-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Jeon, E.S.; Moon, H.J.; Lee, M.J.; Song, H.Y.; Kim, Y.M.; Cho, M.; Suh, D.S.; Yoon, M.S.; Chang, C.L.; Jung, J.S.; et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells 2008, 26, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Barengo, N.; Ladanyi, A.; Lee, J.S.; Marini, F.; Lengyel, E.; Naora, H. Hoxa9 promotes ovarian cancer growth by stimulating cancer-associated fibroblasts. J. Clin. Investig. 2012, 122, 3603–3617. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.F.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Cornil, I.; Theodorescu, D.; Man, S.; Herlyn, M.; Jambrosic, J.; Kerbel, R.S. Fibroblast cell-interactions with human-melanoma cells affect tumor-cell growth as a function of tumor progression. Proc. Natl. Acad. Sci. USA 1991, 88, 6028–6032. [Google Scholar] [CrossRef] [PubMed]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. Mir200-regulated cxcl12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Geng, F.; Liang, S.; Zhao, H.; Liu, M.; Wang, H. Caf-derived hgf promotes cell proliferation and drug resistance by up-regulating the c-met/pi3k/akt and grp78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37, BSR20160470. [Google Scholar]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Henriksson, M.L.; Edin, S.; Dahlin, A.M.; Oldenborg, P.A.; Oberg, A.; Van Guelpen, B.; Rutegard, J.; Stenling, R.; Palmqvist, R. Colorectal cancer cells activate adjacent fibroblasts resulting in fgf1/fgfr3 signaling and increased invasion. Am. J. Pathol. 2011, 178, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. Cxcl8/il-8 and cxcl12/sdf-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.S.; Chan, L.K.; Wong, E.C.; Hui, C.W.; Sneddon, K.; Cheung, T.H.; Yim, S.F.; Lee, J.H.; Yeung, C.S.; Chung, T.K.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via tnfalpha-tgfalpha-egfr. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.I.; Engelbrecht, A.M. Metabolic hijacking: A survival strategy cancer cells exploit? Crit. Rev. Oncol. Hematol. 2017, 109, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Ji, G.L.; Le, X.B.; Wang, C.; Xu, L.; Feng, M.; Zhang, Y.G.; Yang, H.L.; Xuan, Y.; Yang, Y.F.; et al. Long noncoding rna linc00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 2017, 77, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumor Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Lederle, W.; Hartenstein, B.; Meides, A.; Kunzelmann, H.; Werb, Z.; Angel, P.; Mueller, M.M. Mmp13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis 2010, 31, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Garinchesa, P.; Old, L.J.; Rettig, W.J. Cell-surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef]
- Yang, W.; Han, W.; Ye, S.; Liu, D.; Wu, J.; Liu, H.; Li, C.; Chen, H. Fibroblast activation protein-alpha promotes ovarian cancer cell proliferation and invasion via extracellular and intracellular signaling mechanisms. Exp. Mol. Pathol. 2013, 95, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Mhawech-Fauceglia, P.; Yan, L.; Sharifian, M.; Ren, X.; Liu, S.; Kim, G.; Gayther, S.A.; Pejovic, T.; Lawrenson, K. Stromal expression of fibroblast activation protein alpha (fap) predicts platinum resistance and shorter recurrence in patients with epithelial ovarian cancer. Cancer Microenviron. 2015, 8, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012, 33, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ertl, H.C.J. Depletion of fap(+) cells reduces immunosuppressive cells and improves metabolism and functions cd8(+)t cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.D.; Valianou, M.; Canutescu, A.A.; Jaffe, E.K.; Lee, H.O.; Wang, H.; Lai, J.H.; Bachovchin, W.W.; Weiner, L.M. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol. Cancer Ther. 2005, 4, 351–360. [Google Scholar] [PubMed]
- Kelly, T. Fibroblast activation protein-alpha and dipeptidyl peptidase iv (cd26): Cell-surface proteases that activate cell signaling and are potential targets for cancer therapy. Drug Resist. Updat. 2005, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, C.T.; Ma, T.T.; Li, Z.Y.; Zhou, L.N.; Mu, B.; Leng, F.; Shi, H.S.; Li, Y.O.; Wei, Y.Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. Tgf-beta signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. Targeting tgf-beta signaling for therapeutic gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [PubMed]
- Abendstein, B.; Stadlmann, S.; Knabbe, C.; Buck, M.; Muller-Holzner, E.; Zeimet, A.G.; Marth, C.; Obrist, P.; Krugmann, J.; Offner, F.A. Regulation of transforming growth factor-beta secretion by human peritoneal mesothelial and ovarian carcinoma cells. Cytokine 2000, 12, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Matsumura, N.; Mandai, M.; Huang, Z.; Oura, T.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Kang, H.S.; Okamoto, T.; et al. The activated transforming growth factor-beta signaling pathway in peritoneal metastases is a potential therapeutic target in ovarian cancer. Int. J. Cancer 2012, 130, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Peille, A.L.; Vuaroqueaux, V.; Lahn, M. Anti-tumor activity of the tgf-beta receptor kinase inhibitor galunisertib (ly2157299 monohydrate) in patient-derived tumor xenografts. Cell Oncol. (Dordr.) 2015, 38, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Shan, N.; Zhao, C.; Wang, Y.; Xu, F.; Li, J.; Yu, X.; Gao, L.; Yi, Z. Ly2109761 enhances cisplatin antitumor activity in ovarian cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4923–4932. [Google Scholar] [PubMed]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernandez, M.S.; Romero, F.L.; Sepulveda-Sanchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac safety of tgf-beta receptor i kinase inhibitor ly2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.J.; Liu, T.B.; Xia, B.R.; Yang, C.Y.; Hou, S.Y.; Xie, W.L.; Lou, G. Platelet-derived growth factor d is a prognostic biomarker and is associated with platinum resistance in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2018, 28, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Chang, D.D.; Jeng, M.H. Imatinib mesylate (gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and akt inactivation. Clin. Cancer Res. 2004, 10, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine pdgf signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef] [PubMed]
- Haubeiss, S.; Schmid, J.O.; Murdter, T.E.; Sonnenberg, M.; Friedel, G.; van der Kuip, H.; Aulitzky, W.E. Dasatinib reverses cancer-associated fibroblasts (cafs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol. Cancer 2010, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Emerson, R.E.; Schilder, J.; Menning, N.; Baldridge, L.A.; Johnson, C.S.; Breen, T.; McClean, J.; Stephens, D.; Whalen, C.; et al. Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A hoosier oncology group trial. Cancer 2008, 113, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Sill, M.W.; Lankes, H.A.; DeGeest, K.; Bristow, R.E.; Mutch, D.; Yamada, S.D.; Cohn, D.; Calvert, V.; Farley, J.; et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: A gynecologic oncology group trial. J. Clin. Oncol. 2011, 29, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Zetter, B.R. Angiogenesis and tumor metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, H.C.; Kohn, E.C.; Steinberg, S.M.; Rothenberg, M.L.; Merino, M.J. Tumor angiogenesis in advanced stage ovarian carcinoma. Am. J. Pathol. 1995, 147, 33–41. [Google Scholar] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Ko, S.Y.; Naora, H. Therapeutic strategies for targeting the ovarian tumor stroma. World J. Clin. Cases 2014, 2, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, J.; Kuniyasu, H.; Crispens, M.A.; Price, J.E.; Bucana, C.D.; Fidler, I.J. Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice. J. Natl. Cancer Inst. 1998, 90, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.P.; Cai, A.; Wendt, K.; Young, M.; Gamero, A.; De Cesare, S. Angiogenic protein expression in advanced epithelial ovarian cancer. Clin. Cancer Res. 1997, 3, 1579–1586. [Google Scholar] [PubMed]
- Kraft, A.; Weindel, K.; Ochs, A.; Marth, C.; Zmija, J.; Schumacher, P.; Unger, C.; Marme, D.; Gastl, G. Vascular endothelial growth factor in the sera and effusions of patients with malignant and nonmalignant disease. Cancer 1999, 85, 178–187. [Google Scholar] [CrossRef]
- Mesiano, S.; Ferrara, N.; Jaffe, R.B. Role of vascular endothelial growth factor in ovarian cancer: Inhibition of ascites formation by immunoneutralization. Am. J. Pathol. 1998, 153, 1249–1256. [Google Scholar] [CrossRef]
- Shen, G.H.; Ghazizadeh, M.; Kawanami, O.; Shimizu, H.; Jin, E.; Araki, T.; Sugisaki, Y. Prognostic significance of vascular endothelial growth factor expression in human ovarian carcinoma. Br. J. Cancer 2000, 83, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.J.; Al-Attar, A.; Rolland, P.; Scott, I.V.; Deen, S.; Liu, D.T.; Spendlove, I.; Durrant, L.G. Vascular endothelial growth factor expression in ovarian cancer: A model for targeted use of novel therapies? Clin. Cancer Res. 2008, 14, 3030–3035. [Google Scholar] [CrossRef] [PubMed]
- Graybill, W.; Sood, A.K.; Monk, B.J.; Coleman, R.L. State of the science: Emerging therapeutic strategies for targeting angiogenesis in ovarian cancer. Gynecol. Oncol. 2015, 138, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Glajzer, J.; Kulbe, H.; Trillsch, F.; Mustea, A.; et al. Characterisation of tumour microvessel density during progression of high-grade serous ovarian cancer: Clinico-pathological impact (an octips consortium study). Br. J. Cancer 2018, 119, 330–338. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.; Zachary, I. The vascular endothelial growth factor (vegf) family: Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (vegf) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Kshirsagar, S.; Li, W.; Gui, L.; Ramakrishnan, S.; Gupta, P.; Law, P.Y.; Hebbel, R.P. Vegf prevents apoptosis of human microvascular endothelial cells via opposing effects on mapk/erk and sapk/jnk signaling. Exp. Cell Res. 1999, 247, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Cebe-Suarez, S.; Zehnder-Fjallman, A.; Ballmer-Hofer, K. The role of vegf receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006, 63, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Eskander, R.N.; Randall, L.M. Bevacizumab in the treatment of ovarian cancer. Biologics 2011, 5, 1–5. [Google Scholar] [PubMed]
- Mabuchi, S.; Terai, Y.; Morishige, K.; Tanabe-Kimura, A.; Sasaki, H.; Kanemura, M.; Tsunetoh, S.; Tanaka, Y.; Sakata, M.; Burger, R.A.; et al. Maintenance treatment with bevacizumab prolongs survival in an in vivo ovarian cancer model. Clin. Cancer Res. 2008, 14, 7781–7789. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (nrg oncology/gynecologic oncology group study gog-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef]
- Burger, R.A.; Sill, M.W.; Monk, B.J.; Greer, B.E.; Sorosky, J.I. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: A gynecologic oncology group study. J. Clin. Oncol. 2007, 25, 5165–5171. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The aurelia open-label randomized phase iii trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Moroney, J.W.; Sood, A.K.; Coleman, R.L. Aflibercept in epithelial ovarian carcinoma. Future Oncol. 2009, 5, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.S.; Jin, K.T.; He, K.F.; Zhang, J.; Wang, H.H.; Cao, J. Clinical applications of vegf-trap (aflibercept) in cancer treatment. J. Chin. Med. Assoc. 2010, 73, 449–456. [Google Scholar] [CrossRef]
- Byrne, A.T.; Ross, L.; Holash, J.; Nakanishi, M.; Hu, L.; Hofmann, J.I.; Yancopoulos, G.D.; Jaffe, R.B. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin. Cancer Res. 2003, 9, 5721–5728. [Google Scholar] [PubMed]
- Colombo, N.; Mangili, G.; Mammoliti, S.; Kalling, M.; Tholander, B.; Sternas, L.; Buzenet, G.; Chamberlain, D. A phase ii study of aflibercept in patients with advanced epithelial ovarian cancer and symptomatic malignant ascites. Gynecol. Oncol. 2012, 125, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Berlin, S.; Ivy, P.; Tyburski, K.; Krasner, C.; Zarwan, C.; Berkenblit, A.; Campos, S.; Horowitz, N.; Cannistra, S.A.; et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J. Clin. Oncol. 2009, 27, 5601–5606. [Google Scholar] [CrossRef] [PubMed]
- Hirte, H.; Lheureux, S.; Fleming, G.F.; Sugimoto, A.; Morgan, R.; Biagi, J.; Wang, L.; McGill, S.; Ivy, S.P.; Oza, A.M. A phase 2 study of cediranib in recurrent or persistent ovarian, peritoneal or fallopian tube cancer: A trial of the princess margaret, chicago and california phase ii consortia. Gynecol. Oncol. 2015, 138, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Hackshaw, A.; Kaye, S.; Jayson, G.; Gabra, H.; McNeish, I.; Earl, H.; Perren, T.; Gore, M.; Persic, M.; et al. Randomized phase ii placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor bibf 1120 after chemotherapy for relapsed ovarian cancer. J. Clin. Oncol. 2011, 29, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, A.; Kristensen, G.; Ray-Coquard, I.; Reuss, A.; Pignata, S.; Colombo, N.; Denison, U.; Vergote, I.; del Campo, J.M.; Ottevanger, P.; et al. Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (ago-ovar 12): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2016, 17, 78–89. [Google Scholar] [CrossRef]
- Pignata, S.; Lorusso, D.; Scambia, G.; Sambataro, D.; Tamberi, S.; Cinieri, S.; Mosconi, A.M.; Orditura, M.; Bartolini, S.; Arcangeli, V.; et al. Mito-11: A randomized multicenter phase ii trial testing the addition of pazopanib to weekly paclitaxel in platinum-resistant or -refractory advanced ovarian cancer (aoc). J. Clin. Oncol. 2014, 32. [Google Scholar]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of e-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Davidowitz, R.A.; Selfors, L.M.; Iwanicki, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef] [PubMed]
- Lessan, K.; Aguiar, D.J.; Oegema, T.; Siebenson, L.; Skubitz, A.P. Cd44 and beta1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am. J. Pathol. 1999, 154, 1525–1537. [Google Scholar] [CrossRef]
- Kenny, H.A.; Chiang, C.Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Janiak, A.; Zemskov, E.A.; Belkin, A.M. Cell surface transglutaminase promotes rhoa activation via integrin clustering and suppression of the src-p190rhogap signaling pathway. Mol. Biol. Cell 2006, 17, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, M.; Cao, L.; Pincheira, R.; Emerson, R.; Bigsby, R.; Nakshatri, H.; Matei, D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Cao, L.; Shen, C.; Satpathy, M.; Chelladurai, B.; Bigsby, R.M.; Nakshatri, H.; Matei, D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. [Google Scholar] [CrossRef] [PubMed]
- Condello, S.; Cao, L.; Matei, D. Tissue transglutaminase regulates beta-catenin signaling through a c-src-dependent mechanism. FASEB J. 2013, 27, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Condello, S.; Sima, L.; Ivan, C.; Cardenas, H.; Schiltz, G.; Mishra, R.K.; Matei, D. Tissue tranglutaminase regulates interactions between ovarian cancer stem cells and the tumor niche. Cancer Res. 2018, 78, 2990–3001. [Google Scholar] [CrossRef] [PubMed]
- Strobel, T.; Cannistra, S.A. Beta1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol. Oncol. 1999, 73, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A.; Kansas, G.S.; Niloff, J.; DeFranzo, B.; Kim, Y.; Ottensmeier, C. Binding of ovarian cancer cells to peritoneal mesothelium in vitro is partly mediated by cd44h. Cancer Res. 1993, 53, 3830–3838. [Google Scholar] [PubMed]
- Strobel, T.; Swanson, L.; Cannistra, S.A. In vivo inhibition of cd44 limits intra-abdominal spread of a human ovarian cancer xenograft in nude mice: A novel role for cd44 in the process of peritoneal implantation. Cancer Res. 1997, 57, 1228–1232. [Google Scholar] [PubMed]
- Slack-Davis, J.K.; Atkins, K.A.; Harrer, C.; Hershey, E.D.; Conaway, M. Vascular cell adhesion molecule-1 is a regulator of ovarian cancer peritoneal metastasis. Cancer Res. 2009, 69, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as therapeutic targets: Successes and cancers. Cancers (Basel) 2017, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase ii, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Trikha, M.; Zhou, Z.; Nemeth, J.A.; Chen, Q.; Sharp, C.; Emmell, E.; Giles-Komar, J.; Nakada, M.T. Cnto 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int. J. Cancer 2004, 110, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Manning, C.D.; Millar, H.; McCabe, F.L.; Ferrante, C.; Sharp, C.; Shahied-Arruda, L.; Doshi, P.; Nakada, M.T.; Anderson, G.M. Cnto 95, a fully human anti alphav integrin antibody, inhibits cell signaling, migration, invasion, and spontaneous metastasis of human breast cancer cells. Clin. Exp. Metastasis 2008, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Mullamitha, S.A.; Ton, N.C.; Parker, G.J.; Jackson, A.; Julyan, P.J.; Roberts, C.; Buonaccorsi, G.A.; Watson, Y.; Davies, K.; Cheung, S.; et al. Phase i evaluation of a fully human anti-alphav integrin monoclonal antibody (cnto 95) in patients with advanced solid tumors. Clin. Cancer Res. 2007, 13, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage iv metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Dolgos, H.; Freisleben, A.; Wimmer, E.; Scheible, H.; Kratzer, F.; Yamagata, T.; Gallemann, D.; Fluck, M. In vitro and in vivo drug disposition of cilengitide in animals and human. Pharmacol. Res. Perspect. 2016, 4, e00217. [Google Scholar] [CrossRef] [PubMed]
- Soffietti, R.; Trevisan, E.; Ruda, R. What have we learned from trials on antiangiogenic agents in glioblastoma? Expert Rev. Neurother. 2014, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kenny, H.A.; Jagadeeswaran, S.; Zillhardt, M.R.; Montag, A.G.; Kistner, E.; Yamada, S.D.; Mitra, A.K.; Lengyel, E. {beta}3-integrin expression on tumor cells inhibits tumor progression, reduces metastasis, and is associated with a favorable prognosis in patients with ovarian cancer. Am. J. Pathol. 2009, 175, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Lengyel, E. Mmp-2 functions as an early response protein in ovarian cancer metastasis. Cell Cycle 2009, 8, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Jones, K.C.; Hunter, T. Multiple grb2-mediated integrin-stimulated signaling pathways to erk2/mitogen-activated protein kinase: Summation of both c-src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol. Cell Biol. 1998, 18, 2571–2585. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, M.W.; Price, L.S.; Schwartz, M.A. Focal adhesion kinase mediates the integrin signaling requirement for growth factor activation of map kinase. J. Cell Biol. 1999, 147, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.K.; Tancioni, I.; Lawson, C.; Miller, N.L.; Jean, C.; Chen, X.L.; Uryu, S.; Kim, J.; Tarin, D.; Stupack, D.G.; et al. Inhibition of focal adhesion kinase (fak) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin. Exp. Metastasis 2013, 30, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Graeber, T.G.; Baldwin, R.L.; Karlan, B.Y.; Rao, J.; Chang, D.D. Gene expression in epithelial ovarian carcinoma. Oncogene 2002, 21, 6289–6298. [Google Scholar] [CrossRef] [PubMed]
- Yakubov, B.; Chelladurai, B.; Schmitt, J.; Emerson, R.; Turchi, J.J.; Matei, D. Extracellular tissue transglutaminase activates noncanonical nf-kappab signaling and promotes metastasis in ovarian cancer. Neoplasia 2013, 15, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, M.; Shao, M.; Emerson, R.; Donner, D.B.; Matei, D. Tissue transglutaminase regulates matrix metalloproteinase-2 in ovarian cancer by modulating camp-response element-binding protein activity. J. Biol. Chem. 2009, 284, 15390–15399. [Google Scholar] [CrossRef] [PubMed]
- Khanna, M.; Chelladurai, B.; Gavini, A.; Li, L.; Shao, M.; Courtney, D.; Turchi, J.J.; Matei, D.; Meroueh, S. Targeting ovarian tumor cell adhesion mediated by tissue transglutaminase. Mol. Cancer Ther. 2011, 10, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Yakubov, B.; Chen, L.; Belkin, A.M.; Zhang, S.; Chelladurai, B.; Zhang, Z.Y.; Matei, D. Small molecule inhibitors target the tissue transglutaminase and fibronectin interaction. PLoS ONE 2014, 9, e89285. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta. 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. Ne. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.; Tinker, A.V.; Lee, C.H.; Subramanian, S.; van de Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial t cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and brca1 loss. Mod. Pathol. 2009, 22, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial cd8+ tumor-infiltrating lymphocytes and a high cd8+/regulatory t cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Santoiemma, P.P.; Reyes, C.; Wang, L.P.; McLane, M.W.; Feldman, M.D.; Tanyi, J.L.; Powell, D.J., Jr. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol. Oncol. 2016, 143, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J., Jr. Cd137 accurately identifies and enriches for naturally occurring tumor-reactive t cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017, 170, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Bosmuller, H.C.; Wagner, P.; Peper, J.K.; Schuster, H.; Pham, D.L.; Greif, K.; Beschorner, C.; Rammensee, H.G.; Stevanovic, S.; Fend, F.; et al. Combined immunoscore of cd103 and cd3 identifies long-term survivors in high-grade serous ovarian cancer. Int. J. Gynecol. Cancer 2016, 26, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, H.; Fujita, K.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; Tanaka, K. Immunomodulation in patients with epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Cancer Res. 1994, 54, 190–196. [Google Scholar] [PubMed]
- Kandalaft, L.E.; Chiang, C.L.; Tanyi, J.; Motz, G.; Balint, K.; Mick, R.; Coukos, G. A phase i vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, bms-936558, ono-4538) in patients with previously treated advanced non-small-cell lung cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: Pd-1/pd-l1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating cd8+ t lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Krempski, J.; Karyampudi, L.; Behrens, M.D.; Erskine, C.L.; Hartmann, L.; Dong, H.; Goode, E.L.; Kalli, K.R.; Knutson, K.L. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 2011, 186, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Darb-Esfahani, S.; Kunze, C.A.; Kulbe, H.; Sehouli, J.; Wienert, S.; Lindner, J.; Budczies, J.; Bockmayr, M.; Dietel, M.; Denkert, C.; et al. Prognostic impact of programmed cell death-1 (pd-1) and pd-ligand 1 (pd-l1) expression in cancer cells and tumor-infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget 2016, 7, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. Pd-l1 expression is associated with tumor-infiltrating t cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic pd-1 pathway blockade augments with other modalities of immunotherapy t-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Pihapaul, S.A.; Ott, P.A.; Mehnert, J.M.; Bertonrigaud, D.; Johnson, E.A.; Cheng, J.D.; Yuan, S.; Rubin, E.H.; Matei, D.E. Antitumor activity and safety of pembrolizumab in patients (pts) with pd-l1 positive advanced ovarian cancer: Interim results from a phase ib study. J. Clin. Oncol. 2015, 33, 5510. [Google Scholar] [CrossRef]
- Disis, M.L.; Patel, M.R.; Pant, S.; Infante, J.R.; Lockhart, A.C.; Kelly, K.; Beck, J.T.; Gordon, M.S.; Weiss, G.J.; Ejadi, S.; et al. Avelumab (msb0010718c), an anti-pd-l1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: A phase ib, open-label expansion trial. J. Clin. Oncol. 2015, 33, 5509. [Google Scholar] [CrossRef]
- Lau, E. Mismatch repair deficiency predicts benefit of anti-pd-1 therapy. Lancet Oncol. 2015, 16, e319. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of brca1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of pd-1/pd-l1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Serafini, P.; Apolloni, E.; Zanovello, P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J. Immunother. 2001, 24, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high m1/m2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Deavers, M.; Patenia, R.; Bassett, R.L., Jr.; Mueller, P.; Ma, Q.; Wang, E.; Freedman, R.S. Monocyte/macrophage and t-cell infiltrates in peritoneum of patients with ovarian cancer or benign pelvic disease. J. Transl. Med. 2006, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via nf-kappa b and jnk. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Ladanyi, A.; Lengyel, E.; Naora, H. Expression of the homeobox gene hoxa9 in ovarian cancer induces peritoneal macrophages to acquire an m2 tumor-promoting phenotype. Am. J. Pathol. 2014, 184, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microrna101 and suppressing the corepressor ctbp2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via cxcr2 ligand upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Bronte, V.; Ugel, S. Tumor-induced myeloid-derived suppressor cells. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Pulaski, H.L.; Spahlinger, G.; Silva, I.A.; McLean, K.; Kueck, A.S.; Reynolds, R.K.; Coukos, G.; Conejo-Garcia, J.R.; Buckanovich, R.J. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J. Transl. Med. 2009, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Penn, C.A.; Yang, K.; Zong, H.; Lim, J.Y.; Cole, A.; Yang, D.; Baker, J.; Goonewardena, S.N.; Buckanovich, R.J. Therapeutic impact of nanoparticle therapy targeting tumor-associated macrophages. Mol. Cancer Ther. 2018, 17, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. Bet bromodomain inhibition promotes anti-tumor immunity by suppressing pd-l1 expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.L.; et al. Epigenetic therapy activates type i interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Wimberger, P.; Kumper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-epcam x anti-cd3 antibody: A phase i/ii study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Course of Treatment | Target | TME Component | Patient Population | Phase Trial Size | Trial Endpoint | Clinical Outcome |
|---|---|---|---|---|---|---|---|
| ICON7 | Chemo ± Bevac | VEGF-A | Endothelium | High risk ovarian cancer, stage IIIC or IV | Phase III N = 1528 | PFS | At 42 months 22.4 vs. 24.1 months p = 0.04 |
| GOG218 | Chemo vs. Chemo + Bevac initiation vs. Chemo + Bevac Throughout | VEGF-A | Endothelium | New Diagnosed Stage III or IV OC | Phase III N = 1873 | PFS, OS | Median PFS; 10.3 vs. 11.2 vs. 14.1 months; OS; ns |
| AURELIA | Chemo ± Bevac | VEGF-A | Endothelium | Recurrent OC PL-R | Phase III N = 361 | PFS, OS | Median PFS; 3.4 vs. 6.7 months. OS; 13.3 vs. 16.6 months |
| OCEANS | Chemo ± Bevac | VEGF-A | Endothelium | Recurrent OC PL-S | Phase III N = 484 | PFS | Median PFS 8.4 vs. 12.4 months |
| GOG213 | Chemo ± Bevac | VEGF-A | Endothelium | Recurrent OC PL-S | Phase III N = 674 | ORR | Median overall survival 37.3 vs. 42.2 months |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwani, N.G.; Sima, L.E.; Nieves-Neira, W.; Matei, D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers 2018, 10, 266. https://doi.org/10.3390/cancers10080266
Nwani NG, Sima LE, Nieves-Neira W, Matei D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers. 2018; 10(8):266. https://doi.org/10.3390/cancers10080266
Chicago/Turabian StyleNwani, Nkechiyere G., Livia E. Sima, Wilberto Nieves-Neira, and Daniela Matei. 2018. "Targeting the Microenvironment in High Grade Serous Ovarian Cancer" Cancers 10, no. 8: 266. https://doi.org/10.3390/cancers10080266
APA StyleNwani, N. G., Sima, L. E., Nieves-Neira, W., & Matei, D. (2018). Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers, 10(8), 266. https://doi.org/10.3390/cancers10080266