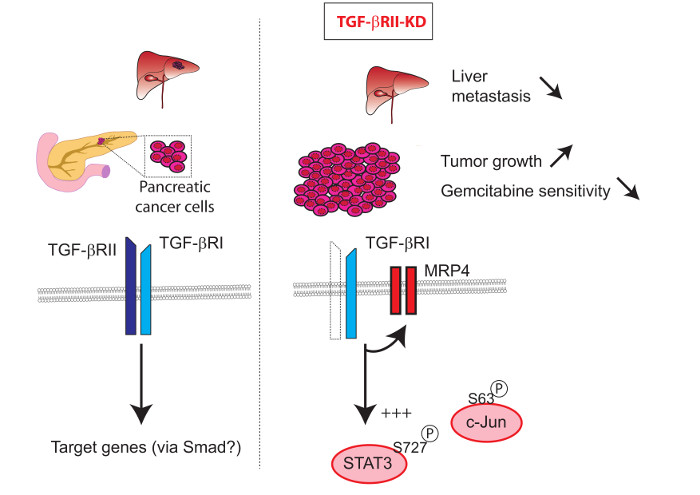
TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727

 , , ,
, , ,  and
and 
Abstract
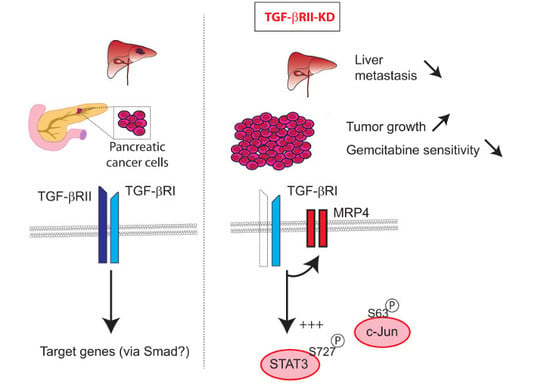
:
1. Introduction
2. Results
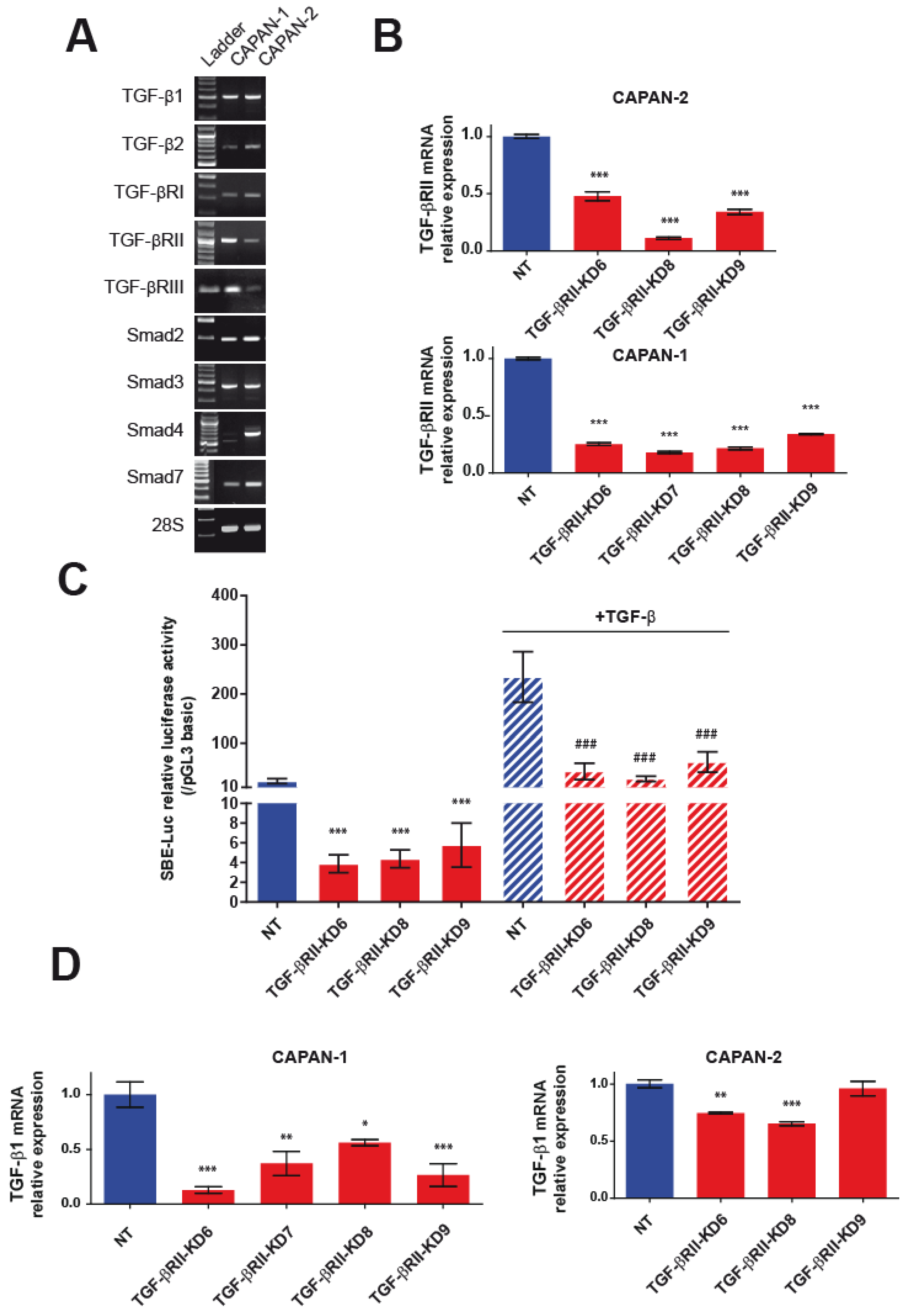
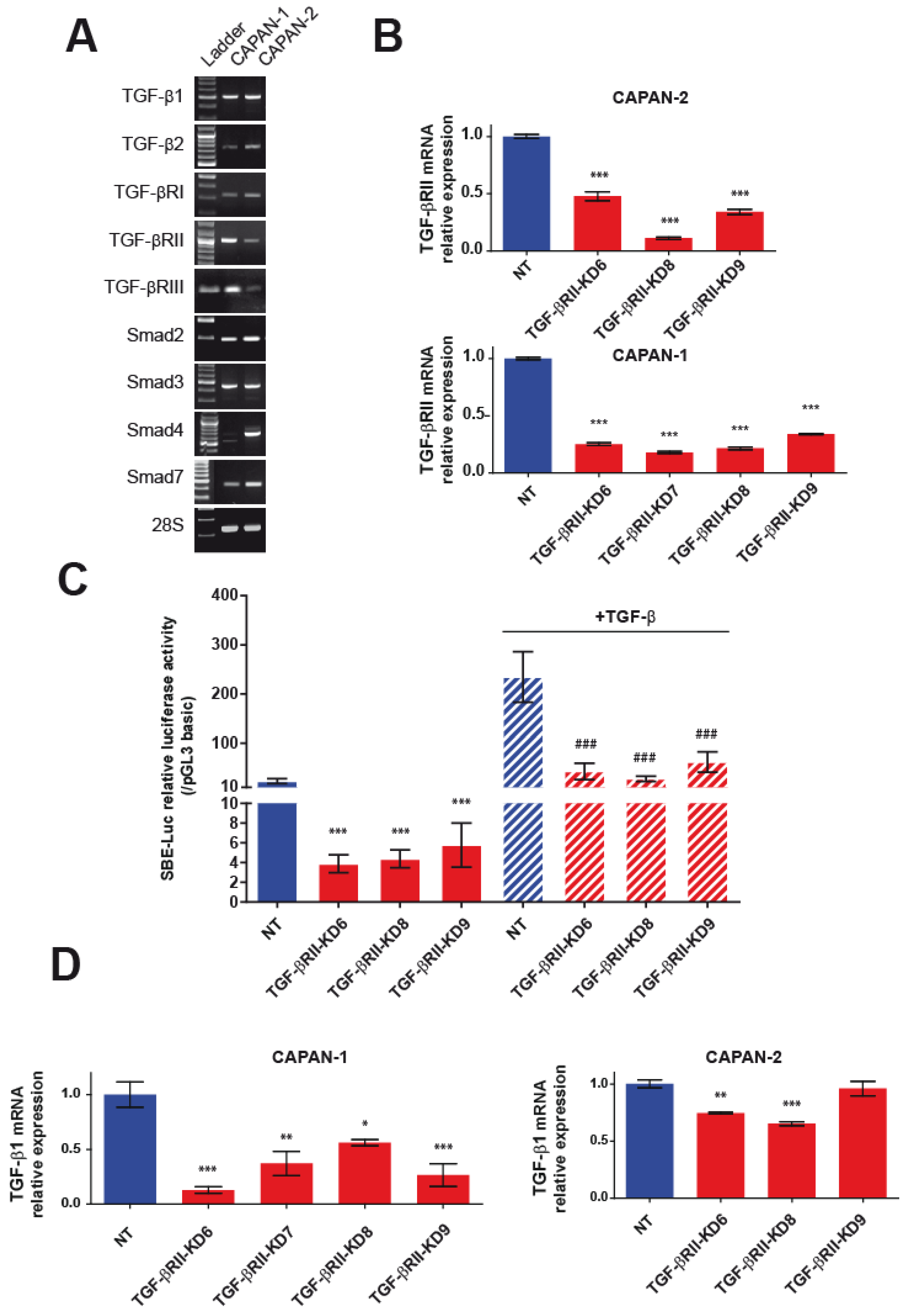
2.1. Generation and Characterization of Stable TGF-βRII-KD Cell Lines
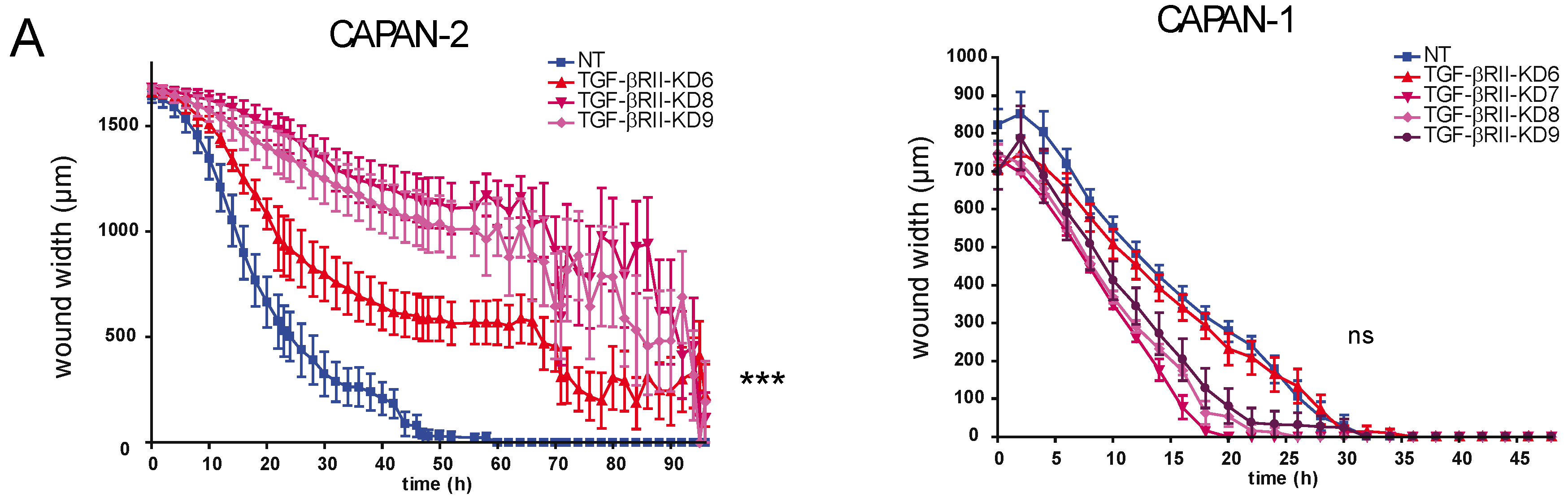
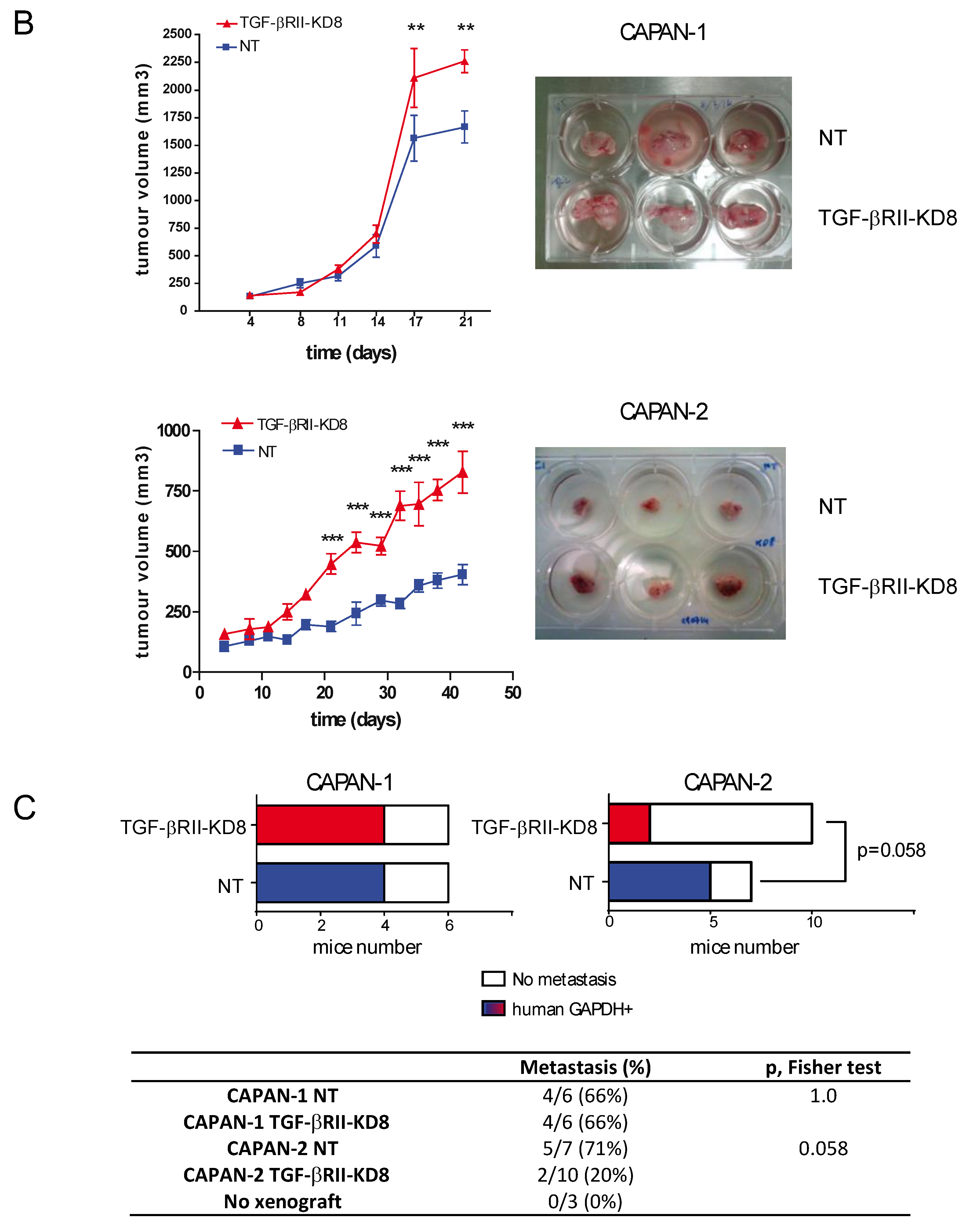
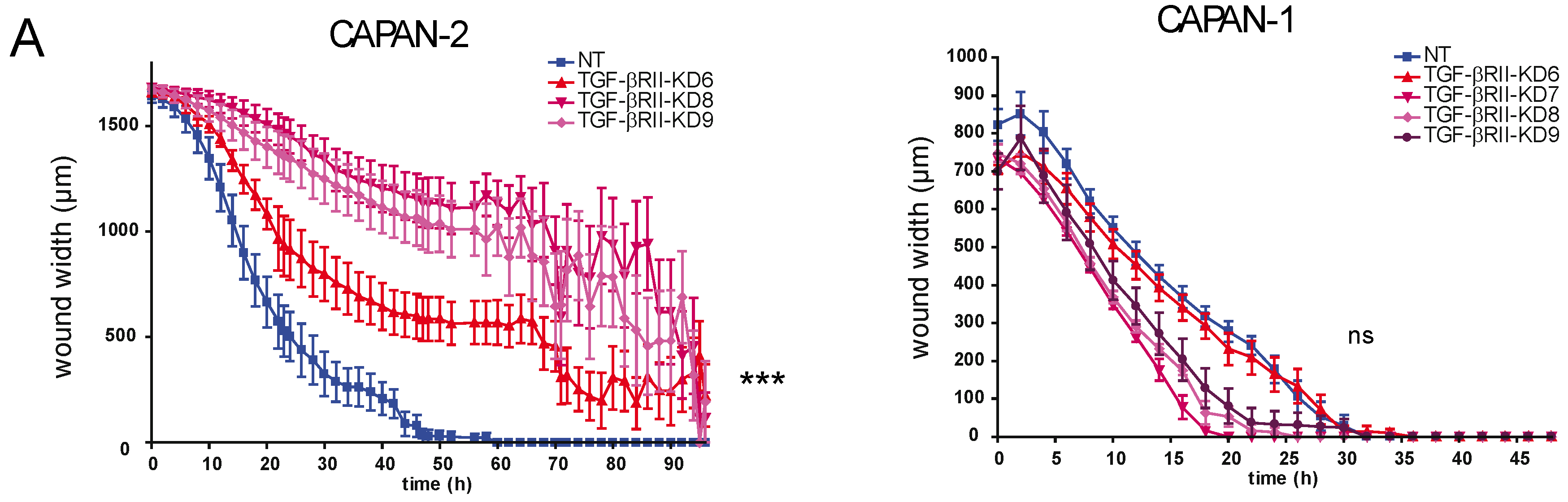
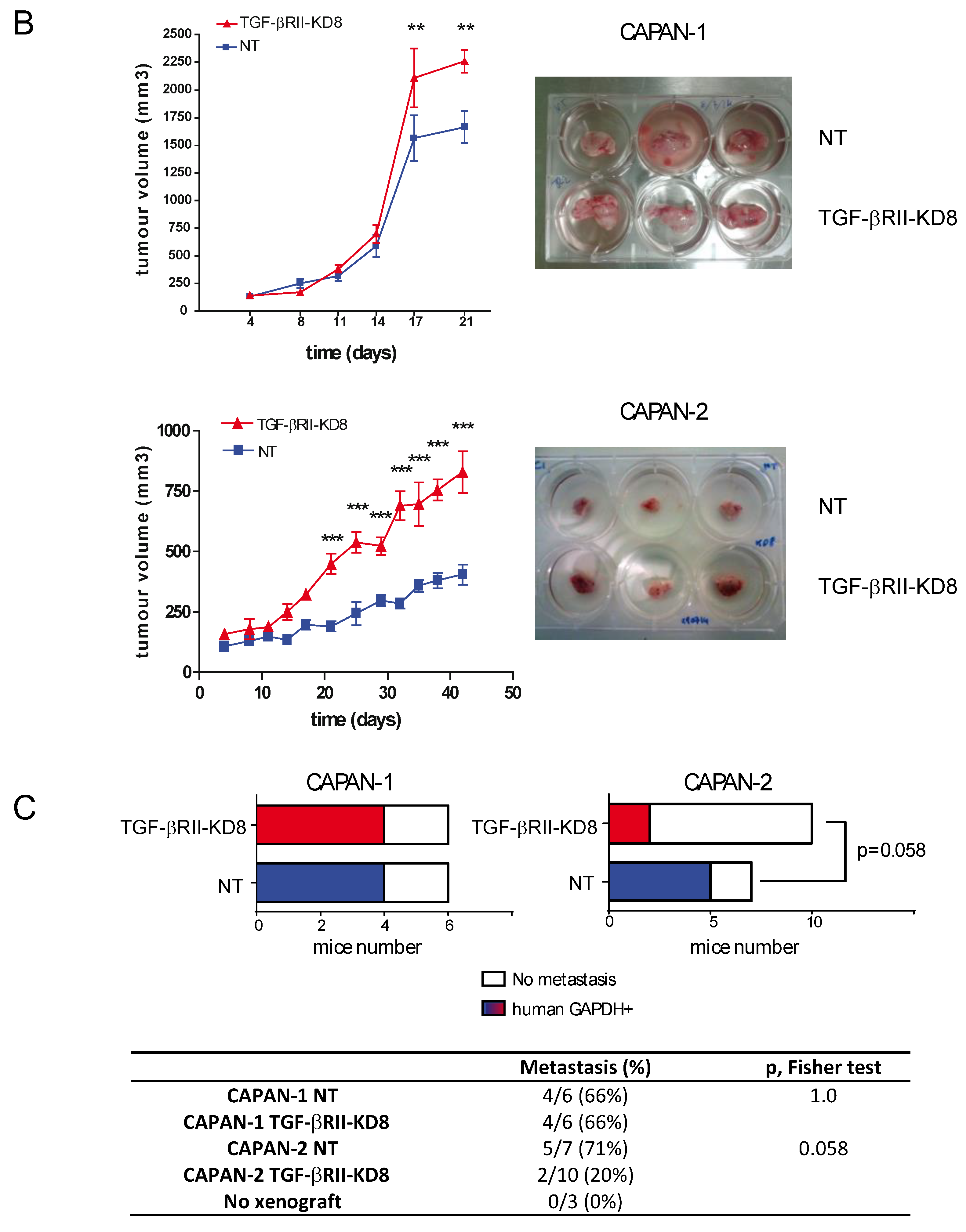
2.2. Involvement of TGF-βRII in PC Cell Biological Properties
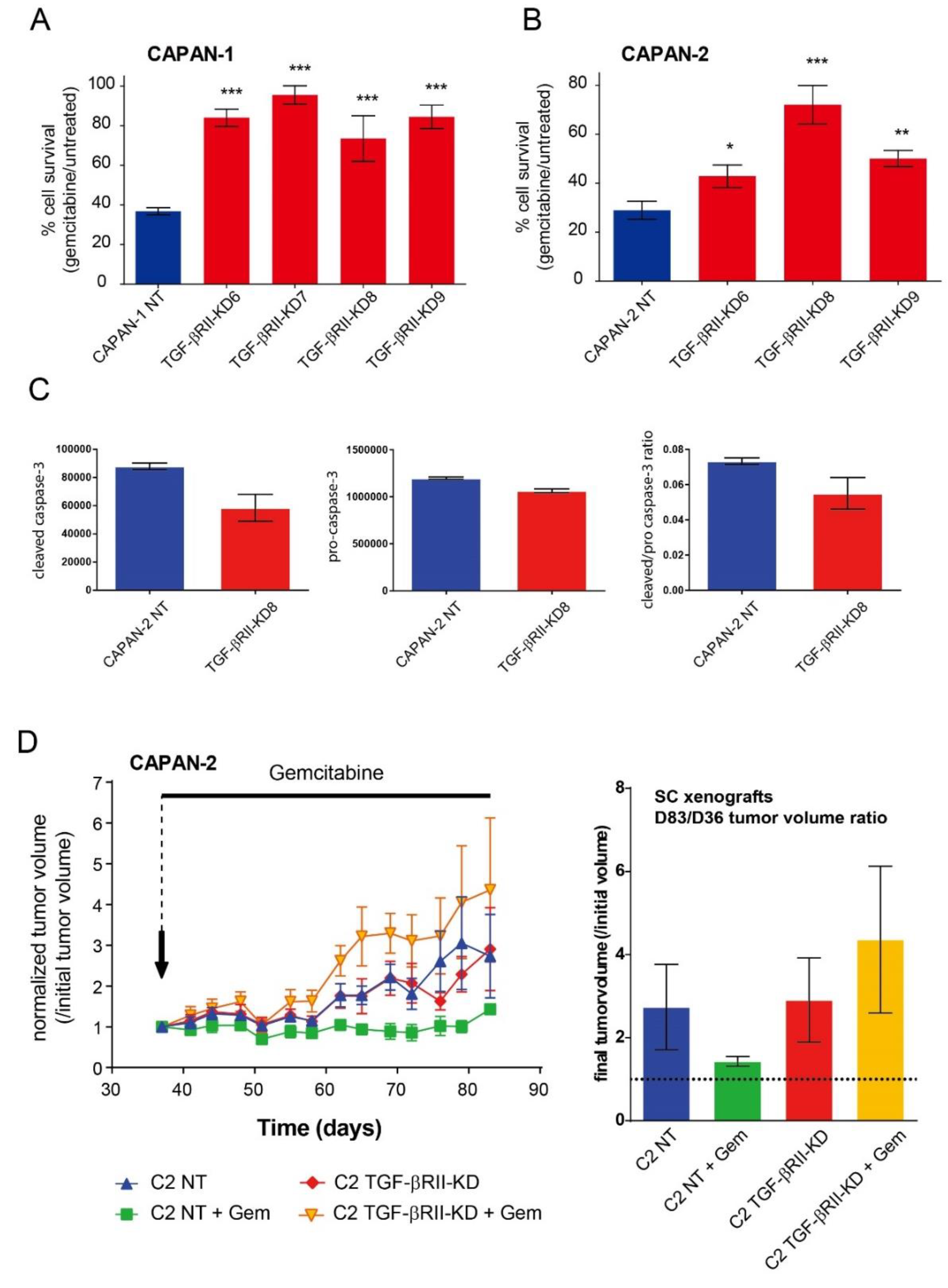
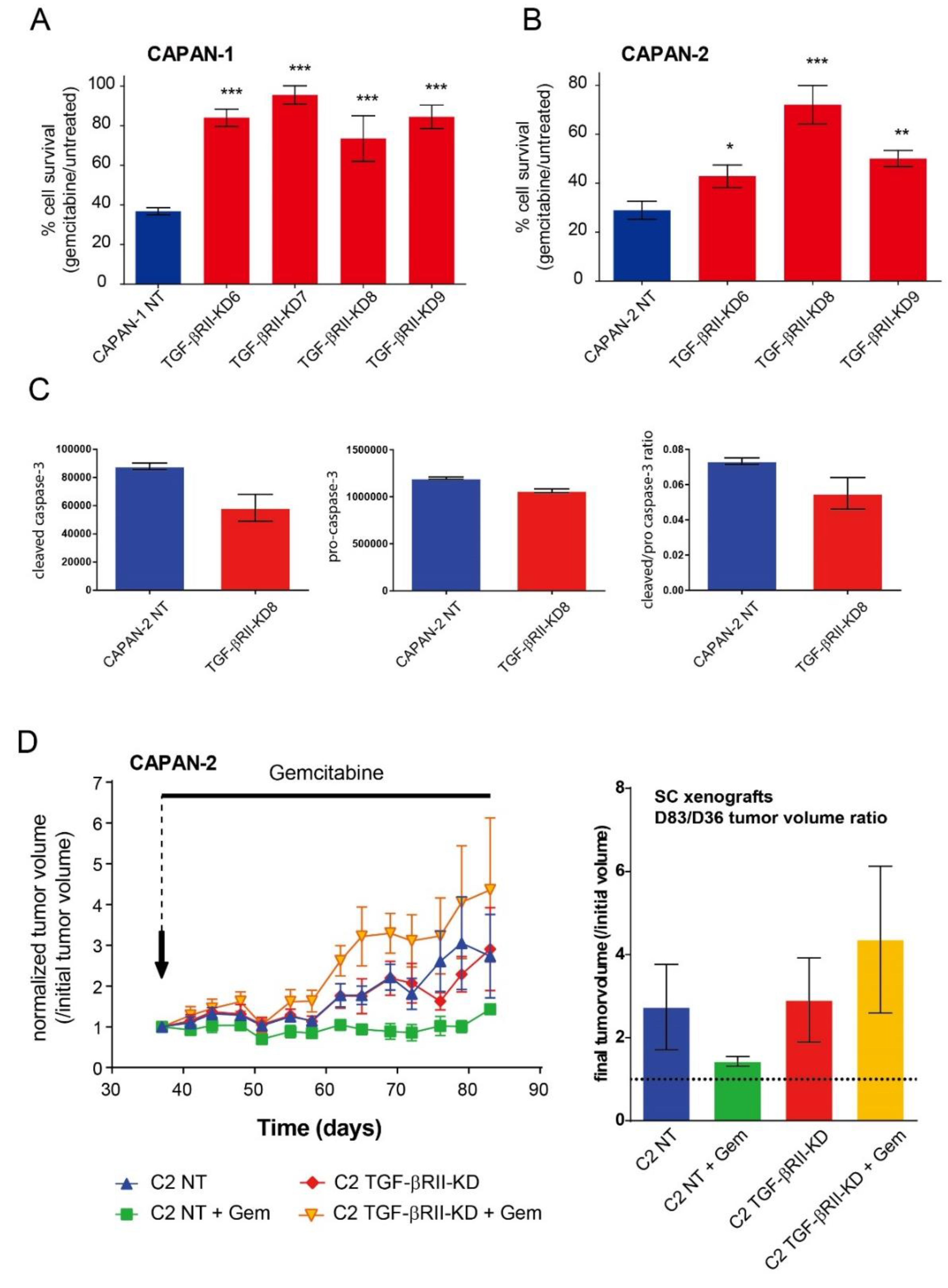
2.3. Role of TGF-βRII on PC Cells Sensitivity to Gemcitabine
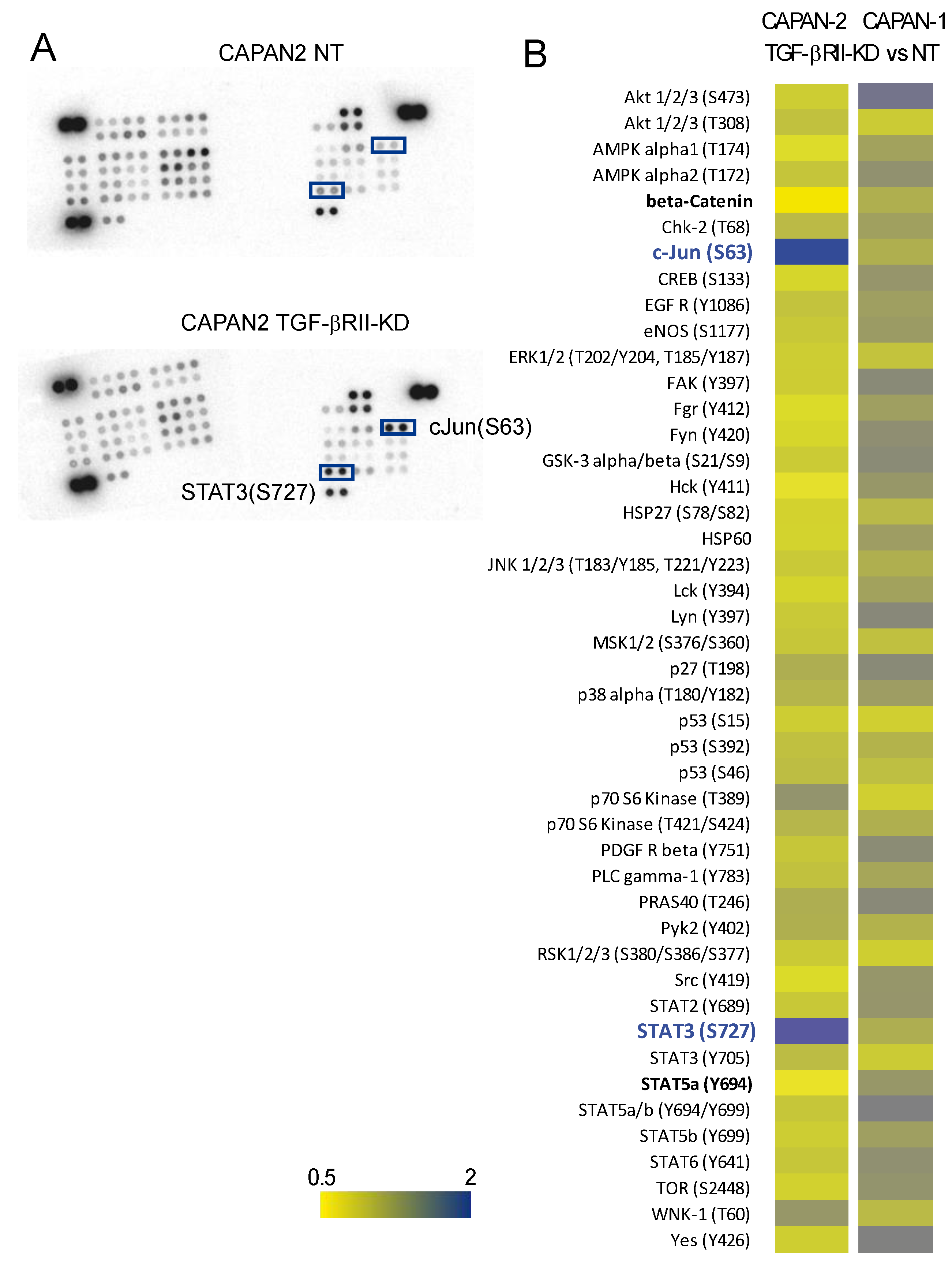
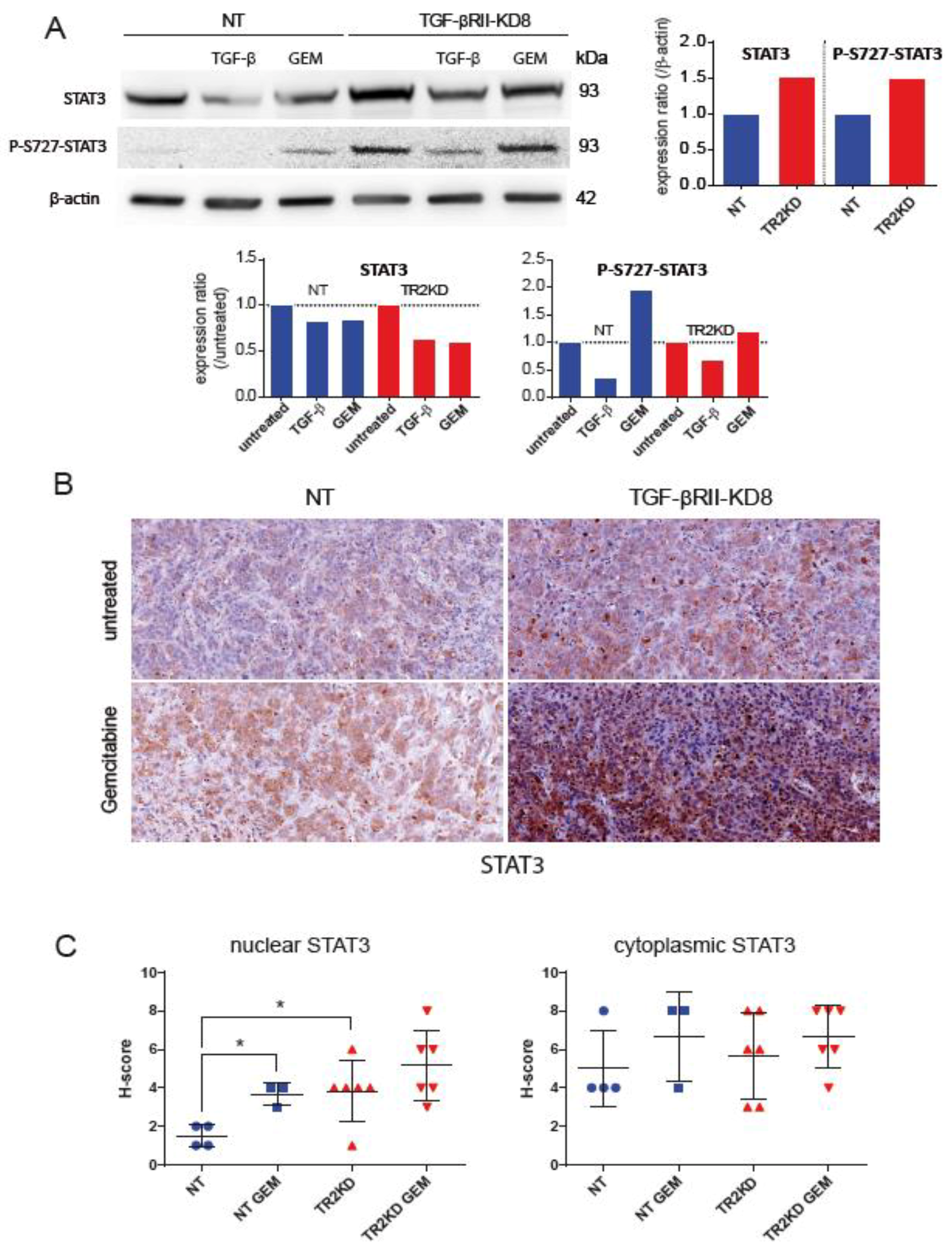
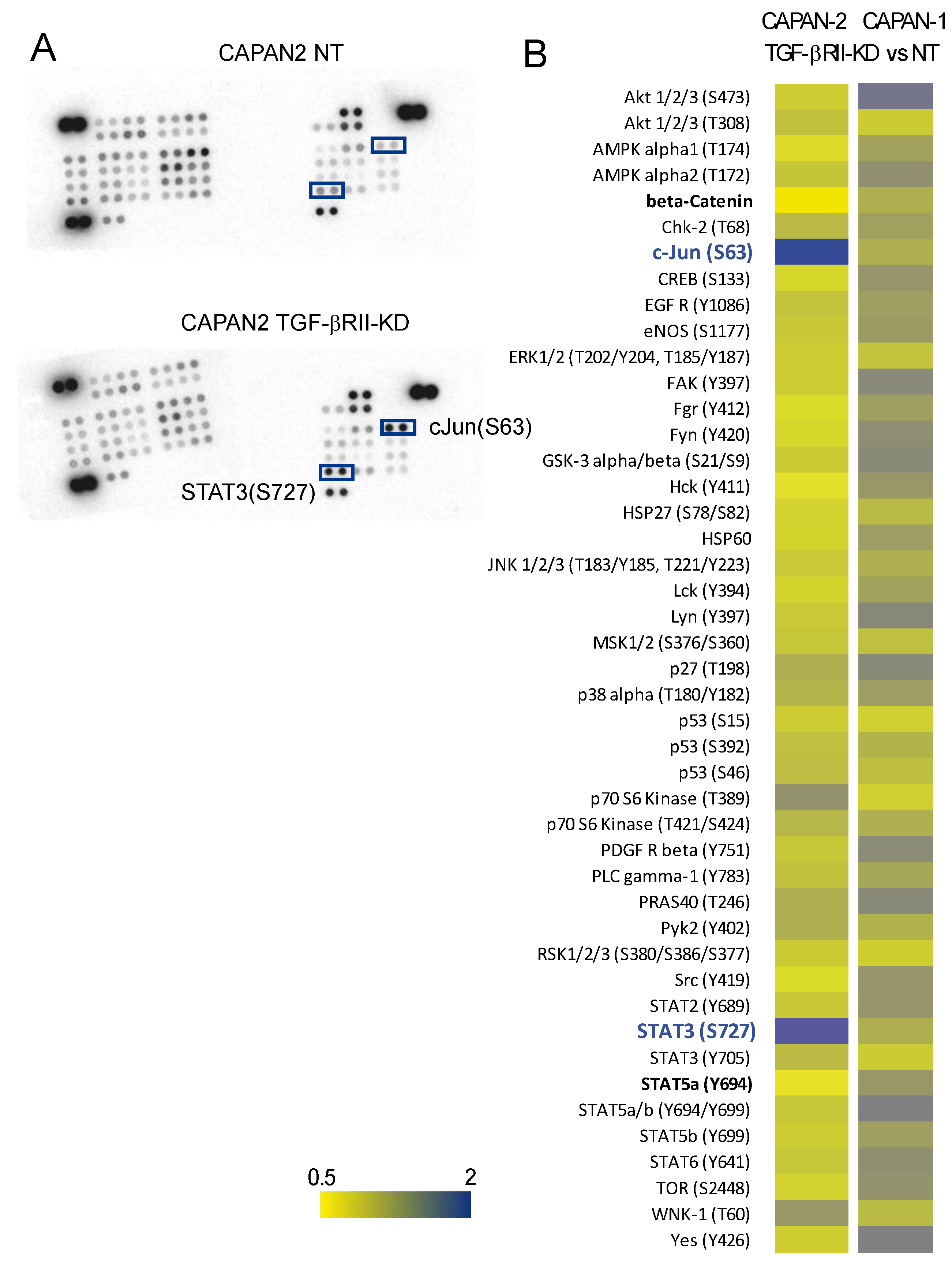
2.4. Identification of Signalling Pathways Altered following TGF-βRII Knocking Down
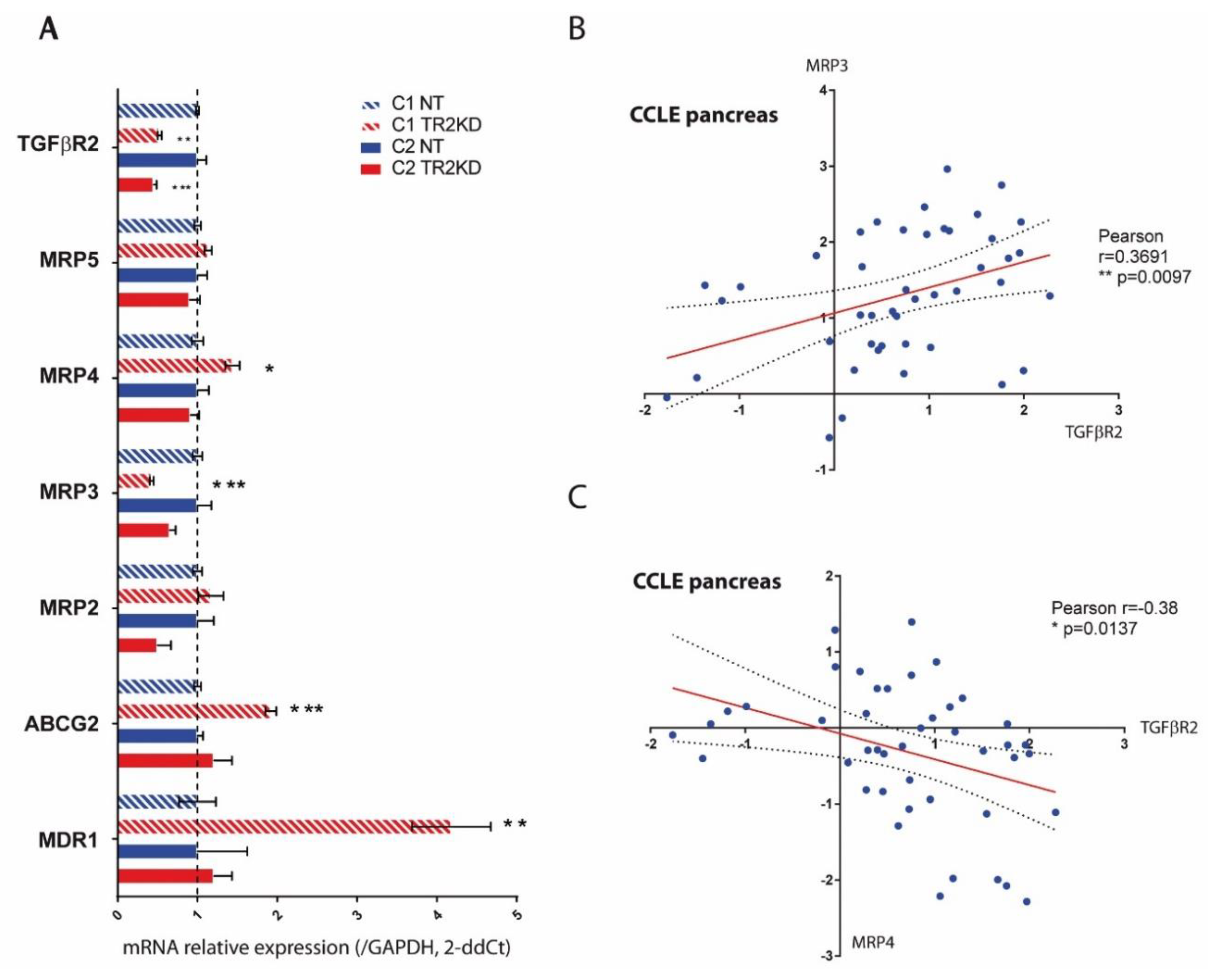
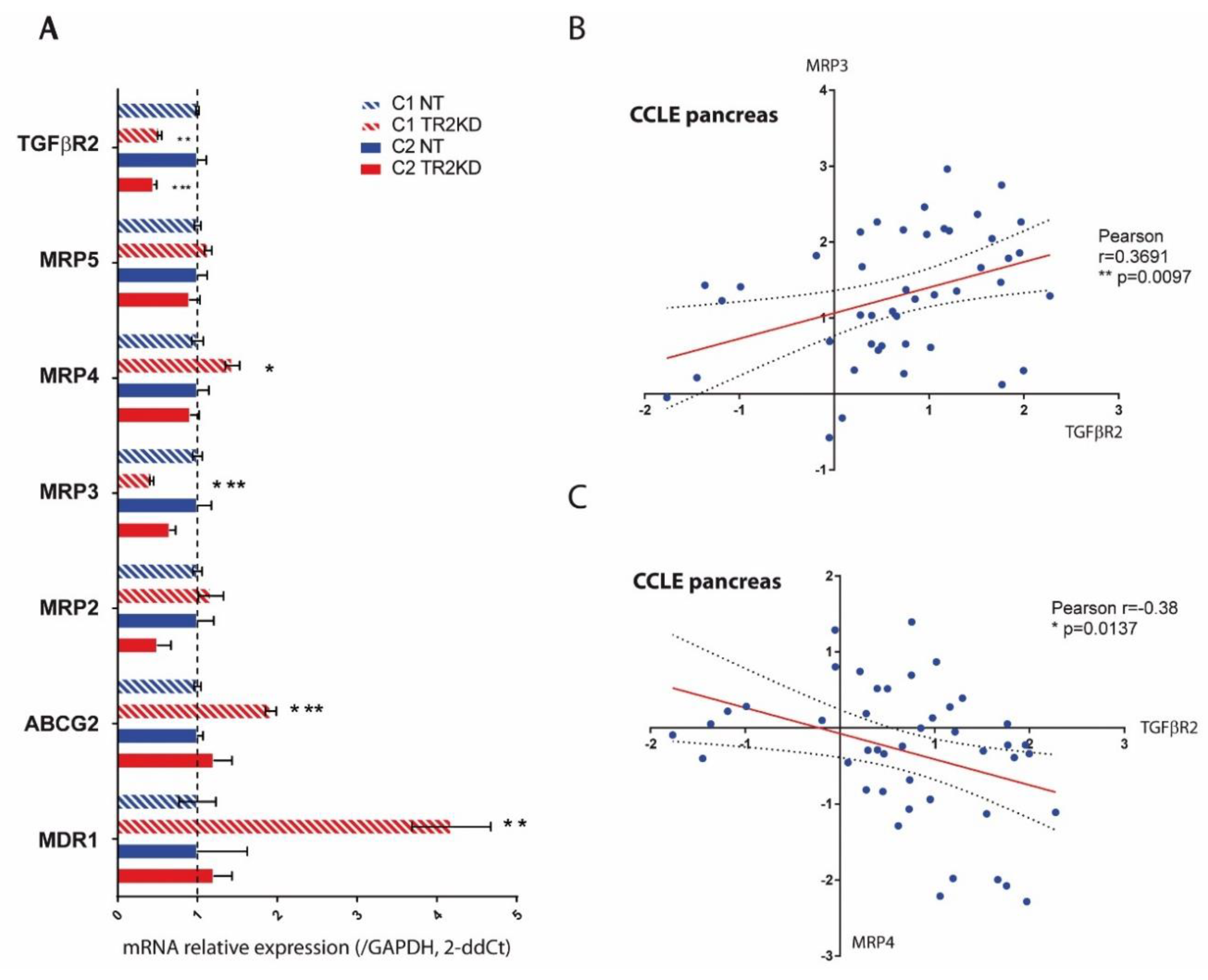
2.5. TGF-βRII Silencing Alters the Expression of ABC Transporters and EMT Markers in PC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. qRT-PCR
4.3. Protein Extraction and Western-Blotting
4.4. Cell Proliferation
4.5. Wound Healing Test
4.6. Cytotoxicity Assay
4.7. Subcutaneous Xenografts
4.8. Immunohistochemistry
4.9. Expression Analysis in CCLE Database
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- TCGA-Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, N.; Perrais, M.; Mariette, C.; Batra, S.K.; Aubert, J.P.; Pigny, P.; van Seuningen, I. A role for human MUC4 mucin gene, the ErbB2 ligand, as a target of TGF-beta in pancreatic carcinogenesis. Oncogene 2004, 23, 5729–5738. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Duchene, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.R.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar] [PubMed]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-beta signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostapoff, K.T.; Cenik, B.K.; Wang, M.; Ye, R.; Xu, X.; Nugent, D.; Hagopian, M.M.; Topalovski, M.; Rivera, L.B.; Carroll, K.D.; et al. Neutralizing murine TGFbetaR2 promotes a differentiated tumor cell phenotype and inhibits pancreatic cancer metastasis. Cancer Res. 2014, 74, 4996–5007. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, H.; Bai, M.; Ning, T.; Wang, C.; Fan, Q.; Wang, Y.; Fu, Z.; Wang, N.; Liu, R.; et al. miR-135b Promotes Cancer Progression by Targeting Transforming Growth Factor Beta Receptor II (TGFBR2) in Colorectal Cancer. PLoS ONE 2015, 10, e0130194. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, P.; Rodriguez, F.; Schmitz, M.; Meurer, S.K.; Qureshi-Baig, K.; Felten, P.; Ginolhac, A.; Antunes, L.; Frasquilho, S.; Zugel, N.; et al. The miR-371 approximately 373 Cluster Represses Colon Cancer Initiation and Metastatic Colonization by Inhibiting the TGFBR2/ID1 Signaling Axis. Cancer Res. 2018, 78, 3793–3808. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.; Chan, E.; Chu, F.; Pham, T.; Koeppen, H.; Forrest, W.; Merchant, M.; Settleman, J. Cotargeting of MEK and PDGFR/STAT3 Pathways to Treat Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.L.; Caprioli, R.; Chen, X.; Walsh, A.J.; Skala, M.C.; Moses, H.L.; et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015, 149, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Duhagon Serrat, M.A.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubbarao, K.; Peterson, L.; Zhao, S.; Hill, P.; Cao, L.; Zhou, Q.; Nawrocki, S.T.; Freeman, J.W. Inhibiting signal transducer and activator of transcription-3 increases response to gemcitabine and delays progression of pancreatic cancer. Mol. Cancer 2013, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabayashi, K.; Ijichi, H.; Mohri, D.; Tada, M.; Yamamoto, K.; Asaoka, Y.; Ikenoue, T.; Tateishi, K.; Nakai, Y.; Isayama, H.; et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res. 2013, 73, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Rosa Rosa, J.M.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Mitsopoulos, C.; Sims, D.; Hakas, J.; Zvelebil, M.; Lord, C.J.; et al. Comprehensive genomic analysis of a BRCA2 deficient human pancreatic cancer. PLoS ONE 2011, 6, e21639. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bryson, B.L.; Junk, D.J.; Cipriano, R.; Jackson, M.W. STAT3-mediated SMAD3 activation underlies Oncostatin M-induced Senescence. Cell Cycle 2017, 16, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Kali, A.; Ostapchuk, Y.O.; Belyaev, N.N. TNFalpha and TGFbeta-1 synergistically increase the cancer stem cell properties of MiaPaCa-2 cells. Oncol. Lett. 2017, 14, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Sims, A.H.; Stal, O.; Ferno, M.; Landberg, G. Loss of TGFbeta Receptor Type 2 Expression Impairs Estrogen Response and Confers Tamoxifen Resistance. Cancer Res. 2015, 75, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta 2017, 1861, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, N.; Skrypek, N.; Merlin, J.; Dessein, A.F.; Dumont, P.; Leteurtre, E.; Harris, A.; Desseyn, J.L.; Susini, C.; Frenois, F.; et al. The mucin MUC4 and its membrane partner ErbB2 regulate biological properties of human CAPAN-2 pancreatic cancer cells via different signalling pathways. PLoS ONE 2012, 7, e32232. [Google Scholar] [CrossRef] [PubMed]
- Van Seuningen, I.; Perrais, M.; Pigny, P.; Porchet, N.; Aubert, J.P. Sequence of the 5′-flanking region and promoter activity of the human mucin gene MUC5B in different phenotypes of colon cancer cells. Biochem. J. 2000, 348 Pt 3, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, P.; Jonckheere, N.; Almeida, R.; Ducourouble, M.P.; Serpa, J.; Silva, E.; Pigny, P.; Silva, F.S.; Reis, C.; Silberg, D.; et al. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. J. Biol. Chem. 2003, 278, 51549–51556. [Google Scholar] [CrossRef] [PubMed]
- Van Seuningen, I.; Ostrowski, J.; Bustelo, X.R.; Sleath, P.R.; Bomsztyk, K. The K protein domain that recruits the interleukin 1-responsive K protein kinase lies adjacent to a cluster of c-Src and Vav SH3-binding sites. Implications that K protein acts as a docking platform. J. Biol. Chem. 1995, 270, 26976–26985. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, N.; Fauquette, V.; Stechly, L.; Saint-Laurent, N.; Aubert, S.; Susini, C.; Huet, G.; Porchet, N.; Van Seuningen, I.; Pigny, P. Tumour growth and resistance to gemcitabine of pancreatic cancer cells are decreased by AP-2alpha overexpression. Br. J. Cancer 2009, 101, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Piessen, G.; Jonckheere, N.; Vincent, A.; Hemon, B.; Ducourouble, M.P.; Copin, M.C.; Mariette, C.; van Seuningen, I. Regulation of the human mucin MUC4 by taurodeoxycholic and taurochenodeoxycholic bile acids in oesophageal cancer cells is mediated by hepatocyte nuclear factor 1alpha. Biochem. J. 2007, 402, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; Melis, M.H.; Jonckheere, N.; Ducourouble, M.P.; Buller, H.A.; Renes, I.; Einerhand, A.W.; van Seuningen, I. The murine Muc2 mucin gene is transcriptionally regulated by the zinc-finger GATA-4 transcription factor in intestinal cells. Biochem. Biophys. Res. Commun. 2004, 325, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Orientation | Sequences of Primers (5′–3′) | T°m (°C) | Expected Size |
|---|---|---|---|---|
| RT-PCR | ||||
| 28S | Forward Reverse | GCAGGGCGAAGCAGAAGGAAACT TGAGATCGTTTCGGCCCCAA | 59 | 231 |
| TGF-β1 | Forward Reverse | GAGGTGACCTGGCCACCATTCAT CCAGCCGAGGTCCTTGCGGA | 60 | 194 |
| TGF-β2 | Forward Reverse | GCTTTTCTGATCCTGCATCTG CAATACCTGCAAATCTTGCTTC | 56 | 823 |
| TGF-βRI | Forward Reverse | CTCTCCTTTTTTCTTCAGATCTGC AATCCAACTCCTTTGCCCTT | 55 | 328 |
| TGF-βRII | Forward Reverse | GCCAACAACATCAACCACAACACA TAGTGTTTAGGGAGCCGTCTTCAG | 61 | 1003 |
| TGF-βRIII | Forward Reverse | TGCCTTACTTCTCTTGCCTTAA GCAAAGTGGCATCATATTATT | 56 | 100 |
| Smad2 | Forward Reverse | GTCCATCTTGCCATTCACG TGGTGATGGCTTTCTCAAGC | 55 | 192 |
| Smad3 | Forward Reverse | GGGCTCCCTCATGTCATCTA GGCTCGCAGTAGGTAACTGG | 60 | 443 |
| SMAD4 | Forward Reverse | CTCCTGAGTATTGGTGTTCC CTAAAGGTTGTGGGTCTGC | 56 | 796 |
| Smad7 | Forward Reverse | GGCTCGCAGTAGGTAACTGG TTGTTGTCCGAATTGAGCTG | 55 | 448 |
| qPCR | ||||
| TGF-β1 | Forward Reverse | CACTCTCAAACCTTTACGAGACC CGTTGCTAGGGGCGAAGATG | 58 | 131 |
| TGF-βRII | Forward Reverse | AGGAGTATGCCTCTTGGAAGAC AGCCAGTATTGTTTCCCCAAC | 58 | 123 |
| Human GADPH | Forward Reverse | CCACATCGCTCAGACACCAT CCAGGCGCCCAATACG | 58 | 70 |
| Mouse GADPH | Forward Reverse | AGGTCGGTGTGAACGGATTTG TGTAGACCATGTAGTTGAGGTCA | 58 | 129 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchêne, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers 2018, 10, 254. https://doi.org/10.3390/cancers10080254
Drubay V, Skrypek N, Cordiez L, Vasseur R, Schulz C, Boukrout N, Duchêne B, Coppin L, Van Seuningen I, Jonckheere N. TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers. 2018; 10(8):254. https://doi.org/10.3390/cancers10080254
Chicago/Turabian StyleDrubay, Vincent, Nicolas Skrypek, Lucie Cordiez, Romain Vasseur, Céline Schulz, Nihad Boukrout, Belinda Duchêne, Lucie Coppin, Isabelle Van Seuningen, and Nicolas Jonckheere. 2018. "TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727" Cancers 10, no. 8: 254. https://doi.org/10.3390/cancers10080254
APA StyleDrubay, V., Skrypek, N., Cordiez, L., Vasseur, R., Schulz, C., Boukrout, N., Duchêne, B., Coppin, L., Van Seuningen, I., & Jonckheere, N. (2018). TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers, 10(8), 254. https://doi.org/10.3390/cancers10080254