TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction

 , ,
, , 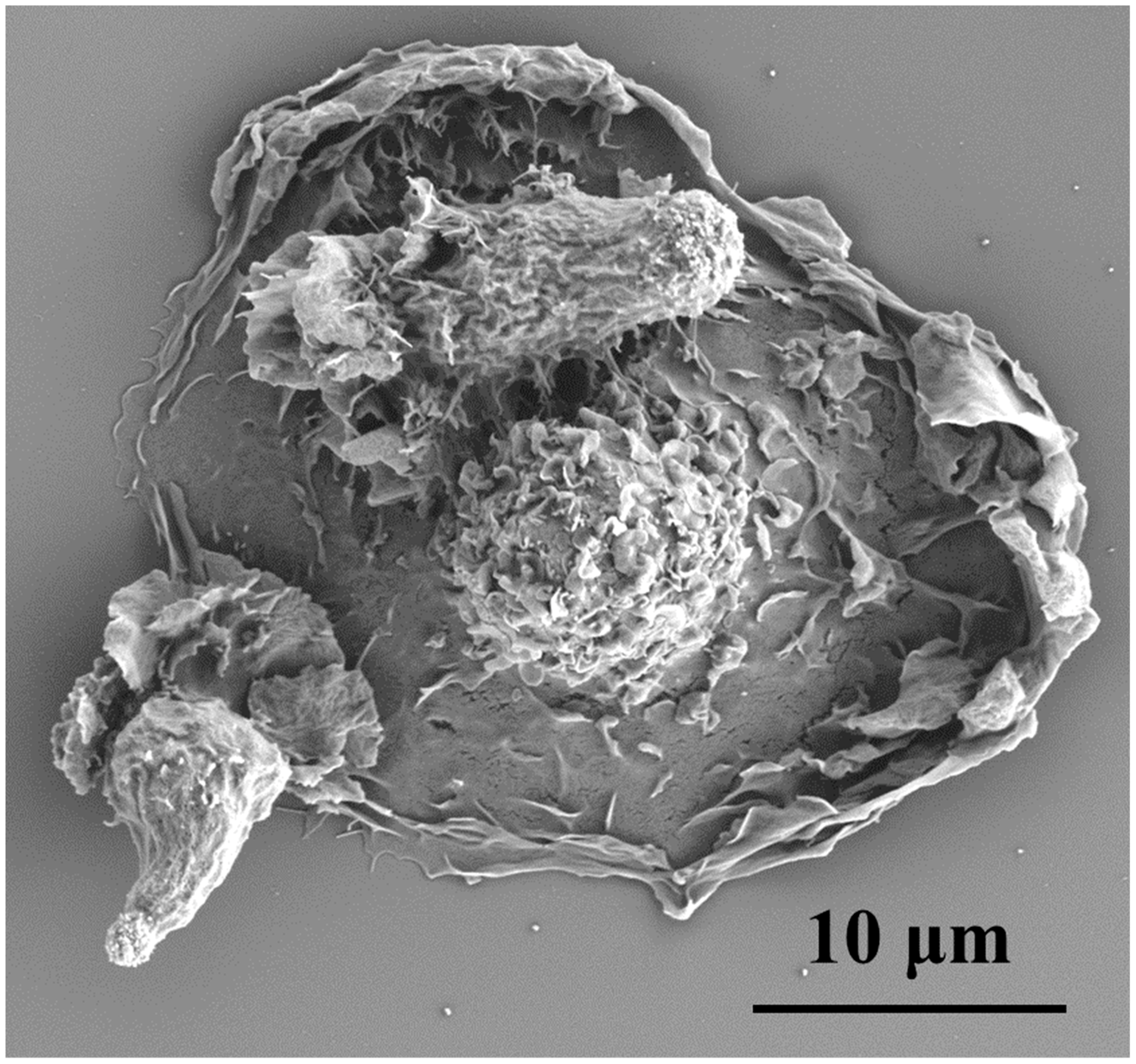{kind=link}
{kind=link}
{kind=link}
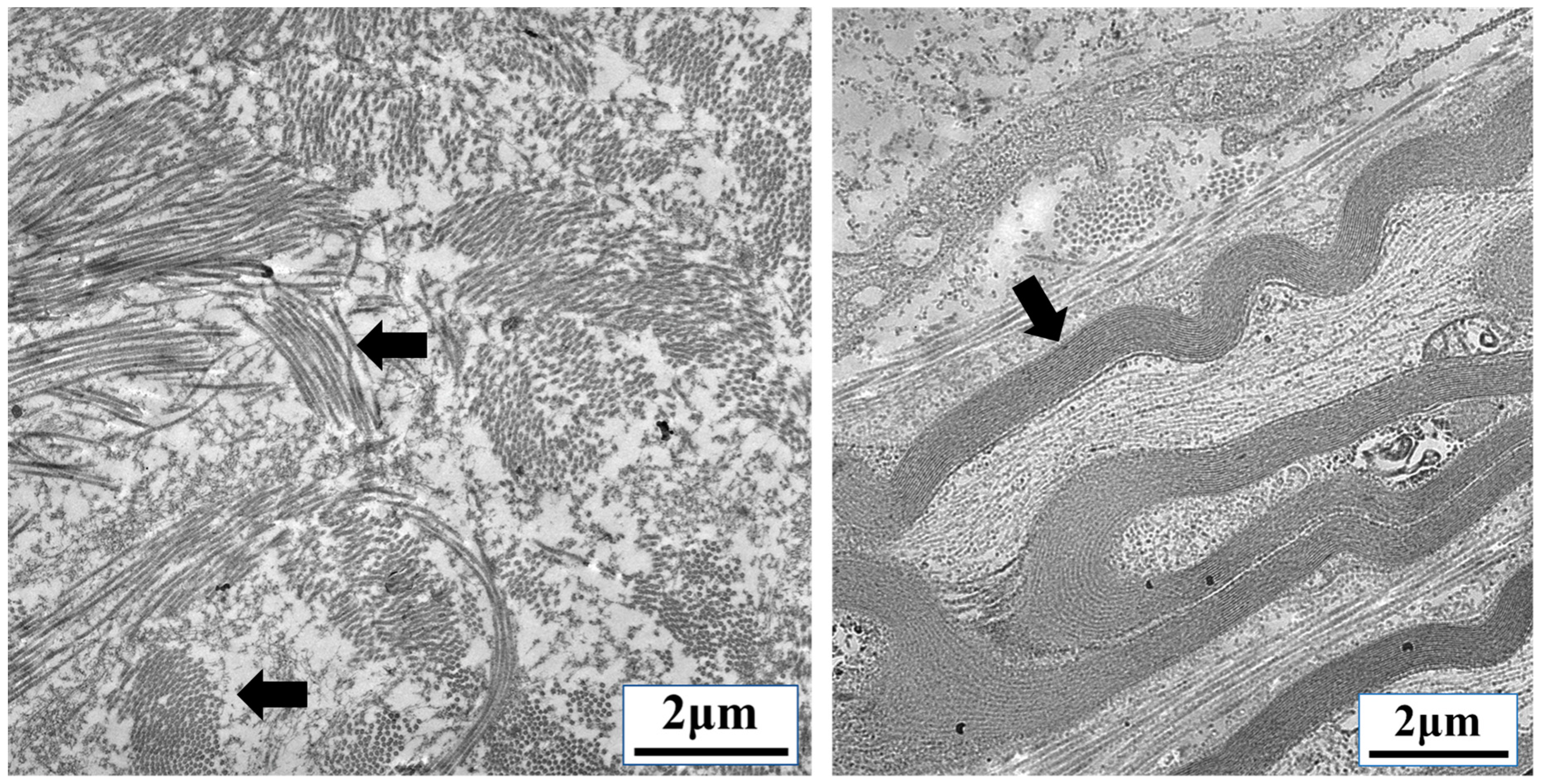{kind=link}
{kind=link}
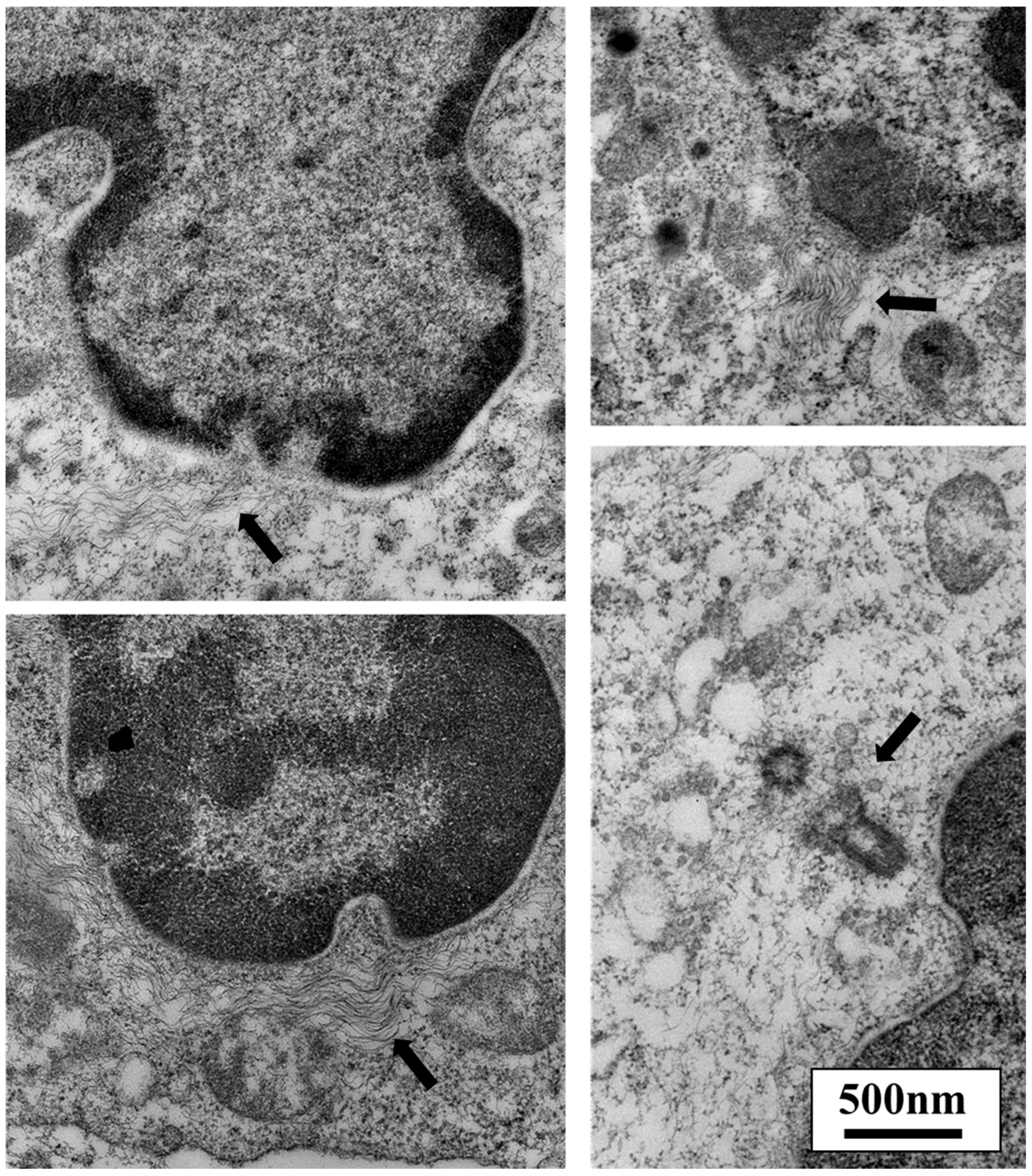{kind=link}
{kind=link}
Abstract
1. Introduction
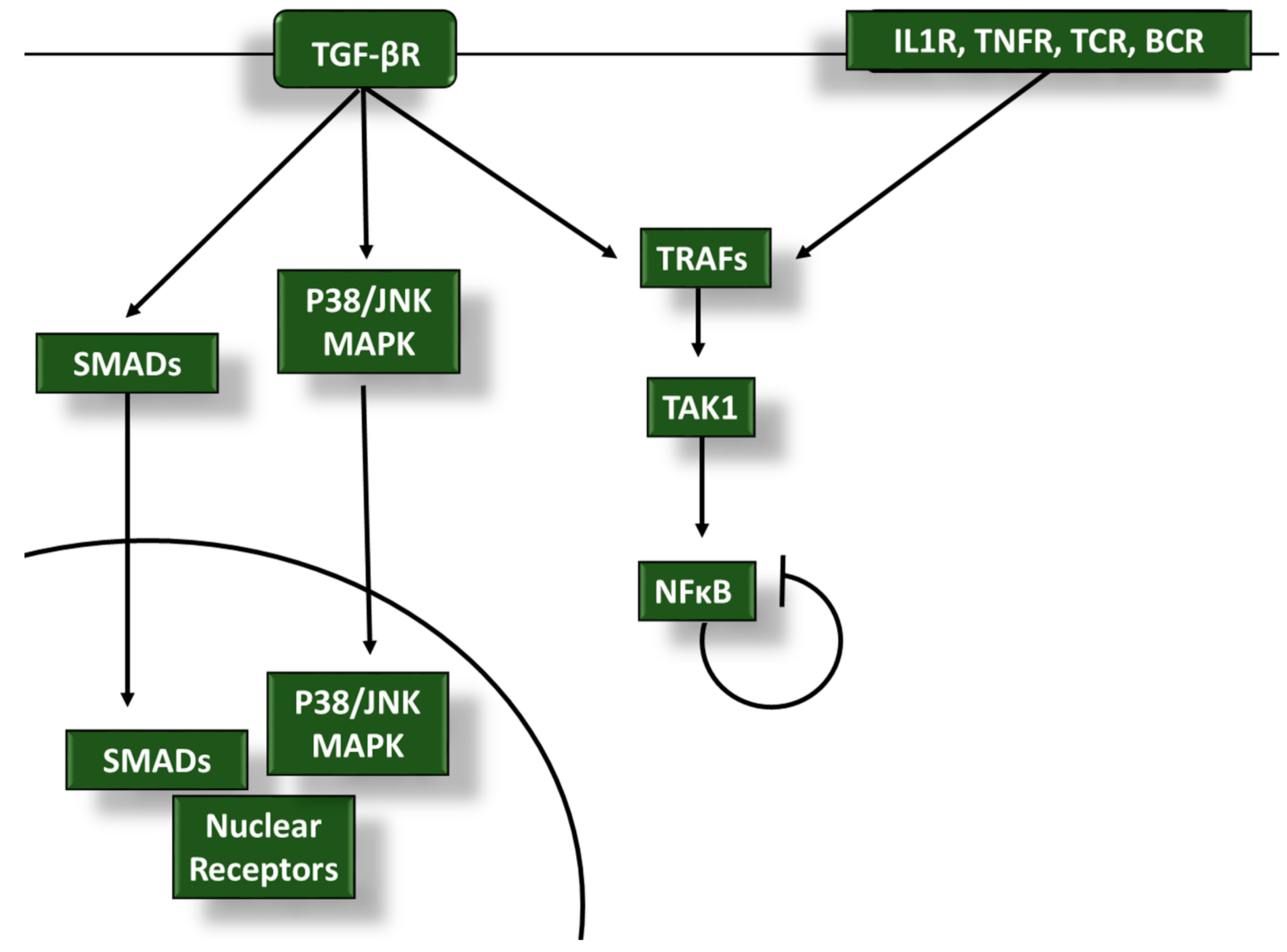
2. TGF-β Inhibits Proinflammatory Signaling in Tumor-Infiltrating Leukocytes
3. TGF-β Signaling Promotes Tumor Growth and Inhibits Inflammation through Mechanobiology
- ECM;
- cell adhesion receptors;
- cytoskeleton;
- nuclear membrane adaptors;
- chromatin.
3.1. TGF-β and ECM
3.2. TGF-β and Nuclear Mechanobiology in Cancer
4. Materials and Methods
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Glossary
References
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the tgfbeta superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The tgfbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- De Caestecker, M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in tgf-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Wilding, G.; Zugmeier, G.; Knabbe, C.; Flanders, K.; Gelmann, E. Differential effects of transforming growth factor beta on human prostate cancer cells in vitro. Mol. Cell. Endocrinol. 1989, 62, 79–87. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. Tgf-beta tumor suppression through a lethal emt. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, S.J.; Bang, Y.J.; Park, J.G.; Kim, N.K.; Roberts, A.B.; Sporn, M.B. Genetic changes in the transforming growth factor beta (tgf-beta) type ii receptor gene in human gastric cancer cells: Correlation with sensitivity to growth inhibition by tgf-beta. Proc. Natl. Acad. Sci. USA 1994, 91, 8772–8776. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant tgf-beta and il-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. Tgf-beta signaling blockade inhibits pthrp secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Tgfbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Cassar, L.; Nicholls, C.; Pinto, A.R.; Chen, R.; Wang, L.; Li, H.; Liu, J.P. Tgf-beta receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence. Protein. Cell 2017, 8, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Acar, A.; Magnusson, Y.; Gregersson, P.; Ryden, L.; Landberg, G. Tgf-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene 2015, 34, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Zarzynska, J.M. Two faces of tgf-beta1 in breast cancer. Med. Inflamm. 2014, 2014, 141747. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, S.H.; Cheng, H.; Li, J.; Feng, R. The effect of tgf-beta-induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in t47d and mcf-7 human breast cancer cells. Arch. Biochem. Biophys. 2015, 583, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Boone, S.D.; Baumgartner, K.B.; Baumgartner, R.N.; Connor, A.E.; Pinkston, C.M.; John, E.M.; Hines, L.M.; Stern, M.C.; Giuliano, A.R.; Torres-Mejia, G.; et al. Associations between genetic variants in the tgf-beta signaling pathway and breast cancer risk among hispanic and non-hispanic white women. Breast Cancer Res. Treat. 2013, 141, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Z.; Cui, R.; He, H.; Lin, X.; Sheng, Y.; Zhang, H. Co-expression of parathyroid hormone related protein and tgf-beta in breast cancer predicts poor survival outcome. BMC Cancer 2015, 15, 925. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a tgf-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chang, Y.H.; Lin, W.Y.; Chen, W.C.; Chen, M.F. Tgf beta1 expression correlates with survival and tumor aggressiveness of prostate cancer. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S1587–S1593. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. Tgf-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-beta and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Kast, W.M. Effects of tgf-beta on the immune system: Implications for cancer immunotherapy. Leukemia 1999, 13, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.; Jobling, M.F.; Pajares, M.J.; Ravani, S.A.; Glick, A.B.; Lavin, M.J.; Koslov, S.; Shiloh, Y.; Barcellos-Hoff, M.H. Inhibition of transforming growth factor-beta 1 signaling attenuates ataxia telanglectasia mutated activity in response to genotoxic stress. Cancer Res. 2006, 66, 10861–10869. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Bouquet, S.; Lo, C.H.; Pellicciotta, I.; Bolourchi, S.; Parry, R.; Barcellos-Hoff, M.H. Attenuation of the DNA damage response by transforming growth factor-beta inhibitors enhances radiation sensitivity of non-small-cell lung cancer cells in vitro and in vivo. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the tgfbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Transforming growth factor-beta and the immune response to malignant disease. Clin. Cancer Res. 2007, 13, 6247–6251. [Google Scholar] [CrossRef] [PubMed]
- Furler, R.L.; Uittenbogaart, C.H. Gli2 regulates tgf-beta1 in human cd4+ t cells: Implications in cancer and hiv pathogenesis. PLoS ONE 2012, 7, e40874. [Google Scholar] [CrossRef] [PubMed]
- Strassmann, G.; Bertolini, D.R.; Eidelman, O. Inhibition of immune reactions in vivo by liposome associated transforming growth factor (tgf) type beta 1. Clin. Exp. Immunol. 1991, 86, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Li, M.O. Tgf-beta: Guardian of t cell function. J. Immunol. 2013, 191, 3973–3979. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, S.; Sha, H.; Liu, Z.; Yang, L.; Xue, Z.; Chen, H.; Qi, L. Emerging roles for xbp1, a super transcription factor. Gene Exp. 2010, 15, 13–25. [Google Scholar] [CrossRef]
- Fujita, T.; Teramoto, K.; Ozaki, Y.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Fujino, S.; Kontani, K.; Ogasawara, K. Inhibition of transforming growth factor-beta-mediated immunosuppression in tumor-draining lymph nodes augments antitumor responses by various immunologic cell types. Cancer Res. 2009, 69, 5142–5150. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed]
- Werner, F.; Jain, M.K.; Feinberg, M.W.; Sibinga, N.E.; Pellacani, A.; Wiesel, P.; Chin, M.T.; Topper, J.N.; Perrella, M.A.; Lee, M.E. Transforming growth factor-beta 1 inhibition of macrophage activation is mediated via smad3. J. Biol. Chem. 2000, 275, 36653–36658. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Che, D.; Yang, F.; Chi, C.; Meng, H.; Shen, J.; Qi, L.; Liu, F.; Lv, L.; Li, Y.; et al. Tumor-associated macrophages promote tumor metastasis via the tgf-beta/sox9 axis in non-small cell lung cancer. Oncotarget 2017, 8, 99801–99815. [Google Scholar] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Ahmed, R. Features of responding t cells in cancer and chronic infection. Curr. Opin. Immunol. 2010, 22, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yi, H.; Xia, X.P.; Zhao, Y. Transforming growth factor-beta: An important role in cd4+cd25+ regulatory t cells and immune tolerance. Autoimmunity 2006, 39, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Flavell, R.A. Tgf-beta and regulatory t cell in immunity and autoimmunity. J. Clin. Immunol. 2008, 28, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Perruche, S.; Li, J. Cd4+cd25+ t regulatory cells and tgf-beta in mucosal immune system: The good and the bad. Curr. Med. Chem. 2007, 14, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Theron, A.J.; Anderson, R.; Rossouw, T.M.; Steel, H.C. The role of transforming growth factor beta-1 in the progression of hiv/aids and development of non-aids-defining fibrotic disorders. Front. Immunol. 2017, 8, 1461. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Flavell, R.A. Regulatory t cells, transforming growth factor-beta, and immune suppression. Proc. Am. Thorac. Soc. 2007, 4, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, M. The tak1-traf6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. Traf6 mediates smad-independent activation of jnk and p38 by tgf-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, G.; Kishida, S.; Hiyama, A.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Ninomiya-Tsuji, J.; Matsumoto, K. Tab2, a novel adaptor protein, mediates activation of tak1 mapkkk by linking tak1 to traf6 in the il-1 signal transduction pathway. Mol. Cell 2000, 5, 649–658. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type i tgf-beta receptor engages traf6 to activate tak1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Iwata, H.; Masuyama, N.; Gotoh, Y.; Yamaguchi, K.; Irie, K.; Matsumoto, K.; Nishida, E.; Ueno, N. Role of tak1 and tab1 in bmp signaling in early xenopus development. EMBO J. 1998, 17, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nagai, S.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. Xiap, a cellular member of the inhibitor of apoptosis protein family, links the receptors to tab1-tak1 in the bmp signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. Traf6 ubiquitinates tgfbeta type i receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. The sumo guards for snail. Oncotarget 2017, 8, 97701–97702. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors snail and slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, X.; Yang, L.; Kisseleva, T.; Brenner, D.A.; Seki, E. Transcriptional repression of the transforming growth factor beta (tgf-beta) pseudoreceptor bmp and activin membrane-bound inhibitor (bambi) by nuclear factor kappab (nf-kappab) p50 enhances tgf-beta signaling in hepatic stellate cells. J. Biol. Chem. 2014, 289, 7082–7091. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ou, Z.; Chen, X.; Zu, X.; Liu, L.; Li, Y.; Cao, Z.; Chen, M.; Chen, Z.; Chen, H.; et al. Lps/tlr4 signaling enhances tgf-beta response through downregulating bambi during prostatic hyperplasia. Sci. Rep. 2016, 6, 27051. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V. Unraveling the mechanobiology of extracellular matrix. Annu. Rev. Physiol. 2018, 80, 353–387. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of yap/taz in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, E.; Nishina, H. Role of hippo-yap/taz signaling pathway in mechanotransduction. Clin. Calcium 2016, 26, 1751–1756. [Google Scholar] [PubMed]
- Mohri, Z.; Del Rio Hernandez, A.; Krams, R. The emerging role of yap/taz in mechanotransduction. J. Thorac. Dis. 2017, 9, E507–E509. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in fibrosis: Tgf-beta, wnt, and yap/taz converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, S.E.; Szymaniak, A.D.; Varelas, X. The transcriptional regulators taz and yap direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 2014, 289, 13461–13474. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, K.; Hu, Y.; Guo, X.; Wang, D.; Shi, W.; Li, J.; Song, J. Yap modulates tgf-beta1-induced simultaneous apoptosis and emt through upregulation of the egf receptor. Sci. Rep. 2017, 7, 45523. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. Yap/taz are mechanoregulators of tgf-beta-smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Mechanical control of tissue morphogenesis during embryological development. Int. J. Dev. Biol. 2006, 50, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Tensegrity ii. How structural networks influence cellular information processing networks. J. Cell Sci. 2003, 116, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Tensegrity i. Cell structure and hierarchical systems biology. J. Cell Sci. 2003, 116, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E.; Wang, N.; Stamenovic, D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep. Prog. Phys. 2014, 77, 046603. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; McCune, B.K.; Sporn, M.B. Tgf-beta: Regulation of extracellular matrix. Kidney Int. 1992, 41, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent tgf-beta-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Rys, J.P.; Monteiro, D.A.; Alliston, T. Mechanobiology of tgfbeta signaling in the skeleton. Matrix Biol. 2016, 52–54, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.S.; Jeong, K.W. Role of flightless-i (drosophila) homolog in the transcription activation of type i collagen gene mediated by transforming growth factor beta. Biochem. Biophs. Res. Commun. 2014, 454, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; D’Alfonso, T.M.; Shin, S.J.; Reinhart-King, C.A. Mechanobiology of tumor invasion: Engineering meets oncology. Crit. Rev. Oncol. Hematol. 2012, 83, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Makale, M. Cellular mechanobiology and cancer metastasis. Birth Defects Res. C Embryo Today 2007, 81, 329–343. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A.; Mercurio, A.M.; Stack, M.S.; Chapman, H.A.; Zutter, M.M.; Muschel, R.J.; Raz, A.; Matrisian, L.M.; Sloane, B.F.; Noel, A.; et al. Proteases, extracellular matrix, and cancer: A workshop of the path b study section. Am. J. Pathol. 2004, 164, 1131–1139. [Google Scholar] [CrossRef]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent tgf-beta1 for activation. J. Cell. Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, J.; Fan, Y.; Wang, Z.; Tian, R.; Ji, W.; Zhang, F.; Niu, R. Tgf-beta transactivates egfr and facilitates breast cancer migration and invasion through canonical smad3 and erk/sp1 signaling pathways. Mol. Oncol. 2018, 12, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Gao-Feng Xiong, R.X. Function of cancer cell-derived extracellular matrix in tumor progression. J. Cancer Metastasis Treat. 2016, 2, 357–364. [Google Scholar] [CrossRef]
- Geng, M.M.; Ellenrieder, V.; Wallrapp, C.; Muller-Pillasch, F.; Sommer, G.; Adler, G.; Gress, T.M. Use of representational difference analysis to study the effect of tgfb on the expression profile of a pancreatic cancer cell line. Genes Chrom. Cancer 1999, 26, 70–79. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.A.; Donato, D.M.; Balcioglu, H.E.; Schmidt, T.; Danen, E.H.J.; Koenderink, G.H. A guide to mechanobiology: Where biology and physics meet. BBA-Mol. Cell Res. 2015, 1853, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Altinay, S. Is extracellular matrix a castle against to invasion of cancer cells? Tumor Metastasis 2016, 23–42. [Google Scholar]
- Chiara Liverani, L.M.; Cristofolini, L.; Giordano, E.; Minardi, S.; della Porta, G.; de Vita, A.; Miserocchi, G.; Spadazzi, C.; Tasciotti, E.; Amadori, D.; et al. Investigating the mechanobiology of cancer cell-ecm interaction through collagen-based 3d scaffolds. Cell. Mol. Bioeng. 2017, 10, 223–234. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar] [PubMed]
- Campbell, B.H.; Agarwal, C.; Wang, J.H. Tgf-beta1, tgf-beta3, and pge(2) regulate contraction of human patellar tendon fibroblasts. Biomech. Model. Mechanobiol. 2004, 2, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Jinka, R.; Kapoor, R.; Sistla, P.G.; Raj, T.A.; Pande, G. Alterations in cell-extracellular matrix interactions during progression of cancers. Int. J. Cell Biol. 2012, 2012, 219196. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell. Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Ingber, D.E. Tensegrity and mechanoregulation: From skeleton to cytoskeleton. Osteoarthr. Cartil. 1999, 7, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Guizzardi, S.; Passeri, G.; Macaluso, G.M.; Scandroglio, R. Life on the wire: On tensegrity and force balance in cells. Acta Bio-Med. Atenei Parm. 2005, 76, 5–12. [Google Scholar]
- Ingber, D.E. Integrins, tensegrity, and mechanotransduction. Gravitational and space biology bulletin. Am. Soc. Gravit Space Biol. Bull. 1997, 10, 49–55. [Google Scholar]
- Volokh, K.Y. On tensegrity in cell mechanics. Mol. Cell. Biomech. 2011, 8, 195–214. [Google Scholar] [PubMed]
- Wall, M.; Butler, D.; El Haj, A.; Bodle, J.C.; Loboa, E.G.; Banes, A.J. Key developments that impacted the field of mechanobiology and mechanotransduction. J. Orthop. Res. 2018, 36, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.C.; Hsu, S.F.; Wu, C.L.; Li, T.M.; Kao, S.T.; Tsai, F.J.; Chen, W.C.; Liu, S.C.; Wu, C.M.; Tang, C.H. Transforming growth factor-beta1 increases cell migration and beta1 integrin up-regulation in human lung cancer cells. Lung Cancer 2009, 64, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-tgf-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Radisky, D.C. Extracellular matrix as a contextual determinant of transforming growth factor-beta signaling in epithelial-mesenchymal transition and in cancer. Cell Adhes. Migr. 2014, 8, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, D.; Unseld, M.; Prager, G.W. Integrins in the spotlight of cancer. Int. J. Mol. Sci. 2016, 17, 2037. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in tgfbeta activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Dynamic control of tgf-beta signaling and its links to the cytoskeleton. FEBS Lett. 2008, 582, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Stournaras, C. Regulation of actin organisation by tgf-beta in h-ras-transformed fibroblasts. J. Cell Sci. 1999, 112, 1169–1179. [Google Scholar] [PubMed]
- Edlund, S.; Landstrom, M.; Heldin, C.H.; Aspenstrom, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small gtpases cdc42 and rhoa. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a rhoa-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Rothballer, A.; Kutay, U. The diverse functional lincs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma 2013, 122, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Konde, E.; Bourgeois, B.; Tellier-Lebegue, C.; Wu, W.; Perez, J.; Caputo, S.; Attanda, W.; Gasparini, S.; Charbonnier, J.B.; Gilquin, B.; et al. Structural analysis of the smad2-man1 interaction that regulates transforming growth factor-beta signaling at the inner nuclear membrane. Biochemistry 2010, 49, 8020–8032. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; Voncken, J.W.; Kubben, N.; Broers, J.L.; Duisters, R.; van Leeuwen, R.E.; Crijns, H.J.; Ramaekers, F.C.; Hutchison, C.J.; Pinto, Y.M. A-type lamins are essential for tgf-beta1 induced pp2a to dephosphorylate transcription factors. Hum. Mol. Genet. 2005, 14, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Le Dour, C.; Wu, W.; Bereziat, V.; Capeau, J.; Vigouroux, C.; Worman, H.J. Extracellular matrix remodeling and transforming growth factor-beta signaling abnormalities induced by lamin a/c variants that cause lipodystrophy. J. Lipid Res. 2017, 58, 151–163. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.P.; Bult, C.J.; Cremer, C.; Grunze, M.; Knowles, B.B.; Langowski, J.; McNally, J.; Pederson, T.; Politz, J.C.; Pombo, A.; et al. Genome function and nuclear architecture: From gene expression to nanoscience. Genome. Res. 2003, 13, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Pederson, T. The nuclear physique. Int. Rev. Cell Mol. Biol. 2014, 307, 1–13. [Google Scholar] [PubMed]
- Pederson, T.; King, M.C.; Marko, J.F. Forces, fluctuations, and self-organization in the nucleus. Mol. Biol. Cell 2015, 26, 3915–3919. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-c: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Anzaldo, A.; Dent, M.A.; Martinez-Gomez, A. The higher-order structure in the cells nucleus as the structural basis of the post-mitotic state. Prog. Biophys. Mol. Biol. 2014, 114, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef] [PubMed]
- Gaarenstroom, T.; Hill, C.S. Tgf-beta signaling to chromatin: How smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 2014, 32, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y. DNA damage and apoptosis. Cell Death Differ. 2001, 8, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Cucinotta, F.A. New tricks for an old fox: Impact of tgfbeta on the DNA damage response and genomic stability. Sci. Signal. 2014, 7, re5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, M.R.; Kim, H.J.; An, Y.S.; Yi, J.Y. Tgf-beta1 accelerates the DNA damage response in epithelial cells via smad signaling. Biochem. Biophys. Res. Commun. 2016, 476, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Pertot, A.; Shirole, N.H.; Yao, Z.; Anaparthy, N.; Garvin, T.; Cox, H.; Chang, K.; Rollins, F.; Kendall, J.; et al. Tgf-beta reduces DNA ds-break repair mechanisms to heighten genetic diversity and adaptability of cd44+/cd24- cancer cells. eLife 2017, 6, e21615. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.; Inman, G.J. Crosstalk between p53 and tgf-beta signalling. J. Sig. Transduct. 2012, 2012, 294097. [Google Scholar]
- Centurione, L.; Aiello, F.B. Dna repair and cytokines: Tgf-beta, il-6, and thrombopoietin as different biomarkers of radioresistance. Front. Oncol. 2016, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Diaz, F.J.; Gascard, P.; Balakrishnan, S.K.; Zhao, J.X.; del Rincon, S.V.; Spruck, C.; Tlsty, T.D.; Emerson, B.M. Coordinate transcriptional and translational repression of p53 by tgf-beta 1 impairs the stress response. Mol. Cell 2013, 50, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. The extracellular matrix in cancer progression: Role of hyalectan proteoglycans and adamts enzymes. Cancer Lett. 2017, 385, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furler, R.L.; Nixon, D.F.; Brantner, C.A.; Popratiloff, A.; Uittenbogaart, C.H. TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction. Cancers 2018, 10, 199. https://doi.org/10.3390/cancers10060199
Furler RL, Nixon DF, Brantner CA, Popratiloff A, Uittenbogaart CH. TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction. Cancers. 2018; 10(6):199. https://doi.org/10.3390/cancers10060199
Chicago/Turabian StyleFurler, Robert L., Douglas F. Nixon, Christine A. Brantner, Anastas Popratiloff, and Christel H. Uittenbogaart. 2018. "TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction" Cancers 10, no. 6: 199. https://doi.org/10.3390/cancers10060199
APA StyleFurler, R. L., Nixon, D. F., Brantner, C. A., Popratiloff, A., & Uittenbogaart, C. H. (2018). TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction. Cancers, 10(6), 199. https://doi.org/10.3390/cancers10060199