A New Strategy to Control and Eradicate “Undruggable” Oncogenic K-RAS-Driven Pancreatic Cancer: Molecular Insights and Core Principles Learned from Developmental and Evolutionary Biology

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. As a Central Tumor-Driving Signaling Pathway, Oncogenic K-RAS Activation Remains Largely “Undruggable” in Human Cancer Despite Three Decades of Intense Investigation
3. Targeting and Shutting Down Oncogenic K-RAS Hyperactivation Is the “Holy Grail” in Cancer Biology and Cancer Therapy
4. The Challenges of Targeting K-RAS-Driven Malignant Pancreatic Tumors
5. Developmental Biology and Evolutionary Biology Are the Guiding Light in Cancer Biology
6. Evidence from Developmental Biology and Evolutionary Biology in Support of Cancer Biology
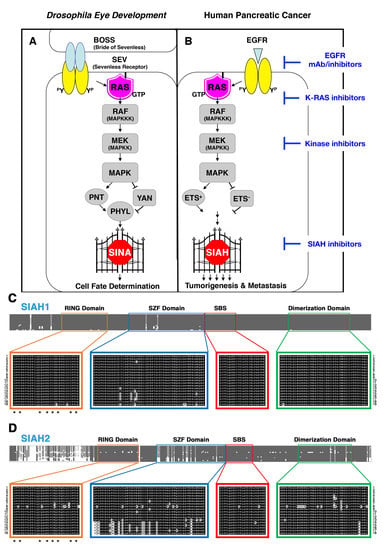
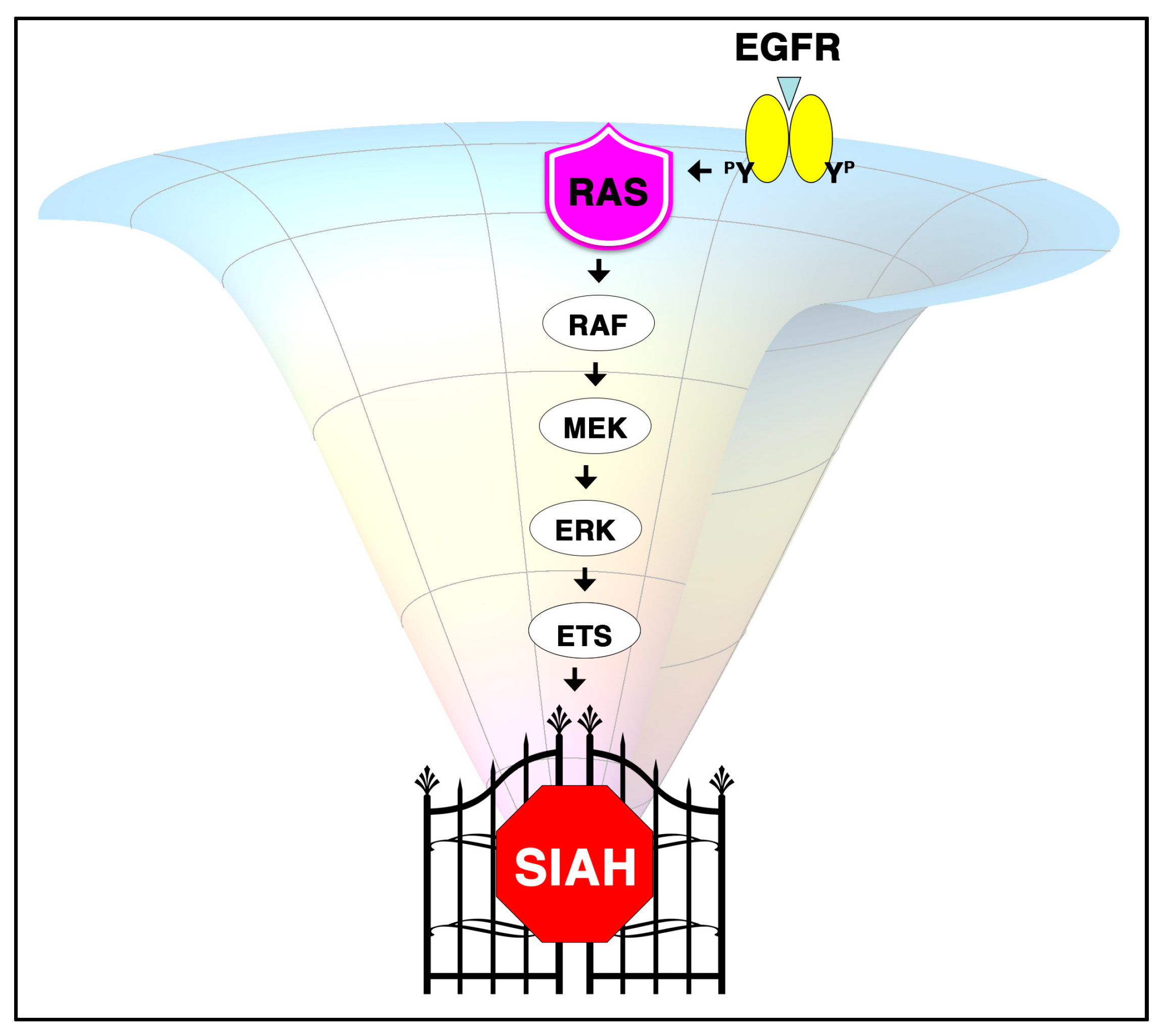
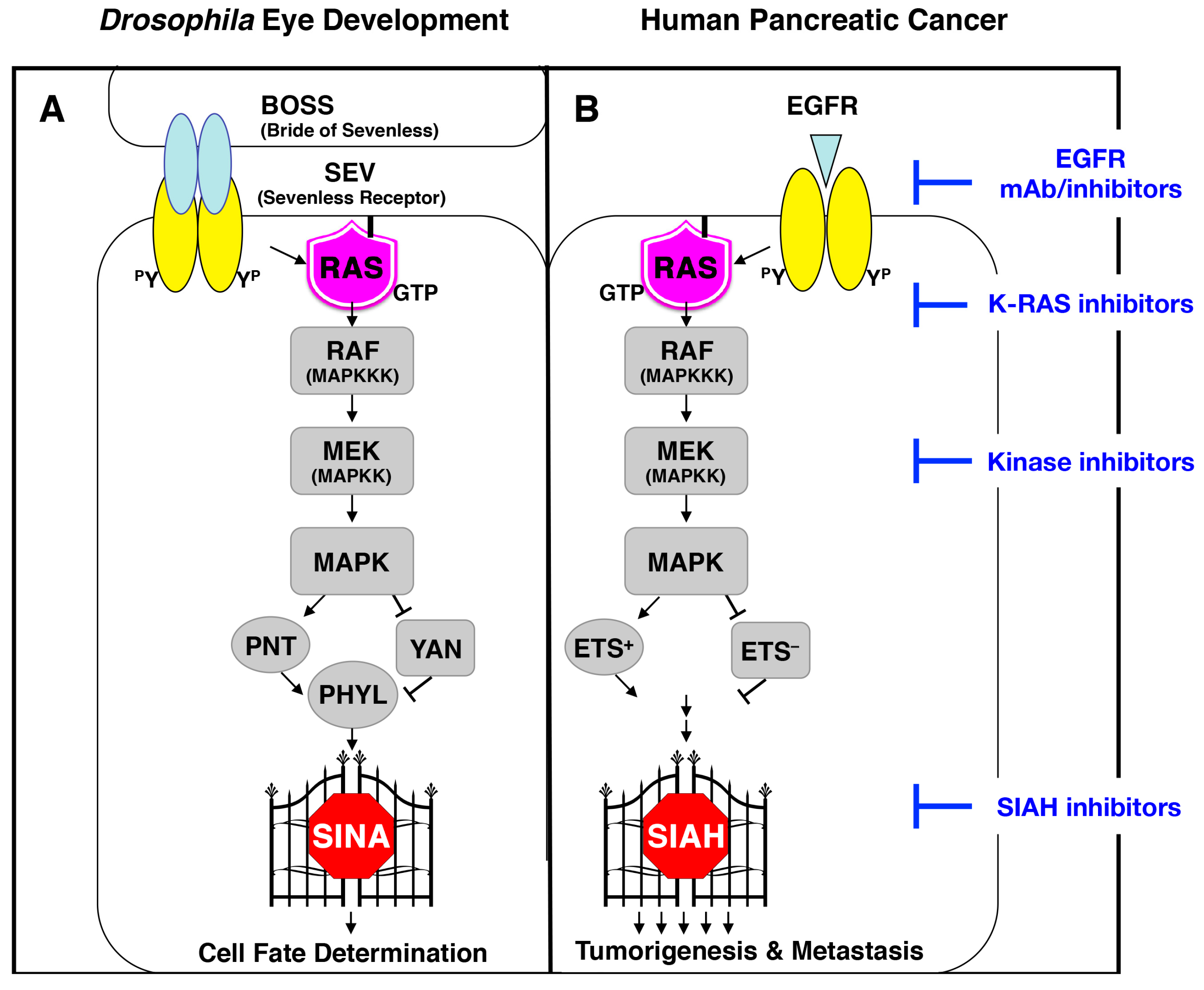
6.1. SIAH Is an Extraordinarily Conserved Signaling Module and the Most Downstream Signaling “Gatekeeper” Indispensable for Proper RAS Signal Transduction in Metazoan Species
6.2. SINA-Mediated Proteolysis Is the Most Downstream Signaling Gatekeeper in the RAS Pathway in Drosophila Eye Development
6.3. The K-RAS and SIAH Signaling Axis Is Highly Conserved in Cancer Biology
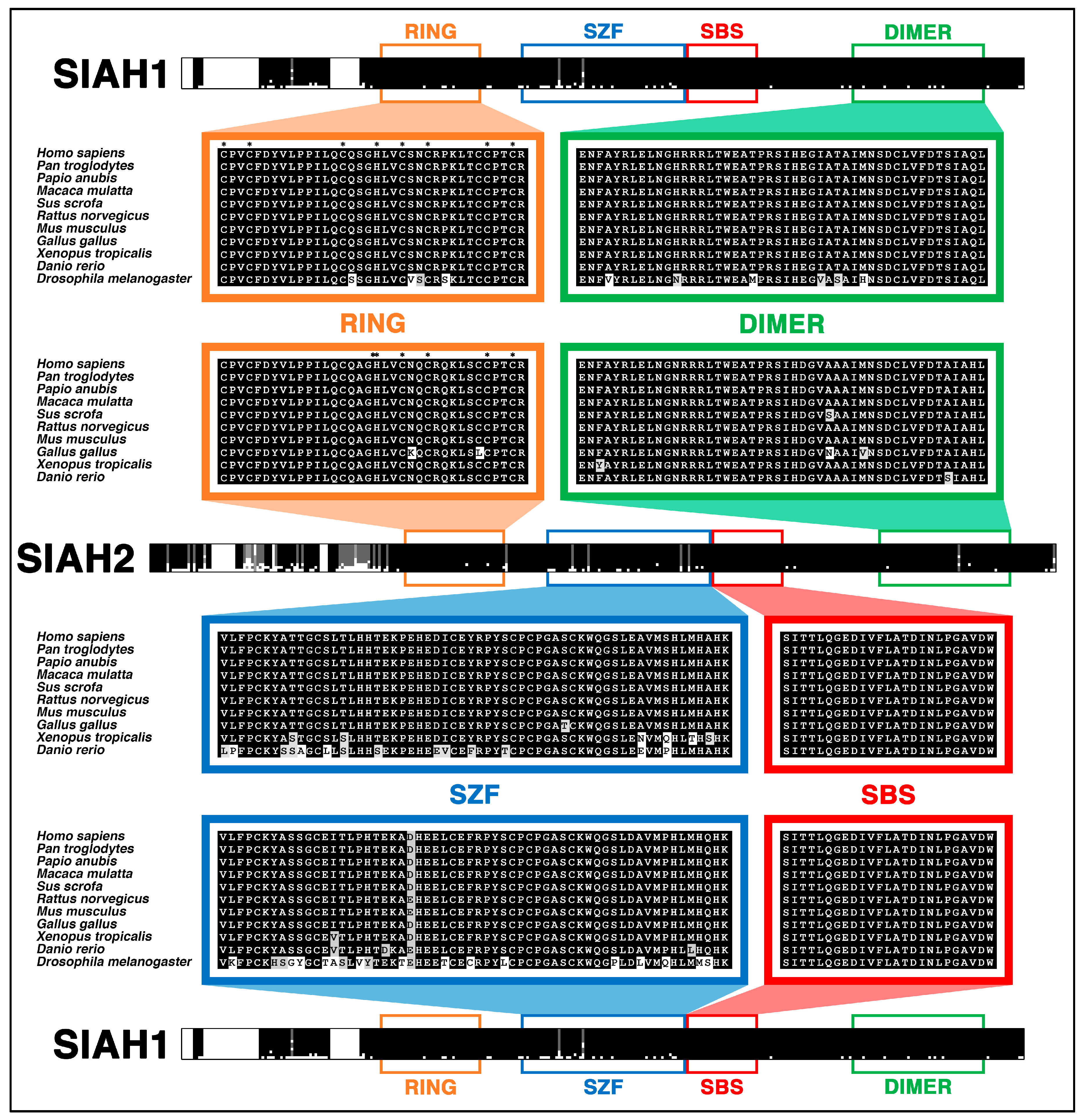
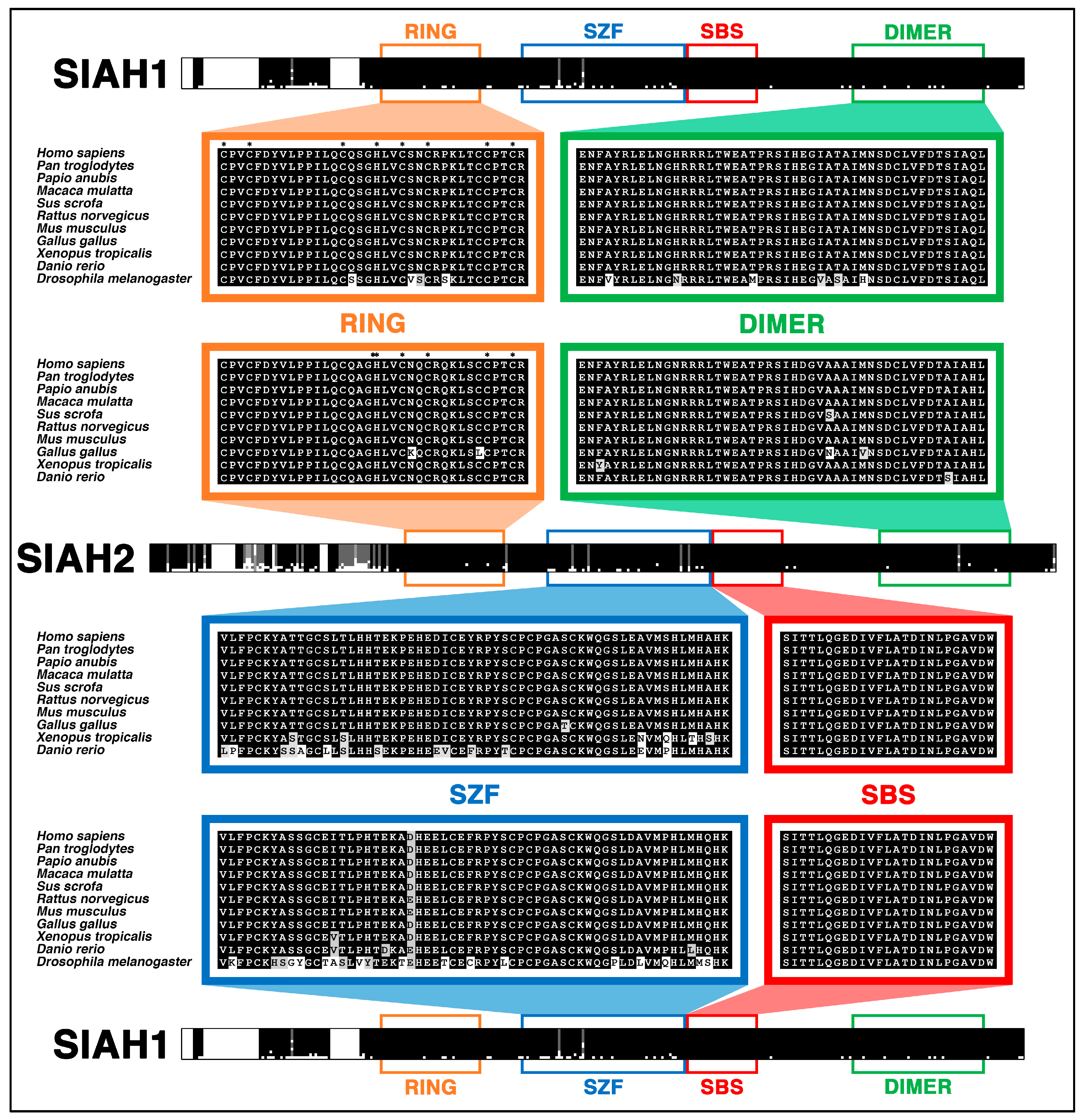
6.4. SIAH Is Extraordinarily and Evolutionarily Conserved in Metazoa
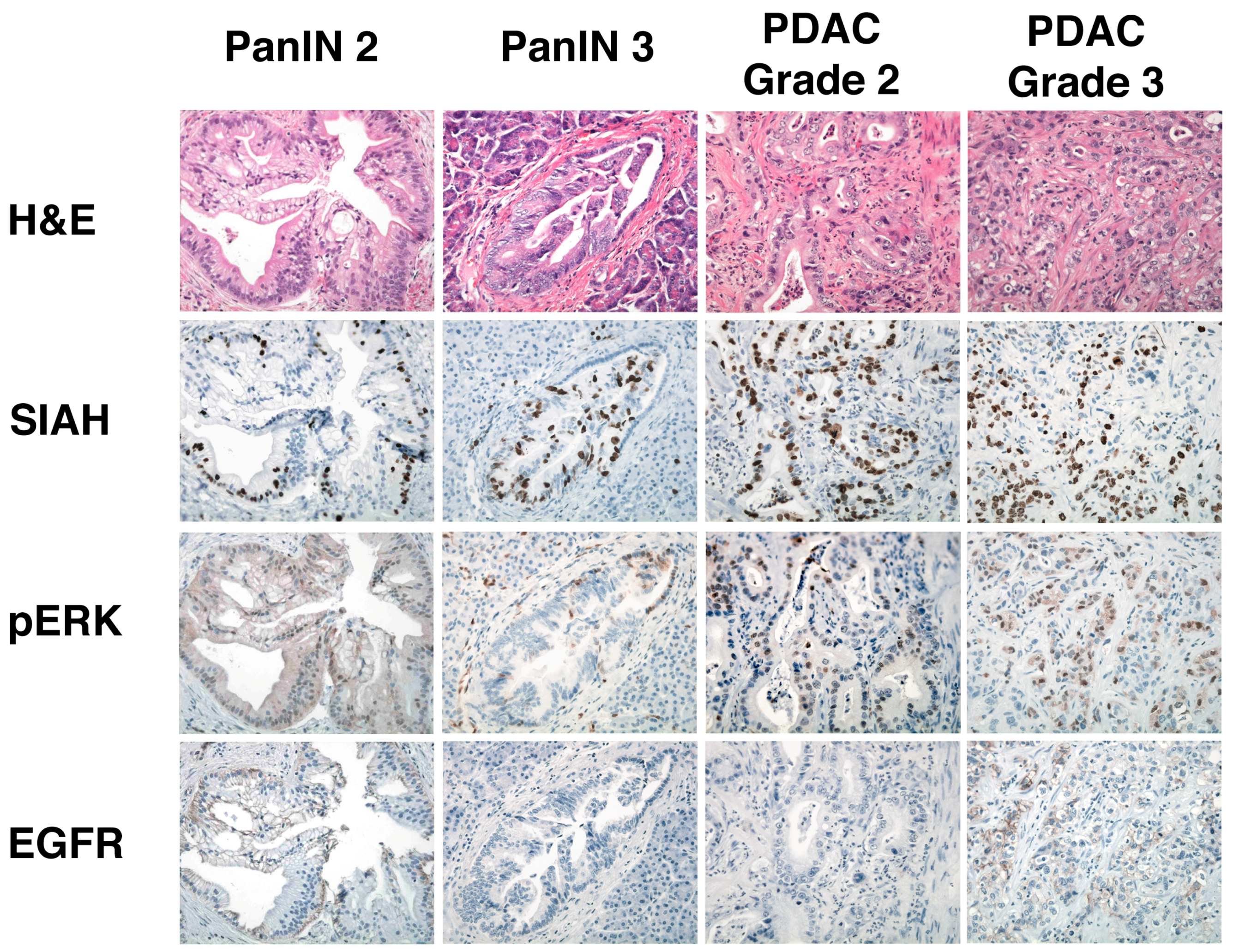
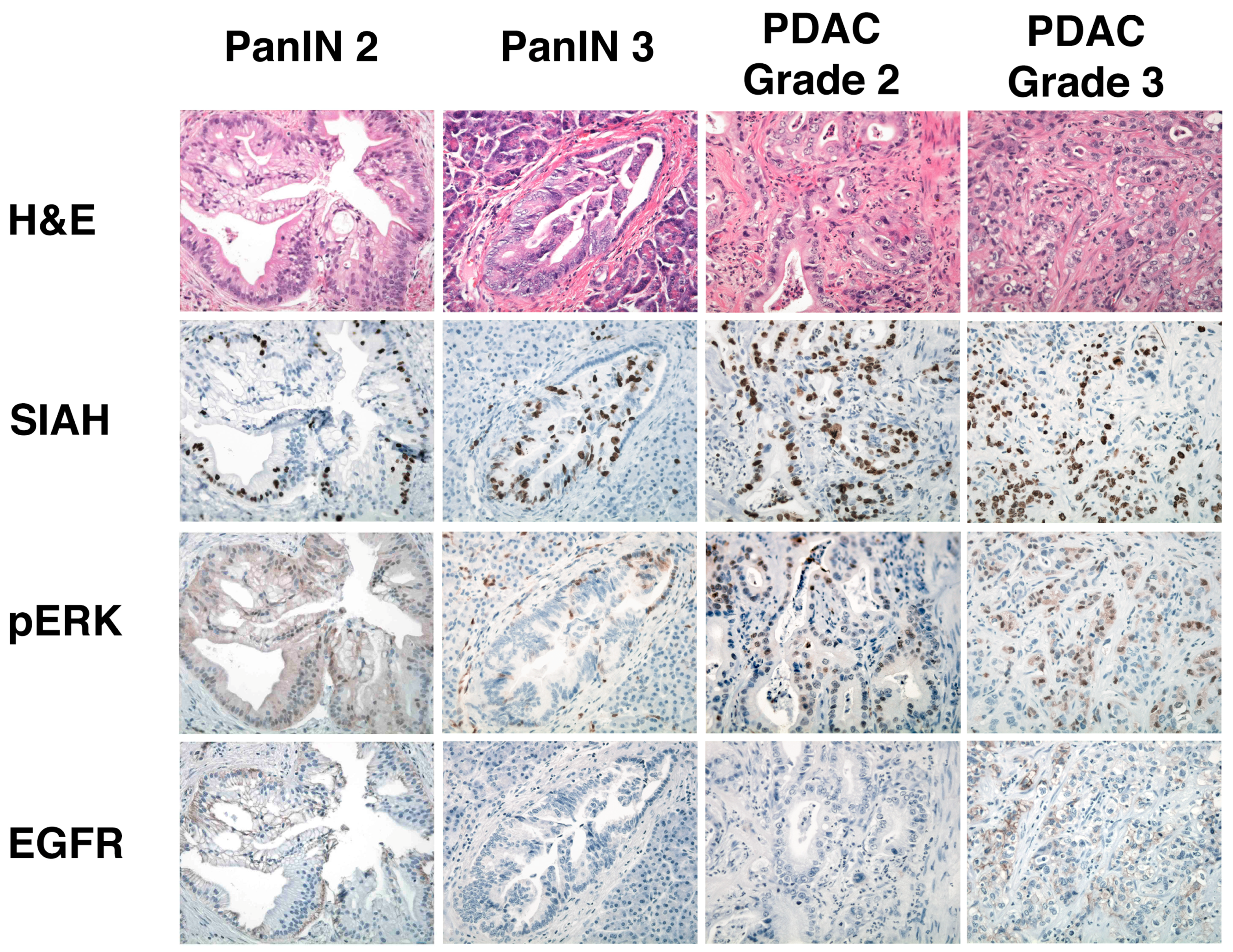
6.5. SIAH Is a Tumor-Specific Biomarker in Human Pancreatic Cancer
6.6. A New Strategy of Anti-SIAH-Based Anti-K-RAS Therapy in Pancreatic Cancer
7. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DIMER | the dimerization domain |
| EGFR | epidermal growth factor receptor |
| ERK | Extracellular signal-Regulated Kinase |
| ETS | E-Twenty-Six |
| FTI | farnesyltransferase inhibitors |
| HER2 | human epidermal growth factor receptor 2 |
| HHMI | Howard Hughes Medical Institute |
| IHC | Immunohistochemical |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | Mitogen-Activated Protein Kinase kinase |
| NCI | National Cancer Institute |
| NIH | National Institutes of Health |
| OS | overall survival |
| PanCAN | Pancreatic Cancer Action Network |
| PanIN | pancreatic intraepithelial neoplasias |
| PDAC | pancreatic ductal adenocarcinoma |
| PDEδ | phosphodiesterase δ |
| PDX | patient-derived xenograft |
| PFS | progression free survival |
| RAF | RAF serine/threonine kinase |
| RAS | rat sarcoma viral oncogene |
| RING | Really Interesting New Gene |
| RTK | Receptor Tyrosine Kinase |
| SBD | substrate-binding domain |
| SBS | substrate-binding site |
| SEV | Sevenless receptor, the Drosophila homolog of mammalian EGFR membrane receptor |
| SIAH | Seven-In-Absentia Homolog |
| SINA | Seven-In-Absentia |
| SOC | standard of care |
| SOS | Son of Sevenless |
| SZF | SIAH-type zinc finger |
| TME | Tumor Microenvironment |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; DePinho, R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed]
- Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Koniaris, L.; Kaushal, S.; Abrams, R.A.; Sauter, P.K.; Coleman, J.; Hruban, R.H.; Lillemoe, K.D. Resected adenocarcinoma of the pancreas-616 patients: Results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 2000, 4, 567–579. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Bockhorn, M.; Uzunoglu, F.G.; Adham, M.; Imrie, C.; Milicevic, M.; Sandberg, A.A.; Asbun, H.J.; Bassi, C.; Buchler, M.; Charnley, R.M.; et al. Borderline resectable pancreatic cancer: A consensus statement by the international study group of pancreatic surgery (ISGPS). Surgery 2014, 155, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Hank, T.; Hinz, U.; Bergmann, F.; Schneider, L.; Springfeld, C.; Jager, D.; Schirmacher, P.; Hackert, T.; Buchler, M.W. Pancreatic cancer surgery: The new r-status counts. Ann. Surg. 2017, 265, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Yeo, T.P.; Hruban, R.H.; Leach, S.D.; Wilentz, R.E.; Sohn, T.A.; Kern, S.E.; Iacobuzio-Donahue, C.A.; Maitra, A.; Goggins, M.; Canto, M.I.; et al. Pancreatic cancer. Curr. Probl. Cancer 2002, 26, 176–275. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.H.; Wang, H.; Fleming, J.B.; Sun, C.C.; Hwang, R.F.; Wolff, R.A.; Varadhachary, G.; Abbruzzese, J.L.; Crane, C.H.; Krishnan, S.; et al. Long-term survival after multidisciplinary management of resected pancreatic adenocarcinoma. Ann. Surg. Oncol. 2009, 16, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Mangu, P.B.; Berlin, J.; Engebretson, A.; Hong, T.S.; Maitra, A.; Mohile, S.G.; Mumber, M.; Schulick, R.; Shapiro, M.; et al. Potentially curable pancreatic cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2016, 34, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Rashid, O.M.; Pimiento, J.M.; Gamenthaler, A.W.; Nguyen, P.; Ha, T.T.; Hutchinson, T.; Springett, G.; Hoffe, S.; Shridhar, R.; Hodul, P.J.; et al. Outcomes of a clinical pathway for borderline resectable pancreatic cancer. Ann. Surg. Oncol. 2016, 23, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.; Combs, S.E.; Springfeld, C.; Hartwig, W.; Hackert, T.; Buchler, M.W. Advanced-stage pancreatic cancer: Therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Philip, P.A.; O’Reilly, E.M.; Uronis, H.E.; Ramanathan, R.K.; Crane, C.H.; Engebretson, A.; et al. Metastatic pancreatic cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2016, 34, 2784–2796. [Google Scholar] [CrossRef] [PubMed]
- Balaban, E.P.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Mukherjee, S.; Crane, C.H.; Javle, M.M.; Eads, J.R.; Allen, P.; Ko, A.H.; et al. Locally advanced, unresectable pancreatic cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2016, 34, 2654–2668. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Palmer, D.; Jackson, R.; Cox, T.; Neoptolemos, J.P.; Ghaneh, P.; Rawcliffe, C.L.; Bassi, C.; Stocken, D.D.; Cunningham, D.; et al. Optimal duration and timing of adjuvant chemotherapy after definitive surgery for ductal adenocarcinoma of the pancreas: Ongoing lessons from the espac-3 study. J. Clin. Oncol. 2014, 32, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs. observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Evans, D.B. Therapeutic advances in localized pancreatic cancer. JAMA Surg. 2016, 151, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Christians, K.K.; Heimler, J.W.; George, B.; Ritch, P.S.; Erickson, B.A.; Johnston, F.; Tolat, P.P.; Foley, W.D.; Evans, D.B.; Tsai, S. Survival of patients with resectable pancreatic cancer who received neoadjuvant therapy. Surgery 2016, 159, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Asare, E.A.; Evans, D.B.; Erickson, B.A.; Aburajab, M.; Tolat, P.; Tsai, S. Neoadjuvant treatment sequencing adds value to the care of patients with operable pancreatic cancer. J. Surg. Oncol. 2016, 114, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Matrisian, L.M.; Berlin, J.D. The past, present, and future of pancreatic cancer clinical trials. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e205–e215. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B.; Erickson, B.A.; Ritch, P. Borderline resectable pancreatic cancer: Definitions and the importance of multimodality therapy. Ann. Surg. Oncol. 2010, 17, 2803–2805. [Google Scholar] [CrossRef] [PubMed]
- Boeck, S.; Haas, M.; Ormanns, S.; Kruger, S.; Siveke, J.T.; Heinemann, V. Neoadjuvant chemotherapy in pancreatic cancer: Innovative, but still difficult. Br. J. Cancer 2014, 111, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B.; Ritch, P.S.; Erickson, B.A. Neoadjuvant therapy for localized pancreatic cancer: Support is growing? Ann. Surg. 2015, 261, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Hruban, R.H. Diagnosis: A step closer to screening for curable pancreatic cancer? Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A.; et al. The early detection of pancreatic cancer: What will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. 2014, 74, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Fleshman, J.M.; Matrisian, L.M.; Berlin, J.D. Evaluation of pancreatic cancer clinical trials and benchmarks for clinically meaningful future trials: A systematic review. JAMA Oncol. 2016, 2, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Joseph Bender, R.; Matrisian, L.M.; Rahib, L.; Hendifar, A.; Hoos, W.A.; Mikhail, S.; Chung, V.; Picozzi, V.; Heartwell, C.; et al. A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the know your tumor (KYT) initiative. Oncotarget 2017, 8, 83446–83456. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. Ras oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force matters: Biomechanical regulation of cell invasion and migration in disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Tuveson, D.A. Untangling the genetics from the epigenetics in pancreatic cancer metastasis. Nat. Genet. 2017, 49, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Jaffee, E.M. Immunotherapy for pancreatic cancer—Science driving clinical progress. Nat. Rev. Cancer 2005, 5, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.T.; Vonderheide, R.H. CD40 stimulation obviates innate sensors and drives t cell immunity in cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. CD47 blockade as another immune checkpoint therapy for cancer. Nat. Med. 2015, 21, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Bajor, D.L.; Winograd, R.; Evans, R.A.; Bayne, L.J.; Beatty, G.L. CD40 immunotherapy for pancreatic cancer. Cancer Immunol. Immunother. 2013, 62, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase i study of an agonist CD40 monoclonal antibody (cp-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; Silvers, M.A.; Ilcheva, M.; Liu, Y.; Moore, Z.R.; Luo, X.; Gao, J.; Anderson, G.; Liu, L.; Sarode, V.; et al. Tumor-selective use of DNA base excision repair inhibition in pancreatic cancer using the nqo1 bioactivatable drug, beta-lapachone. Sci. Rep. 2015, 5, 17066. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Huang, X.; Silvers, M.A.; Gerber, D.E.; Bolluyt, J.; Sarode, V.; Fattah, F.; Deberardinis, R.J.; Merritt, M.E.; Xie, X.J.; et al. Using a novel NQO1 bioactivatable drug, β-Lapachone (arq761), to enhance chemotherapeutic effects by metabolic modulation in pancreatic cancer. J. Surg. Oncol. 2017, 116, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M. Pancreatic adenocarcinoma: The emperor of all cancer maladies. J. Oncol. Pract. 2016, 12, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. Ras genes. Annu. Rev. Biochem. 1987, 56, 779–827. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable ras: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting ras signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Van Sciver, R.E.; Njogu, M.M.; Isbell, A.J.; Odanga, J.J.; Bian, M.; Svyatova, E.; van Reesema, L.L.S.; Zheleva, V.; Eisner, J.L.; Bruflat, J.K.; et al. Blocking siah proteolysis, an important K-RAS vulnerability, to control and eradicate K-RAS-driven metastatic cancer. In Conquering RAS: From Biology to Cancer Therapy; Azmi, A.S., Ed.; Elsevier/AP (Academic Press): Amsterdam, The Netherlands; Boston, MA, USA, 2016. [Google Scholar]
- Pepper, I.J.; Van Sciver, R.E.; Tang, A.H. Phylogenetic analysis of the SINA/SIAH ubiquitin E3 ligase family in metazoa. BMC Evol. Biol. 2017, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Willumsen, B.M. Function and regulation of ras. Annu. Rev. Biochem. 1993, 62, 851–891. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging RAS back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. Kras as a therapeutic target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. Kras alleles: The devil is in the detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. Ras proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.A.; Xu, T. A genetic screen in drosophila for metastatic behavior. Science 2003, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Giehl, K. Oncogenic ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Koop, S.; Schmidt, E.E.; MacDonald, I.C.; Morris, V.L.; Khokha, R.; Grattan, M.; Leone, J.; Chambers, A.F.; Groom, A.C. Independence of metastatic ability and extravasation: Metastatic RAS-transformed and control fibroblasts extravasate equally well. Proc. Natl. Acad. Sci. USA 1996, 93, 11080–11084. [Google Scholar] [CrossRef] [PubMed]
- Varghese, H.J.; Davidson, M.T.; MacDonald, I.C.; Wilson, S.M.; Nadkarni, K.V.; Groom, A.C.; Chambers, A.F. Activated RAS regulates the proliferation/apoptosis balance and early survival of developing micrometastases. Cancer Res. 2002, 62, 887–891. [Google Scholar] [PubMed]
- Gibbs, J.B.; Oliff, A.; Kohl, N.E. Farnesyltransferase inhibitors: RAS research yields a potential cancer therapeutic. Cell 1994, 77, 175–178. [Google Scholar] [CrossRef]
- Kohl, N.E.; Wilson, F.R.; Mosser, S.D.; Giuliani, E.; deSolms, S.J.; Conner, M.W.; Anthony, N.J.; Holtz, W.J.; Gomez, R.P.; Lee, T.J.; et al. Protein farnesyltransferase inhibitors block the growth of RAS-dependent tumors in nude mice. Proc. Natl. Acad. Sci. USA 1994, 91, 9141–9145. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B.; Graham, S.L.; Hartman, G.D.; Koblan, K.S.; Kohl, N.E.; Omer, C.A.; Oliff, A. Farnesyltransferase inhibitors versus ras inhibitors. Curr. Opin. Chem. Biol. 1997, 1, 197–203. [Google Scholar] [CrossRef]
- Caponigro, F.; Casale, M.; Bryce, J. Farnesyl transferase inhibitors in clinical development. Expert Opin. Investig. Drugs 2003, 12, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of RAS for cancer treatment: The search continues. Future Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef] [PubMed]
- Appels, N.M.; Beijnen, J.H.; Schellens, J.H. Development of farnesyl transferase inhibitors: A review. Oncologist 2005, 10, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martin-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as pdedelta inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhuang, C.; Lu, J.; Jiang, Y.; Sheng, C. Discovery of novel kras-pdedelta inhibitors by fragment-based drug design. J. Med. Chem. 2018, 61, 2604–2610. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Wittinghofer, A.; Der, C.J. Selective targeting of the KRAS g12c mutant: Kicking KRAS when it’s down. Cancer Cell 2016, 29, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-RAS(g12c) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-RAS g12c. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic targeting of oncogenic K-RAS by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 2014, 53, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS g12c by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Westover, K.D.; Janne, P.A.; Gray, N.S. Progress on covalent inhibition of KRAS(g12c). Cancer Discov. 2016, 6, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.P.; Jeganathan, S.; Heidrich, A.; Campos, J.; Goody, R.S. Nucleotide based covalent inhibitors of kras can only be efficient in vivo if they bind reversibly with GTP-like affinity. Sci. Rep. 2017, 7, 3687. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent g12c-specific inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. Ras isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Edkins, S.; O’Meara, S.; Parker, A.; Stevens, C.; Reis, M.; Jones, S.; Greenman, C.; Davies, H.; Dalgliesh, G.; Forbes, S.; et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 2006, 5, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Lochhead, P.; Yamauchi, M.; Kuchiba, A.; Qian, Z.R.; Liao, X.; Nishihara, R.; Jung, S.; Wu, K.; Nosho, K.; et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: Cohort study and literature review. Mol. Cancer 2014, 13, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, F. K-ras protein as a drug target. J. Mol. Med. 2016, 94, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Niederhuber, J.E. Developmental biology, self-renewal, and cancer. Lancet Oncol. 2007, 8, 456–457. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Beedle, A.; Hanson, M.A. Principles of Evolutionary Medicine; Oxford University Press: Oxford, UK; New York, NY, USA, 2009; 296p. [Google Scholar]
- Edwards, P.A. The impact of developmental biology on cancer research: An overview. Cancer Metastasis Rev. 1999, 18, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Abbruzzese, J.L. Developmental biology informs cancer: The emerging role of the hedgehog signaling pathway in upper gastrointestinal cancers. Cancer Cell 2003, 4, 245–247. [Google Scholar] [CrossRef]
- Frank, S.A.; Nowak, M.A. Cell biology: Developmental predisposition to cancer. Nature 2003, 422, 494. [Google Scholar] [CrossRef] [PubMed]
- Pepper, J.W.; Scott Findlay, C.; Kassen, R.; Spencer, S.L.; Maley, C.C. Cancer research meets evolutionary biology. Evol. Appl. 2009, 2, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, I.; Rubin, G.M. Making a difference: The role of cell-cell interactions in establishing separate identities for equivalent cells. Cell 1992, 68, 271–281. [Google Scholar] [CrossRef]
- Rubin, G.M.; Chang, H.C.; Karim, F.; Laverty, T.; Michaud, N.R.; Morrison, D.K.; Rebay, I.; Tang, A.; Therrien, M.; Wassarman, D.A. Signal transduction downstream from RAS in drosophila. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; Volume 62, pp. 347–352. [Google Scholar]
- Zipursky, S.L.; Rubin, G.M. Determination of neuronal cell fate: Lessons from the R7 neuron of drosophila. Annu. Rev. Neurosci. 1994, 17, 373–397. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F. Epistasis and quantitative traits: Using model organisms to study gene-gene interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Sackton, T.B.; Hartl, D.L. Genotypic context and epistasis in individuals and populations. Cell 2016, 166, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Rubin, G.M. Seven in absentia, a gene required for specification of R7 cell fate in the drosophila eye. Cell 1990, 63, 561–577. [Google Scholar] [CrossRef]
- Dickson, B.; Hafen, E. Genetics of signal transduction in invertebrates. Curr. Opin. Genet. Dev. 1994, 4, 64–70. [Google Scholar] [CrossRef]
- Fortini, M.E.; Simon, M.A.; Rubin, G.M. Signaling by the sevenless protein tyrosine kinase is mimicked by RAS1 activation. Nature 1992, 355, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.A.; Bowtell, D.D.; Dodson, G.S.; Laverty, T.R.; Rubin, G.M. RAS1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 1991, 67, 701–716. [Google Scholar] [CrossRef]
- Wassarman, D.A.; Therrien, M.; Rubin, G.M. The RAS signaling pathway in drosophila. Curr. Opin. Genet. Dev. 1995, 5, 44–50. [Google Scholar] [CrossRef]
- Raabe, T. The sevenless signaling pathway: Variations of a common theme. Biochim. Biophys. Acta 2000, 1496, 151–163. [Google Scholar] [CrossRef]
- Schmidt, R.L.; Park, C.H.; Ahmed, A.U.; Gundelach, J.H.; Reed, N.R.; Cheng, S.; Knudsen, B.E.; Tang, A.H. Inhibition of RAS-mediated transformation and tumorigenesis by targeting the downstream E3 ubiquitin ligase seven in absentia homologue. Cancer Res. 2007, 67, 11798–11810. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Schmidt, R.L.; Park, C.H.; Reed, N.R.; Hesse, S.E.; Thomas, C.F.; Molina, J.R.; Deschamps, C.; Yang, P.; Aubry, M.C.; et al. Effect of disrupting seven-in-absentia homolog 2 function on lung cancer cell growth. J. Natl. Cancer Inst. 2008, 100, 1606–1629. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The RAS protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Slack, C. Ras signaling in aging and metabolic regulation. Nutr. Healthy Aging 2017, 4, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, R.; Zhang, Q.; Chen, X.; Qian, Y.; Fang, W. The diversification of evolutionarily conserved mapk cascades correlates with the evolution of fungal species and development of lifestyles. Genome Biol. Evol. 2016, 9, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.A.; Bowtell, D.D.; Rubin, G.M. Structure and activity of the sevenless protein: A protein tyrosine kinase receptor required for photoreceptor development in drosophila. Proc. Natl. Acad. Sci. USA 1989, 86, 8333–8337. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.; Sprenger, F.; Morrison, D.; Hafen, E. Raf functions downstream of RAS1 in the sevenless signal transduction pathway. Nature 1992, 360, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Brunner, D.; Oellers, N.; Szabad, J.; Biggs, W.H., III; Zipursky, S.L.; Hafen, E. A gain-of-function mutation in drosophila map kinase activates multiple receptor tyrosine kinase signaling pathways. Cell 1994, 76, 875–888. [Google Scholar] [CrossRef]
- Biggs, W.H., III; Zavitz, K.H.; Dickson, B.; van der Straten, A.; Brunner, D.; Hafen, E.; Zipursky, S.L. The drosophila rolled locus encodes a map kinase required in the sevenless signal transduction pathway. EMBO J. 1994, 13, 1628–1635. [Google Scholar] [PubMed]
- Tang, A.H.; Neufeld, T.P.; Kwan, E.; Rubin, G.M. PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism. Cell 1997, 90, 459–467. [Google Scholar] [CrossRef]
- Hu, G.; Chung, Y.L.; Glover, T.; Valentine, V.; Look, A.T.; Fearon, E.R. Characterization of human homologs of the drosophila seven in absentia (SINA) gene. Genomics 1997, 46, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Z.; Hou, F.; Harding, R.; Huang, X.; Dong, A.; Walker, J.R.; Tong, Y. The substrate binding domains of human SIAH E3 ubiquitin ligases are now crystal clear. Biochim. Biophys. Acta 2017, 1861, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Xie, C.; Xie, W.; Wu, Z.; Wu, M. SIAH-1s, a novel splice variant of SIAH-1 (seven in absentia homolog), counteracts SIAH-1-mediated downregulation of β-catenin. Oncogene 2007, 26, 6319–6331. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; House, C.M.; Traficante, N.; Mackay, J.P.; Relaix, F.; Sassoon, D.A.; Parker, M.W.; Bowtell, D.D. SIAH ubiquitin ligase is structurally related to TRAF and modulates TNF-α signaling. Nat. Struct. Biol. 2002, 9, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, S.; Li, C.; Ni, C.Z.; Takayama, S.; Reed, J.C.; Ely, K.R. Structural analysis of SIAH1 and its interactions with siah-interacting protein (SIP). J. Biol. Chem. 2003, 278, 1837–1840. [Google Scholar] [CrossRef] [PubMed]
- House, C.M.; Hancock, N.C.; Moller, A.; Cromer, B.A.; Fedorov, V.; Bowtell, D.D.; Parker, M.W.; Polekhina, G. Elucidation of the substrate binding site of siah ubiquitin ligase. Structure 2006, 14, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Fearon, E.R. Siah-1 n-terminal ring domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Mol. Cell. Biol. 1999, 19, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Ely, K.R. Degrading liaisons: SIAH structure revealed. Nat. Struct. Biol. 2002, 9, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Santelli, E.; Leone, M.; Li, C.; Fukushima, T.; Preece, N.E.; Olson, A.J.; Ely, K.R.; Reed, J.C.; Pellecchia, M.; Liddington, R.C.; et al. Structural analysis of SIAH1-SIAH-interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. J. Biol. Chem. 2005, 280, 34278–34287. [Google Scholar] [CrossRef] [PubMed]
- Depaux, A.; Regnier-Ricard, F.; Germani, A.; Varin-Blank, N. Dimerization of hsiah proteins regulates their stability. Biochem. Biophys. Res. Commun. 2006, 348, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; House, C.M.; Wong, C.S.; Scanlon, D.B.; Liu, M.C.; Ronai, Z.; Bowtell, D.D. Inhibition of SIAH ubiquitin ligase function. Oncogene 2009, 28, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Kim, H.; Scortegagna, M.; Ronai, Z.A. Regulators and effectors of siah ubiquitin ligases. Cell Biochem. Biophys. 2013, 67, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, R.J.; Toher, J.L.; Yang, X.; Kovalenko, D.; Friesel, R. Regulation of sprouty2 stability by mammalian seven-in-absentia homolog 2. J. Cell. Biochem. 2007, 100, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Sutterluty, H.; Mayer, C.E.; Setinek, U.; Attems, J.; Ovtcharov, S.; Mikula, M.; Mikulits, W.; Micksche, M.; Berger, W. Down-regulation of sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol. Cancer Res. 2007, 5, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, L.J.; Bar-Sagi, D. Spatial regulation of egfr signaling by sprouty2. Curr. Biol. 2007, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Meissner, A.; Dowdle, J.A.; Crowley, D.; Magendantz, M.; Ouyang, C.; Parisi, T.; Rajagopal, J.; Blank, L.J.; Bronson, R.T.; et al. Sprouty-2 regulates oncogenic K-RAS in lung development and tumorigenesis. Genes Dev. 2007, 21, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, G.J.; Vogtlin, A.; Castro, A.; Ferby, I.; Salvagiotto, G.; Ronai, Z.; Lorca, T.; Nebreda, A.R. Meiotic regulation of the cdk activator ringo/speedy by ubiquitin-proteasome-mediated processing and degradation. Nat. Cell Biol. 2006, 8, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Habelhah, H.; Frew, I.J.; Laine, A.; Janes, P.W.; Relaix, F.; Sassoon, D.; Bowtell, D.D.; Ronai, Z. Stress-induced decrease in TRAF2 stability is mediated by SIAH2. EMBO J. 2002, 21, 5756–5765. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. SIAH2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, S.I.; Reed, J.C. SIAH-1, SIP, and EBI collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol. Cell 2001, 7, 915–926. [Google Scholar] [CrossRef]
- Susini, L.; Passer, B.J.; Amzallag-Elbaz, N.; Juven-Gershon, T.; Prieur, S.; Privat, N.; Tuynder, M.; Gendron, M.C.; Israel, A.; Amson, R.; et al. SIAH-1 binds and regulates the function of numb. Proc. Natl. Acad. Sci. USA 2001, 98, 15067–15072. [Google Scholar] [CrossRef] [PubMed]
- Behling, K.C.; Tang, A.; Freydin, B.; Chervoneva, I.; Kadakia, S.; Schwartz, G.F.; Rui, H.; Witkiewicz, A.K. Increased SIAH expression predicts ductal carcinoma in situ (DCIS) progression to invasive carcinoma. Breast Cancer Res. Treat. 2011, 129, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Van Reesema, L.L.; Zheleva, V.; Winston, J.S.; Jansen, R.J.; O’Connor, C.F.; Isbell, A.J.; Bian, M.; Qin, R.; Bassett, P.T.; Hinson, V.J.; et al. SIAH and EGFR, two ras pathway biomarkers, are highly prognostic in locally advanced and metastatic breast cancer. EBioMedicine 2016, 11, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zhang, S.; Vidal, M.; Baer, J.L.; Xu, T.; Fearon, E.R. Mammalian homologs of seven in absentia regulate DCC via the ubiquitin-proteasome pathway. Genes Dev. 1997, 11, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Smyrk, T.C.; Reed, N.R.; Schmidt, R.L.; Schnelldorfer, T.; Chari, S.T.; Petersen, G.M.; Tang, A.H. Combining clinicopathological predictors and molecular biomarkers in the oncogenic K-RAS/KI67/HIF-1α pathway to predict survival in resectable pancreatic cancer. Br. J. Cancer 2015, 112, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sheung, J.; Dong, G.; Coquilla, C.; Daniel-Issakani, S.; Payan, D.G. High-throughput screening for inhibitors of the E3 ubiquitin ligase APC. Methods Enzymol. 2005, 399, 740–754. [Google Scholar] [PubMed]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of MDM2-MDMX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, N.; Aratani, S.; Leach, C.; Amano, T.; Yamano, Y.; Nakatani, K.; Nishioka, K.; Nakajima, T. Ring-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int. J. Mol. Med. 2012, 30, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Rotblat, B.; Ansell, K.; Amelio, I.; Caraglia, M.; Misso, G.; Bernassola, F.; Cavasotto, C.N.; Knight, R.A.; Ciechanover, A.; et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014, 5, e1203. [Google Scholar] [CrossRef] [PubMed]
- Landre, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Fabius, A.W.; Mullenders, J.; Madiredjo, M.; Velds, A.; Kerkhoven, R.M.; Bernards, R.; Beijersbergen, R.L. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat. Chem. Biol. 2006, 2, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Dickens, M.P.; Fitzgerald, R.; Fischer, P.M. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin. Cancer Biol. 2010, 20, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Philip, P.A.; Aboukameel, A.; Wang, Z.; Banerjee, S.; Zafar, S.F.; Goustin, A.S.; Almhanna, K.; Yang, D.; Sarkar, F.H.; et al. Reactivation of p53 by novel MDM2 inhibitors: Implications for pancreatic cancer therapy. Curr. Cancer Drug Targets 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.K.; Fu, H.; Zhou, M. Discovery of dual inhibitors of MDM2 and xiap for cancer treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Vasilena, Z.; Minglei, B.; Justin, J.O.; Monicah, N.; Bruce, E.K.; Amy, H.T. Inhibition of Well-Established Pancreatic and Triple Negative Breast Tumor Growth by Blocking the Most Downstream Signaling Module, SIAH, in the Oncogenic ERBB/K-RAS Signaling Pathway. 2018; in preparation. [Google Scholar]
- Qi, J.; Nakayama, K.; Gaitonde, S.; Goydos, J.S.; Krajewski, S.; Eroshkin, A.; Bar-Sagi, D.; Bowtell, D.; Ronai, Z. The ubiquitin ligase SIAH2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 16713–16718. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Sciver, R.E.; Lee, M.P.; Lee, C.D.; Lafever, A.C.; Svyatova, E.; Kanda, K.; Collier, A.L.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Zheleva, V.; et al. A New Strategy to Control and Eradicate “Undruggable” Oncogenic K-RAS-Driven Pancreatic Cancer: Molecular Insights and Core Principles Learned from Developmental and Evolutionary Biology. Cancers 2018, 10, 142. https://doi.org/10.3390/cancers10050142
Van Sciver RE, Lee MP, Lee CD, Lafever AC, Svyatova E, Kanda K, Collier AL, Siewertsz van Reesema LL, Tang-Tan AM, Zheleva V, et al. A New Strategy to Control and Eradicate “Undruggable” Oncogenic K-RAS-Driven Pancreatic Cancer: Molecular Insights and Core Principles Learned from Developmental and Evolutionary Biology. Cancers. 2018; 10(5):142. https://doi.org/10.3390/cancers10050142
Chicago/Turabian StyleVan Sciver, Robert E., Michael P. Lee, Caroline Dasom Lee, Alex C. Lafever, Elizaveta Svyatova, Kevin Kanda, Amber L. Collier, Lauren L. Siewertsz van Reesema, Angela M. Tang-Tan, Vasilena Zheleva, and et al. 2018. "A New Strategy to Control and Eradicate “Undruggable” Oncogenic K-RAS-Driven Pancreatic Cancer: Molecular Insights and Core Principles Learned from Developmental and Evolutionary Biology" Cancers 10, no. 5: 142. https://doi.org/10.3390/cancers10050142
APA StyleVan Sciver, R. E., Lee, M. P., Lee, C. D., Lafever, A. C., Svyatova, E., Kanda, K., Collier, A. L., Siewertsz van Reesema, L. L., Tang-Tan, A. M., Zheleva, V., Bwayi, M. N., Bian, M., Schmidt, R. L., Matrisian, L. M., Petersen, G. M., & Tang, A. H. (2018). A New Strategy to Control and Eradicate “Undruggable” Oncogenic K-RAS-Driven Pancreatic Cancer: Molecular Insights and Core Principles Learned from Developmental and Evolutionary Biology. Cancers, 10(5), 142. https://doi.org/10.3390/cancers10050142