Bladder Cancer: New Insights into Its Molecular Pathology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
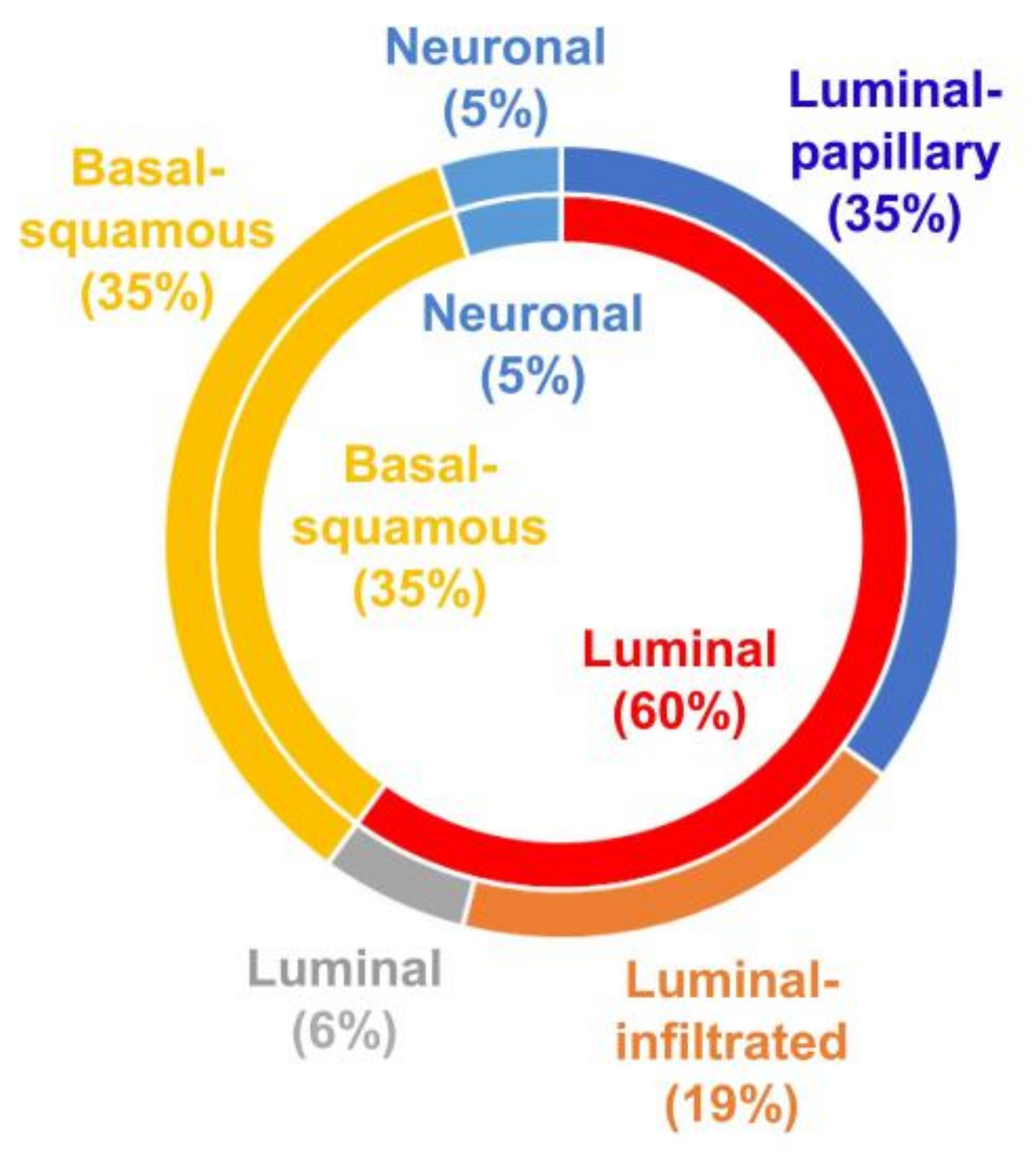{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Biomarkers and Pathways
2.1. Mutation Spectrum
2.2. FGFR3/RAS Pathway
2.3. PIK3/AKT/MTOR Pathway
2.4. TP53/RB1 Pathway
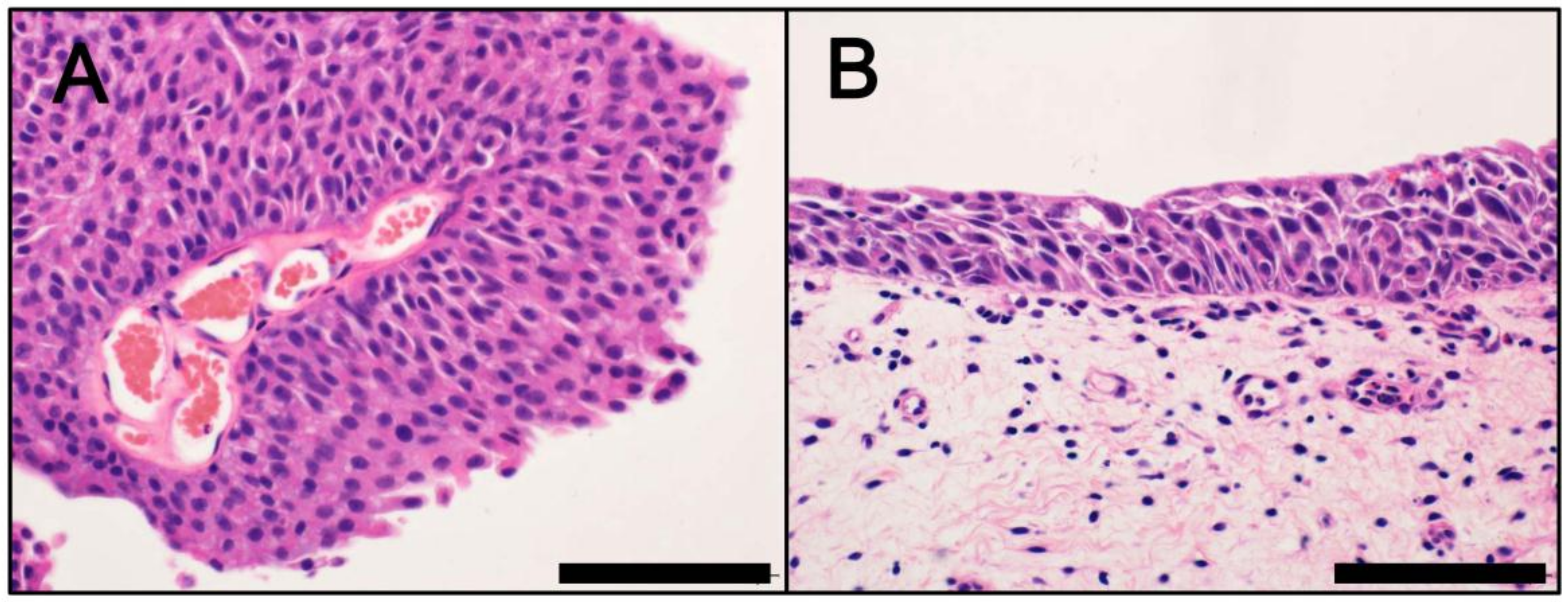
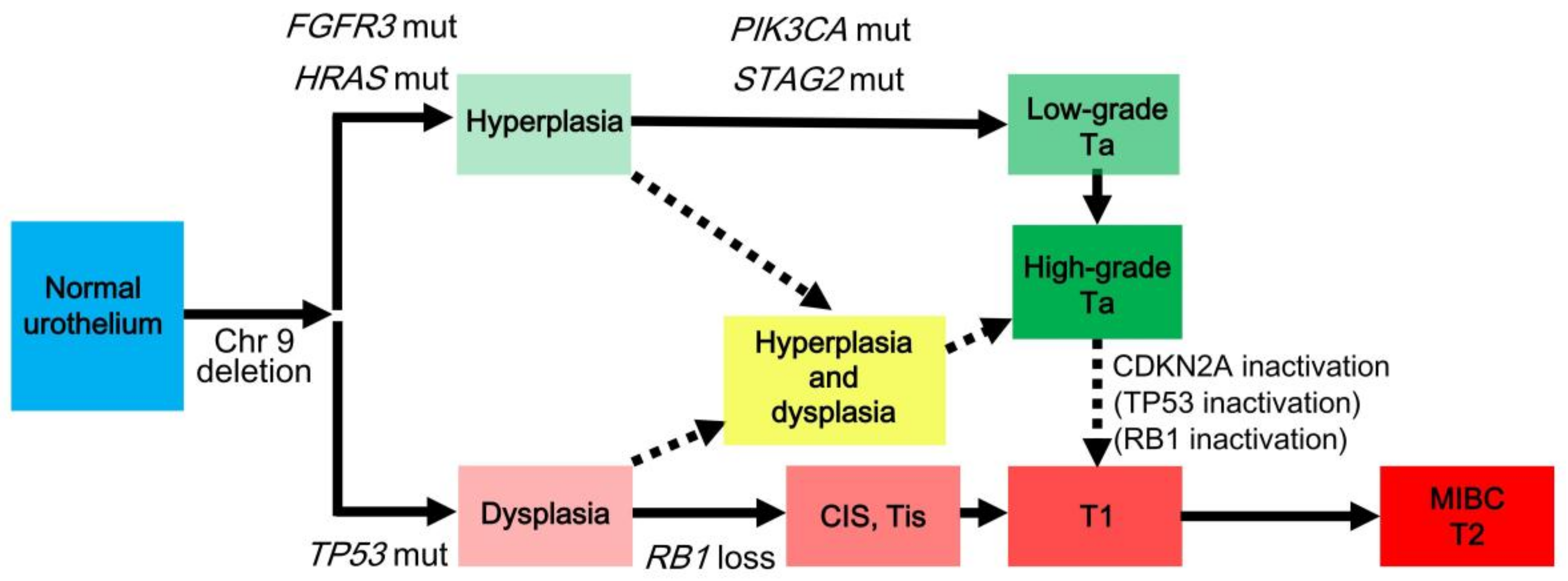
3. Divergent Pathways
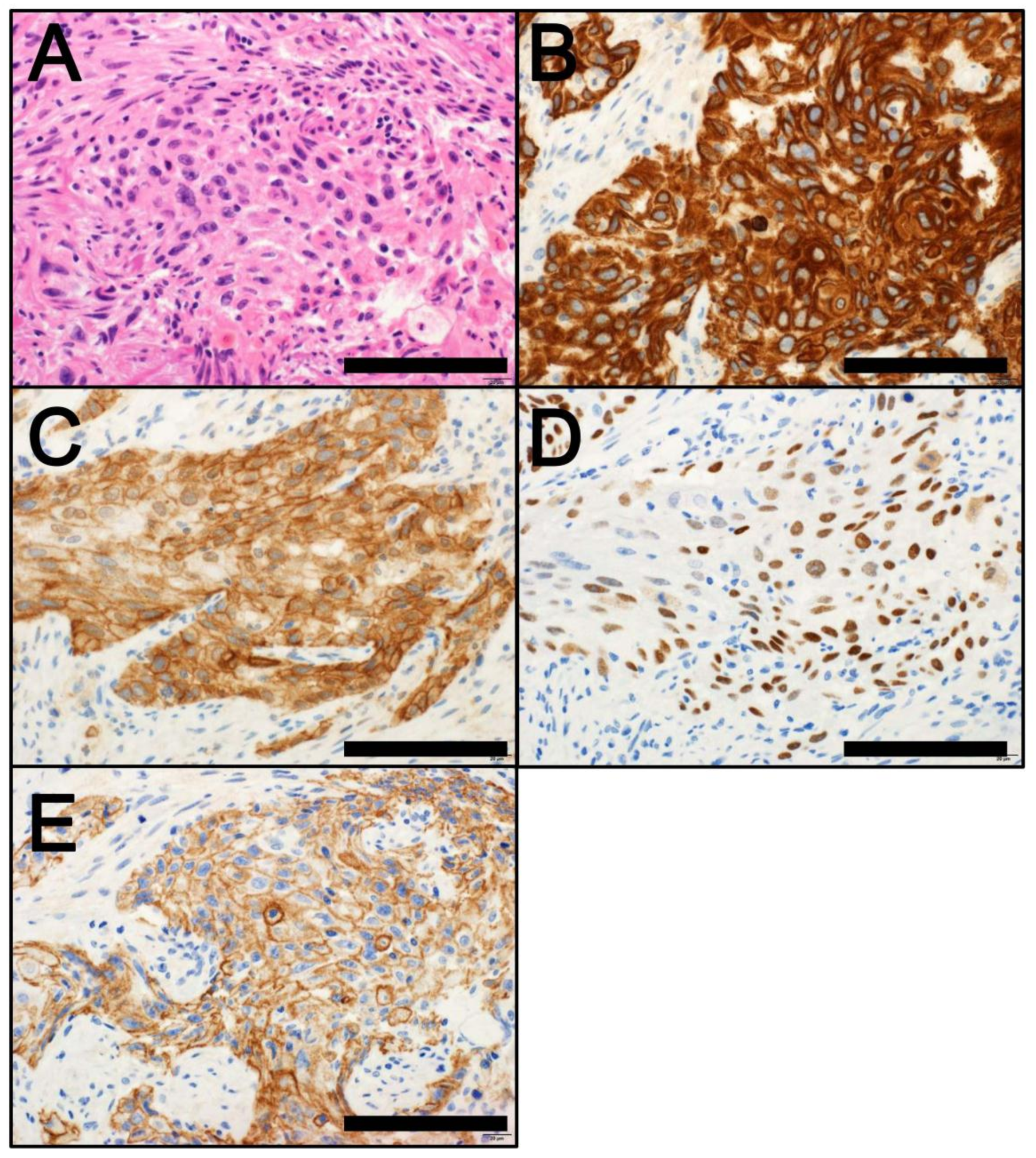
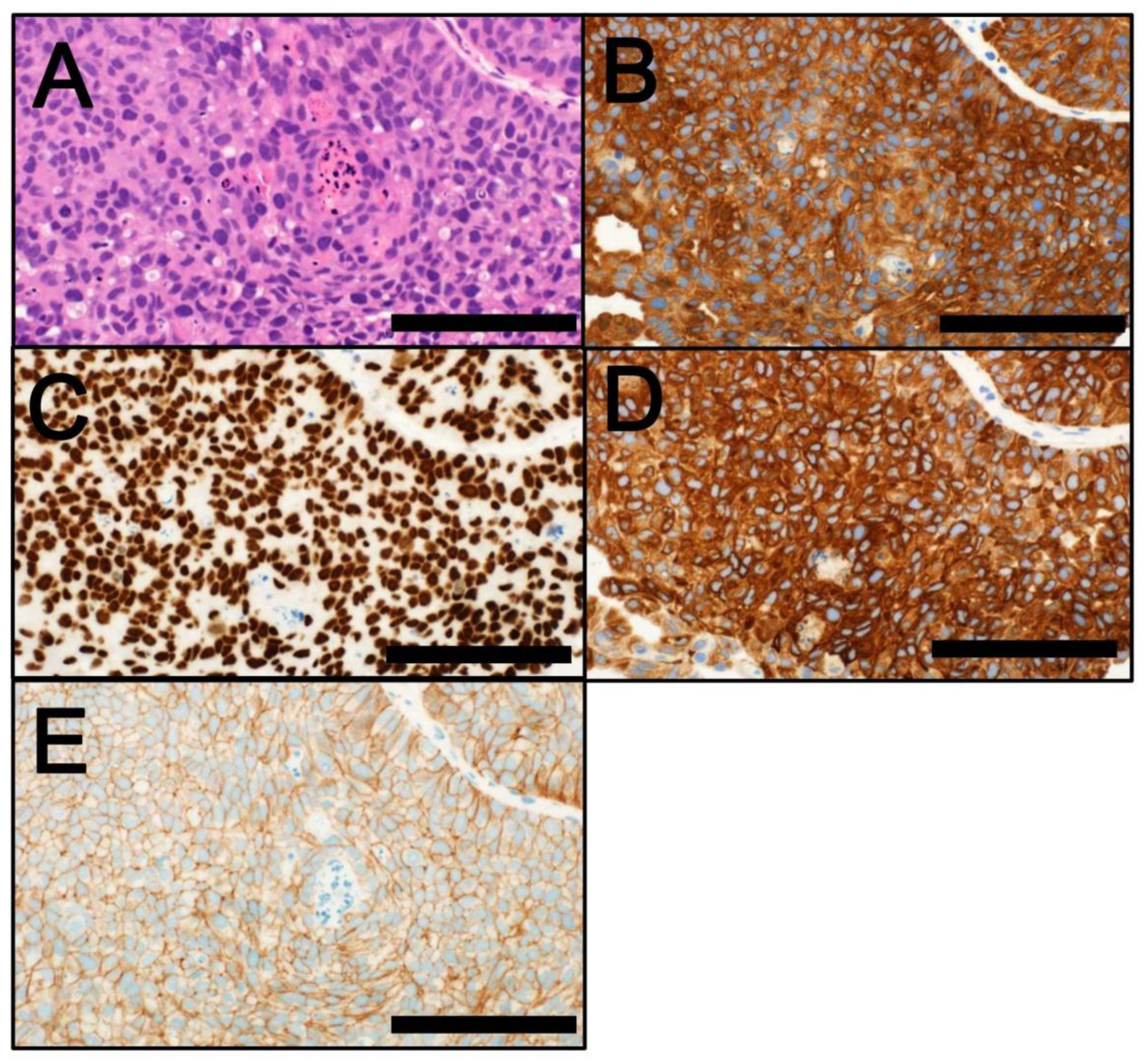
4. Classification by Molecular Subtype
4.1. NMIBC
4.2. MIBC
5. Molecular Alterations and Their Therapeutic Relevance
6. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| CIS | carcinoma in situ |
| EMT | epithelial-mesenchymal transition |
| MIBC | muscle-invasive bladder cancer |
| NAC | neoadjuvant chemotherapy |
| NMIBC | non-muscle-invasive bladder cancer |
| SHH | sonic hedgehog |
| TCGA | The Cancer Genome Atlas |
References
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Prim. 2017, 3, 17022. [Google Scholar] [CrossRef] [PubMed]
- Babjuk, M.; Burger, M.; Zigeuner, R.; Shariat, S.F.; van Rhijn, B.W.; Comperat, E.; Sylvester, R.J.; Kaasinen, E.; Bohle, A.; Palou Redorta, J.; et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: Update 2013. Eur. Urol. 2013, 64, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.L.; Dyrskjot, L.; Netto, G.J. Toward personalized management in bladder cancer: The promise of novel molecular taxonomy. Virchows Arch. 2017, 471, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Humphrey, P.A.; Ulbright, T.M.; Reuter, V.E. WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; IARC Press: Lyon, France, 2016. [Google Scholar]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Akbani, R.; Creighton, C.J.; Lerner, S.P.; Weinstein, J.N.; Getz, G.; Kwiatkowski, D.J. Invasive Bladder Cancer: Genomic Insights and Therapeutic Promise. Clin. Cancer Res. 2015, 21, 4514–4524. [Google Scholar] [CrossRef] [PubMed]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, D.; Frigyesi, A.; Gudjonsson, S.; Sjodahl, G.; Hallden, C.; Chebil, G.; Veerla, S.; Ryden, T.; Mansson, W.; Liedberg, F.; et al. Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer Res. 2010, 70, 3463–3472. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, D.; Sjodahl, G.; Lauss, M.; Staaf, J.; Chebil, G.; Lovgren, K.; Gudjonsson, S.; Liedberg, F.; Patschan, O.; Mansson, W.; et al. Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS ONE 2012, 7, e38863. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, J.; Lamy, P.; Nordentoft, I.; Algaba, F.; Hoyer, S.; Ulhoi, B.P.; Vang, S.; Reinert, T.; Hermann, G.G.; Mogensen, K.; et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 2016, 30, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sjodahl, G.; Eriksson, P.; Liedberg, F.; Hoglund, M. Molecular classification of urothelial carcinoma: Global mRNA classification versus tumour-cell phenotype classification. J. Pathol. 2017, 242, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Balbas-Martinez, C.; Sagrera, A.; Carrillo-de-Santa-Pau, E.; Earl, J.; Marquez, M.; Vazquez, M.; Lapi, E.; Castro-Giner, F.; Beltran, S.; Bayes, M.; et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013, 45, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Gerullis, H.; Otto, T.; Ecke, T.H. Targeted agents in second-line bladder cancer therapy. Anticancer Drugs 2012, 23, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, J.S.; Bondaruk, J.; Shariat, S.F.; Wang, Z.F.; Elkahloun, A.G.; Ozawa, T.; Gerard, J.; Zhuang, D.; Zhang, S.; et al. Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet. 2013, 45, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Ecke, T.H.; Stier, K.; Weickmann, S.; Zhao, Z.; Buckendahl, L.; Stephan, C.; Kilic, E.; Jung, K. miR-199a-3p and miR-214-3p improve the overall survival prediction of muscle-invasive bladder cancer patients after radical cystectomy. Cancer Med. 2017, 6, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Pop-Bica, C.; Gulei, D.; Cojocneanu-Petric, R.; Braicu, C.; Petrut, B.; Berindan-Neagoe, I. Understanding the Role of Non-Coding RNAs in Bladder Cancer: From Dark Matter to Valuable Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 1514. [Google Scholar] [CrossRef] [PubMed]
- Conconi, D.; Sala, E.; Bovo, G.; Strada, G.; Dalpra, L.; Lavitrano, M.; Bentivegna, A. Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma. Int. J. Mol. Sci. 2016, 17, 271. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.A. Cytogenetics and molecular genetics of bladder cancer: A personal view. Am. J. Med. Genet. 2002, 115, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.R. Urothelial tumorigenesis: A tale of divergent pathways. Nat. Rev. Cancer 2005, 5, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Schlake, G.; Zaak, D.; Hungerhuber, E.; Hofstetter, A.; Hofstaedter, F.; Knuechel, R. Occurrence of chromosome 9 and p53 alterations in multifocal dysplasia and carcinoma in situ of human urinary bladder. Cancer Res. 2002, 62, 809–818. [Google Scholar] [PubMed]
- Obermann, E.C.; Junker, K.; Stoehr, R.; Dietmaier, W.; Zaak, D.; Schubert, J.; Hofstaedter, F.; Knuechel, R.; Hartmann, A. Frequent genetic alterations in flat urothelial hyperplasias and concomitant papillary bladder cancer as detected by CGH, LOH, and FISH analyses. J. Pathol. 2003, 199, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Allory, Y.; Beukers, W.; Sagrera, A.; Flandez, M.; Marques, M.; Marquez, M.; van der Keur, K.A.; Dyrskjot, L.; Lurkin, I.; Vermeij, M.; et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: High frequency across stages, detection in urine, and lack of association with outcome. Eur. Urol. 2014, 65, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Huang, C.Y.; Jhuang, Y.L.; Chen, C.C.; Jeng, Y.M. Biological significance of TERT promoter mutation in papillary urothelial neoplasm of low malignant potential. Histopathology 2018, 72, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Gunes, C.; Rudolph, K.L. The role of telomeres in stem cells and cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Hurst, C.D.; Knowles, M.A. Bladder cancer: Multi-omic profiling refines the molecular view. Nat. Rev. Clin. Oncol. 2018, 15, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D. Bladder cancer in 2017: Advancing care through genomics and immune checkpoint blockade. Nat. Rev. Urol. 2018, 15, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Lopez-Knowles, E.; Lloreta, J.; Kogevinas, M.; Amoros, A.; Tardon, A.; Carrato, A.; Serra, C.; Malats, N.; Real, F.X. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J. Clin. Oncol. 2006, 24, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Cappellen, D.; De Oliveira, C.; Ricol, D.; de Medina, S.; Bourdin, J.; Sastre-Garau, X.; Chopin, D.; Thiery, J.P.; Radvanyi, F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 1999, 23, 18–20. [Google Scholar] [PubMed]
- Van Rhijn, B.W.; van der Kwast, T.H.; Liu, L.; Fleshner, N.E.; Bostrom, P.J.; Vis, A.N.; Alkhateeb, S.S.; Bangma, C.H.; Jewett, M.A.; Zwarthoff, E.C.; et al. The FGFR3 mutation is related to favorable pT1 bladder cancer. J. Urol. 2012, 187, 310–314. [Google Scholar] [PubMed]
- Kompier, L.C.; Lurkin, I.; van der Aa, M.N.; van Rhijn, B.W.; van der Kwast, T.H.; Zwarthoff, E.C. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS ONE 2010, 5, e13821. [Google Scholar] [CrossRef] [PubMed]
- Jebar, A.H.; Hurst, C.D.; Tomlinson, D.C.; Johnston, C.; Taylor, C.F.; Knowles, M.A. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene 2005, 24, 5218–5225. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.H.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Zabor, E.C.; Scott, S.N.; Ostrovnaya, I.; Ramirez, R.; Sun, A.; Shah, R.; et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur. Urol. 2015, 67, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Esrig, D.; Spruck, C.H., 3rd; Nichols, P.W.; Chaiwun, B.; Steven, K.; Groshen, S.; Chen, S.C.; Skinner, D.G.; Jones, P.A.; Cote, R.J. p53 nuclear protein accumulation correlates with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am. J. Pathol. 1993, 143, 1389–1397. [Google Scholar] [PubMed]
- Nordentoft, I.; Lamy, P.; Birkenkamp-Demtroder, K.; Shumansky, K.; Vang, S.; Hornshoj, H.; Juul, M.; Villesen, P.; Hedegaard, J.; Roth, A.; et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014, 7, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.D.; Wang, M.; Eble, J.N.; MacLennan, G.T.; Lopez-Beltran, A.; Zhang, S.; Cocco, A.; Cheng, L. Molecular evidence supporting field effect in urothelial carcinogenesis. Clin. Cancer Res. 2005, 11, 6512–6519. [Google Scholar] [CrossRef] [PubMed]
- Van Batavia, J.; Yamany, T.; Molotkov, A.; Dan, H.; Mansukhani, M.; Batourina, E.; Schneider, K.; Oyon, D.; Dunlop, M.; Wu, X.R.; et al. Bladder cancers arise from distinct urothelial sub-populations. Nat. Cell Biol. 2014, 16, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Chow, N.H.; Cairns, P.; Eisenberger, C.F.; Schoenberg, M.P.; Taylor, D.C.; Epstein, J.I.; Sidransky, D. Papillary urothelial hyperplasia is a clonal precursor to papillary transitional cell bladder cancer. Int. J. Cancer 2000, 89, 514–518. [Google Scholar] [CrossRef]
- Mo, L.; Zheng, X.; Huang, H.Y.; Shapiro, E.; Lepor, H.; Cordon-Cardo, C.; Sun, T.T.; Wu, X.R. Hyperactivation of Ha-ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J. Clin. Investig. 2007, 117, 314–325. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Oers, J.M.; Adam, C.; Denzinger, S.; Stoehr, R.; Bertz, S.; Zaak, D.; Stief, C.; Hofstaedter, F.; Zwarthoff, E.C.; van der Kwast, T.H.; et al. Chromosome 9 deletions are more frequent than FGFR3 mutations in flat urothelial hyperplasias of the bladder. Int. J. Cancer 2006, 119, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.P.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017, 14, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Downes, M.R.; Weening, B.; van Rhijn, B.W.; Have, C.L.; Treurniet, K.M.; van der Kwast, T.H. Analysis of papillary urothelial carcinomas of the bladder with grade heterogeneity: Supportive evidence for an early role of CDKN2A deletions in the FGFR3 pathway. Histopathology 2017, 70, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeong, J.; Majewski, T.; Kram, A.; Yoon, D.S.; Zhang, R.D.; Li, J.Z.; Ptaszynski, K.; Kuang, T.C.; Zhou, J.H.; et al. Evidence for alternative candidate genes near RB1 involved in clonal expansion of in situ urothelial neoplasia. Lab. Investig. 2006, 86, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Wu, C.; Eslami, Z.; Tanguay, S.; Aprikian, A.; Kassouf, W.; Brimo, F. The role of immunohistochemistry in the diagnosis of flat urothelial lesions: A study using CK20, CK5/6, P53, Cd138, and Her2/Neu. Ann. Diagn. Pathol. 2014, 18, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.B.; McConkey, D.J.; Dinney, C.P. New strategies in muscle-invasive bladder cancer: On the road to personalized medicine. Clin. Cancer Res. 2011, 17, 2608–2612. [Google Scholar] [CrossRef] [PubMed]
- Grossman, H.B.; Natale, R.B.; Tangen, C.M.; Speights, V.O.; Vogelzang, N.J.; Trump, D.L.; deVere White, R.W.; Sarosdy, M.F.; Wood, D.P., Jr.; Raghavan, D.; et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N. Engl. J. Med. 2003, 349, 859–866. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Choi, W.; Dinney, C.P. New insights into subtypes of invasive bladder cancer: Considerations of the clinician. Eur. Urol. 2014, 66, 609–610. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.G.; Catto, J.W.F. Re: Comprehensive Molecular Characterization of Muscle Invasive Bladder Cancer. Eur. Urol. 2018, 73, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]






© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inamura, K. Bladder Cancer: New Insights into Its Molecular Pathology. Cancers 2018, 10, 100. https://doi.org/10.3390/cancers10040100
Inamura K. Bladder Cancer: New Insights into Its Molecular Pathology. Cancers. 2018; 10(4):100. https://doi.org/10.3390/cancers10040100
Chicago/Turabian StyleInamura, Kentaro. 2018. "Bladder Cancer: New Insights into Its Molecular Pathology" Cancers 10, no. 4: 100. https://doi.org/10.3390/cancers10040100
APA StyleInamura, K. (2018). Bladder Cancer: New Insights into Its Molecular Pathology. Cancers, 10(4), 100. https://doi.org/10.3390/cancers10040100