Role of Pattern Recognition Receptors in KSHV Infection



Abstract
1. Introduction
2. KSHV Infections, Diagnosis and Treatment
2.1. KSHV Genome and Associated Malignancies
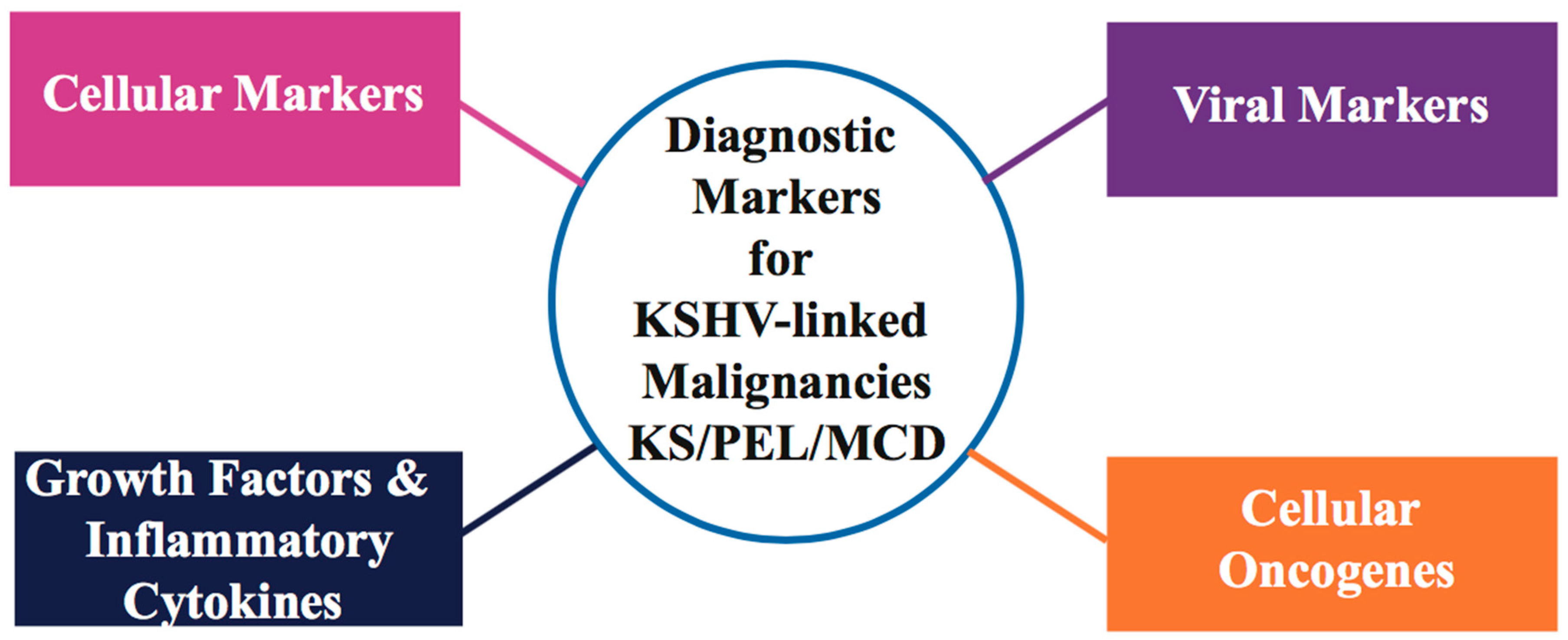
2.2. KS Diagnostic Markers and Therapeutic Approaches
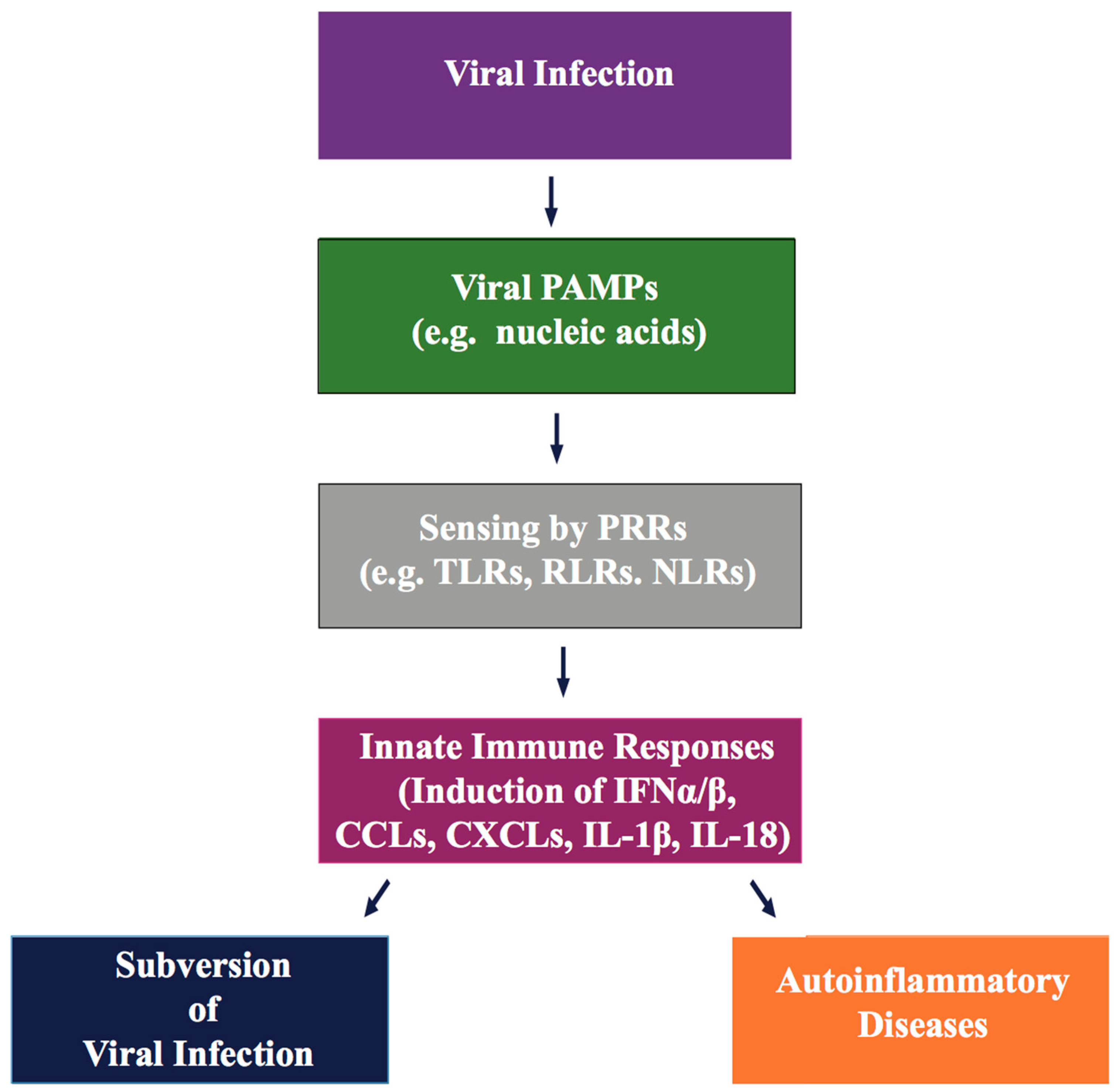
3. KSHV Inhibition of PRR-Dependent Immune Responses
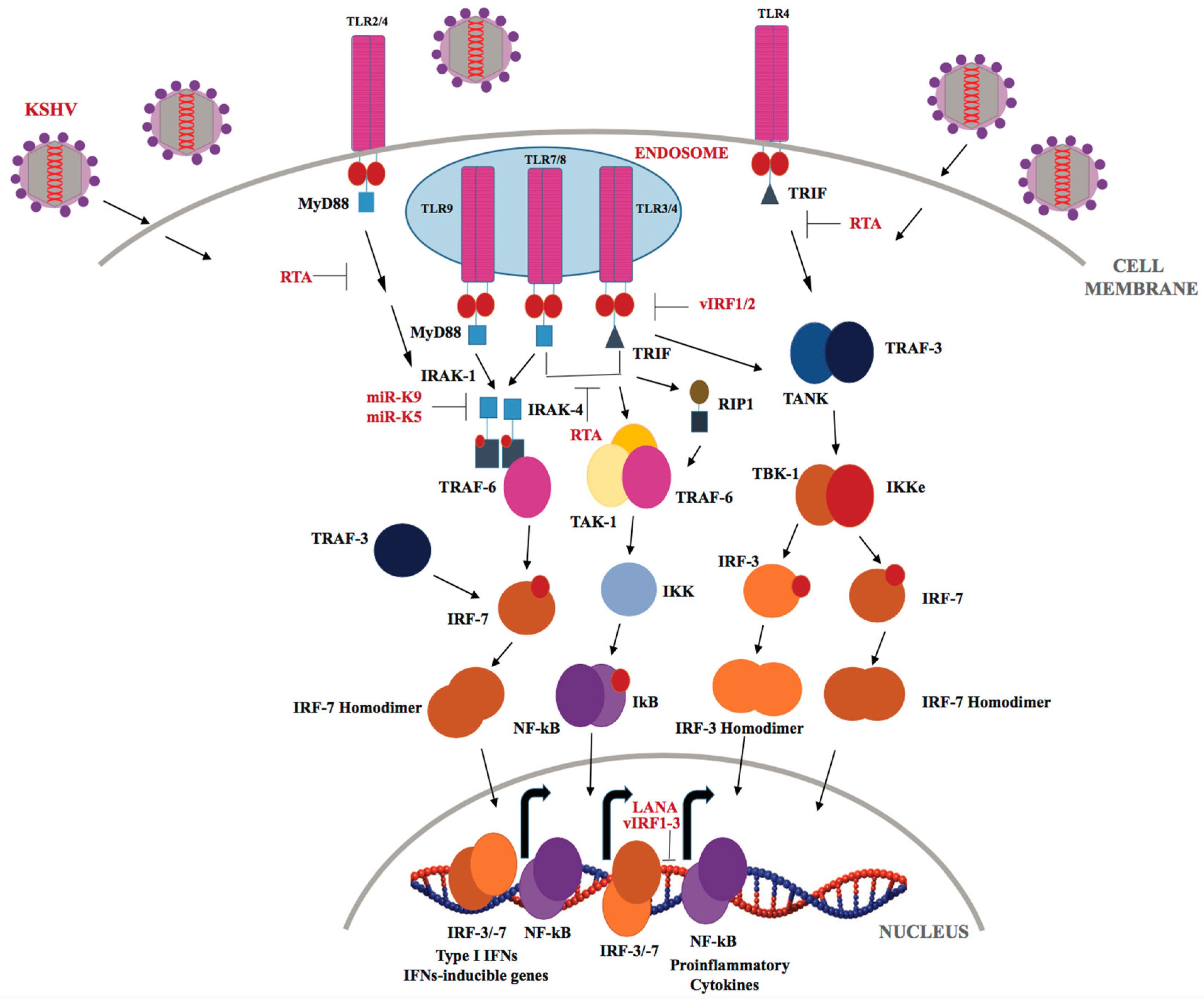
3.1. TLRs in KSHV Infection
3.2. Intracellular NLRs, RLRs, and Cytosolic DNA Receptors in KSHV Infection
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Zur Hausen, H. The search for infectious causes of human cancers: Where and why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Oncogenic DNA viruses. Oncogene 2001, 20, 7820–7823. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, 33S–40S. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Page, A.R.; Hansen, A.E.; Good, R.A. Occurrence of leukemia and lymphoma in patients with agammaglobulinemia. Blood 1963, 21, 197–206. [Google Scholar] [PubMed]
- Boder, E.; Sedgwick, R.P. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 1958, 21, 526–554. [Google Scholar] [PubMed]
- Gatti, R.A.; Good, R.A. Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer 1971, 28, 89–98. [Google Scholar] [CrossRef]
- Rabkin, C.S.; Janz, S.; Lash, A.; Coleman, A.E.; Musaba, E.; Liotta, L.; Biggar, R.J.; Zhuang, Z. Monoclonal origin of multicentric Kaposi’s sarcoma lesions. N. Engl. J. Med. 1997, 336, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Masood, R.; Cai, J.; Law, R.; Gill, P. AIDS-associated Kaposi’s sarcoma pathogenesis, clinical features, and treatment. Curr. Opin. Oncol. 1993, 5, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.; Alfieri, C.; Preiksaitis, J.; Humar, A.; Moore, D.; Tapiero, B.; Tellier, R.; Green, M.; Davies, D.; Hébert, D.; et al. Epstein-Barr virus infection in transplant recipients: Summary of a workshop on surveillance, prevention and treatment. Can. J. Infect. Dis. 2002, 13, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Gbabe, O.F.; Okwundu, C.I.; Dedicoat, M.; Freeman, E.E. Treatment of severe or progressive Kaposi’s sarcoma in HIV-infected adults. Cochrane Database Syst. Rev. 2014, CD003256. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cornali, E.; Zietz, C.; Benelli, R.; Weninger, W.; Masiello, L.; Breier, G.; Tschachler, E.; Albini, A.; Sturzl, M. Vascular endothelial growth factor regulates angiogenesis and vascular permeability in Kaposi’s sarcoma. Am. J. Pathol. 1996, 149, 1851–1869. [Google Scholar] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002, 2, 281–292. [Google Scholar] [CrossRef]
- Borkovic, S.P.; Schwartz, R.A. Kaposi’s sarcoma presenting in the homosexual man—A new and striking phenomenon! Ariz. Med. 1981, 38, 902–904. [Google Scholar] [PubMed]
- Gottlieb, G.J.; Ragaz, A.; Vogel, J.V.; Friedman-Kien, A.; Rywlin, A.M.; Weiner, E.A.; Ackerman, A.B. A preliminary communication on extensively disseminated Kaposi’s sarcoma in young homosexual men. Am. J. Dermatopathol. 1981, 3, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Hymes, K.B.; Cheung, T.; Greene, J.B.; Prose, N.S.; Marcus, A.; Ballard, H.; William, D.C.; Laubenstein, L.J. Kaposi’s sarcoma in homosexual men-a report of eight cases. Lancet 1981, 2, 598–600. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [PubMed]
- Saeed-Abdul-Rahman, I.; Al-Amri, A.M. Castleman disease. Korean J. Hematol. 2012, 47, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B. Early events in Kaposi’s sarcoma-associated herpesvirus infection of target cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Kuhne, K.; Ye, F.; Chen, J.; Zhou, F.; Lei, X.; Gao, S.J. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat. Res. 2007, 133, 69–127. [Google Scholar] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [PubMed]
- Purushothaman, P.; Thakker, S.; Verma, S.C. Transcriptome analysis of Kaposi’s sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J. Virol. 2015, 89, 3093–3111. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Duprez, R. Spindle cells and their role in Kaposi’s sarcoma. Int. J. Biochem. Cell Biol. 2005, 37, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, R.; Enbom, M.; Bidoli, E.; Linde, A.; De Paoli, P.; Dillner, J. Viral load of human herpesvirus 8 in peripheral blood of human immunodeficiency virus-infected patients with Kaposi’s sarcoma. J. Clin. Microbiol. 2001, 39, 4269–4273. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts encoding K12, v-FLIP, v-Cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, L.; Platt, G.M.; Simpson, G.R.; Sarid, R.; Gao, S.J.; Stoiber, H.; Herrington, C.S.; Moore, P.S.; Schulz, T.F. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by ORF73 and is a component of the latency-associated nuclear antigen. J. Virol. 1997, 71, 5915–5921. [Google Scholar] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M. Molecular diagnosis of infection-related cancers in dermatopathology. Semin. Cutan. Med. Surg. 2012, 31, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yarchoan, R.; Wyvill, K.; Okamoto, S.; Little, R.F.; Tosato, G. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 2001, 97, 2173–2176. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Sturzl, M. Kaposi’s sarcoma: A result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 1998, 9, 63–83. [Google Scholar] [CrossRef]
- Ensoli, B.; Sgadari, C.; Barillari, G.; Sirianni, M.C.; Sturzl, M.; Monini, P. Biology of Kaposi’s sarcoma. Eur. J. Cancer 2001, 37, 1251–1269. [Google Scholar] [CrossRef]
- Carbone, A.; Cilia, A.M.; Gloghini, A.; Capello, D.; Fassone, L.; Perin, T.; Rossi, D.; Canzonieri, V.; De Paoli, P.; Vaccher, E.; et al. Characterization of a novel HHV-8-positive cell line reveals implications for the pathogenesis and cell cycle control of primary effusion lymphoma. Leukemia 2000, 14, 1301–1309. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carroll, P.A.; Brazeau, E.; Lagunoff, M. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 2004, 328, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Weninger, W.; Partanen, T.A.; Breiteneder-Geleff, S.; Mayer, C.; Kowalski, H.; Mildner, M.; Pammer, J.; Sturzl, M.; Kerjaschki, D.; Alitalo, K.; et al. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab. Investig. 1999, 79, 243–251. [Google Scholar] [PubMed]
- Marchio, S.; Primo, L.; Pagano, M.; Palestro, G.; Albini, A.; Veikkola, T.; Cascone, I.; Alitalo, K.; Bussolino, F. Vascular endothelial growth factor-C stimulates the migration and proliferation of Kaposi’s sarcoma cells. J. Boil. Chem. 1999, 274, 27617–27622. [Google Scholar] [CrossRef]
- Rosado, F.G.; Itani, D.M.; Coffin, C.M.; Cates, J.M. Utility of immunohistochemical staining with FLI1, D2-40, CD31, and CD34 in the diagnosis of acquired immunodeficiency syndrome-related and non-acquired immunodeficiency syndrome-related Kaposi sarcoma. Arch. Pathol. Lab. Med. 2012, 136, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Faris, M.; Ensoli, B.; Kokot, N.; Nel, A.E. Inflammatory cytokines induce the expression of basic fibroblast growth factor (bFGF) isoforms required for the growth of Kaposi’s sarcoma and endothelial cells through the activation of AP-1 response elements in the bFGF promoter. AIDS 1998, 12, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.X.; Antakly, T.; Cadotte, M.; Kachra, Z.; Kunkel, L.; Masood, R.; Gill, P. Expression and cytokine regulation of glucocorticoid receptors in Kaposi’s sarcoma. Am. J. Pathol. 1996, 148, 1999–2008. [Google Scholar] [PubMed]
- Cai, J.; Gill, P.S.; Masood, R.; Chandrasoma, P.; Jung, B.; Law, R.E.; Radka, S.F. Oncostatin-M is an autocrine growth factor in Kaposi’s sarcoma. Am. J. Pathol. 1994, 145, 74–79. [Google Scholar] [PubMed]
- Liu, Z.; Fang, Q.; Zhou, S.; Minhas, V.; Wood, C.; He, N.; Zhang, T. Seroprevalence of Kaposi’s sarcoma-associated herpesvirus among HIV-infected uygurs in Xinjiang, china. J. Med. Virol. 2017, 89, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.; Cope, A.; Gill, J.; Bourboulia, D.; Hayes, P.; Imami, N.; Kubo, T.; Marcelin, A.; Calvez, V.; Weiss, R.; et al. Identification of Kaposi’s sarcoma-associated herpesvirus (KSHV)-specific cytotoxic T-lymphocyte epitopes and evaluation of reconstitution of KSHV-specific responses in human immunodeficiency virus type 1-infected patients receiving highly active antiretroviral therapy. J. Virol. 2002, 76, 2634–2640. [Google Scholar] [PubMed]
- Pereira, P.F.; Cuzzi, T.; Galhardo, M.C. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An. Bras. Dermatol. 2013, 88, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Dezube, B.J.; Pinkus, G.S.; Tahan, S.R. Histological characterization of regression in acquired immunodeficiency syndrome-related Kaposi’s sarcoma. J. Cutan. Pathol. 2004, 31, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, W.; Wong, K.O.; Wong, C.S.; Dinkel, J.E.; Ben-Dor, D.; Chan, J.K. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am. J. Clin. Pathol. 2004, 121, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Long, E.; Ilie, M.; Hofman, V.; Havet, K.; Selva, E.; Butori, C.; Lacour, J.P.; Nelson, A.M.; Cathomas, G.; Hofman, P. LANA-1, Bcl-2, Mcl-1 and HIF-1α protein expression in HIV-associated Kaposi sarcoma. Virchows Arch. 2009, 455, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Tiainen, M.; Salven, P.; Veikkola, T.; Castanos-Velez, E.; Sarid, R.; Biberfeld, P.; Makela, T.P. Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 1999, 59, 4984–4989. [Google Scholar] [PubMed]
- Nagata, N.; Igari, T.; Shimbo, T.; Sekine, K.; Akiyama, J.; Hamada, Y.; Yazaki, H.; Ohmagari, N.; Teruya, K.; Oka, S.; et al. Diagnostic value of endothelial markers and HHV-8 staining in gastrointestinal Kaposi sarcoma and its difference in endoscopic tumor staging. World J. Gastroenterol. 2013, 19, 3608–3614. [Google Scholar] [CrossRef] [PubMed]
- Russell Jones, R.; Orchard, G.; Zelger, B.; Wilson Jones, E. Immunostaining for CD31 and CD34 in Kaposi sarcoma. J. Clin. Pathol. 1995, 48, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Goldblum, J.R.; Hsi, E.D. Immunohistochemical detection of human herpes virus-8 latent nuclear antigen-1 is useful in the diagnosis of Kaposi sarcoma. Mod. Pathol. 2004, 17, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Schwartz, E.J.; Dezube, B.J.; Kohler, S.; Dorfman, R.F.; Tahan, S.R. C-kit (CD117) expression in AIDS-related, classic, and African endemic Kaposi sarcoma. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.V.; Jarvis, M.A.; Raggo, C.; Bell, Y.C.; Ruhl, R.; Luukkonen, B.G.; Griffith, D.J.; Wait, C.L.; Druker, B.J.; Heinrich, M.C.; et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J. Virol. 2002, 76, 8383–8399. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.M.; Biddolph, S.; Lucas, S.B.; Howells, D.D.; Picton, S.; McGee, J.O.; Silva, I.; Uhlmann, V.; Luttich, K.; O’Leary, J.J. Cyclin D1 expression and HHV8 in Kaposi sarcoma. J. Clin. Pathol. 1999, 52, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Koopal, S.; Furuhjelm, J.H.; Jarviluoma, A.; Jaamaa, S.; Pyakurel, P.; Pussinen, C.; Wirzenius, M.; Biberfeld, P.; Alitalo, K.; Laiho, M.; et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007, 3, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Sin, S.H.; Dittmer, D.P. Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus lymphomas: Implications for therapy. J. Virol. 2007, 81, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Chandran, K.; Patel, K.; Veettil, M.V.; Marginean, A. The Kaposi’s sarcoma-associated herpesvirus (KSHV)-induced 5-lipoxygenase-leukotriene B4 cascade plays key roles in KSHV latency, monocyte recruitment, and lipogenesis. J. Virol. 2014, 88, 2131–2156. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Schulz, T.F.; Kennedy, M.M.; Graham, A.K.; Fisher, C.; Thomas, A.; McGee, J.O.; Weiss, R.A.; O’Leary, J.J. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1995, 1, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Sturzl, M.; Hohenadl, C.; Zietz, C.; Castanos-Velez, E.; Wunderlich, A.; Ascherl, G.; Biberfeld, P.; Monini, P.; Browning, P.J.; Ensoli, B. Expression of K13/v-FLIP gene of human herpesvirus 8 and apoptosis in Kaposi’s sarcoma spindle cells. J. Natl. Cancer Inst. 1999, 91, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Pise-Masison, C.A.; Brady, J.N.; Tosato, G. Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J. Virol. 2009, 83, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, M.G.; Cesarman, E.; Wang, X.; Linkov, I.; Prieto, V.G.; Louie, D.C. Cyclin D1 and retinoblastoma protein expression in Kaposi’s sarcoma. J. Cutan. Pathol. 1997, 24, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Verma, S.C.; Robertson, E.S. ORF73 of herpesvirus saimiri, a viral homolog of Kaposi’s sarcoma-associated herpesvirus, modulates the two cellular tumor suppressor proteins p53 and pRb. J. Virol. 2004, 78, 10336–10347. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, H.; Nagai, M.; Shibanushi, T.; Ohmori, M.; Kawakami, K.; Asakura, H. CD138-positive and Kaposi’s sarcoma-associated herpesvirus (KSHV)-negative B-cell lymphoma with serosal spreading of the body cavity and lymphadenopathy: An autopsy case. Hum. Pathol. 2000, 31, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Deloose, S.T.; Smit, L.A.; Pals, F.T.; Kersten, M.J.; van Noesel, C.J.; Pals, S.T. High incidence of Kaposi sarcoma-associated herpesvirus infection in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma. Leukemia 2005, 19, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, W.; Gao, S.J.; Lu, C. KSHV microRNAs: Tricks of the devil. Trends Microbiol. 2017, 25, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, Y.; Mo, D.; Huang, P.; Sun, R.; Huang, L.; Pan, S.; Xu, J. Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS ONE 2014, 9, e93478. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. In-depth analysis of Kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 2010, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Arai, E.; Kuramochi, A.; Tsuchida, T.; Tsuneyoshi, M.; Kage, M.; Fukunaga, M.; Ito, T.; Tada, T.; Izumi, M.; Shimizu, K.; et al. Usefulness of D2-40 immunohistochemistry for differentiation between kaposiform hemangioendothelioma and tufted angioma. J. Cutan. Pathol. 2006, 33, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kahn, H.J.; Bailey, D.; Marks, A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi’s sarcoma and a subset of angiosarcomas. Mod. Pathol. 2002, 15, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Gregory, S.M.; West, J.A.; Wollish, A.C.; Bennett, C.L.; Blackbourn, D.J.; Heise, M.T.; Damania, B. The viral interferon regulatory factors of Kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 2013, 87, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef]
- Gutierrez, K.D.; Morris, V.A.; Wu, D.; Barcy, S.; Lagunoff, M. Ets-1 is required for the activation of VEGFR3 during latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 2013, 87, 6758–6768. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Sharma-Walia, N.; Kerur, N.; Paul, A.G.; Sadagopan, S.; Cannon, M.; Chandran, B. Kaposi sarcoma-associated herpes virus (KSHV) G protein-coupled receptor (vGPCR) activates the ORF50 lytic switch promoter: A potential positive feedback loop for sustained ORF50 gene expression. Virology 2009, 392, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Svojanovsky, S.R.; Bloomer, C.; Mathur, S.; Chandran, B. Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: Insights into modulation events early during infection. Cancer Res. 2004, 64, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.C.; Miles, S.; Kumar, G.; Nel, A.E. Oncostatin-M stimulates tyrosine protein phosphorylation in parallel with the activation of p42MAPK/ERK-2 in Kaposi’s cells. Evidence that this pathway is important in Kaposi cell growth. J. Clin. Investig. 1993, 92, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Simonart, T.; Van Vooren, J.P. Interleukin-1 β increases the BCL-2/BAX ratio in Kaposi’s sarcoma cells. Cytokine 2002, 19, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Richards, K.; Damania, B. Treatment of Kaposi sarcoma-associated herpesvirus-associated cancers. Front. Microbiol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Yanik, E.L.; Napravnik, S.; Cole, S.R.; Achenbach, C.J.; Gopal, S.; Olshan, A.; Dittmer, D.P.; Kitahata, M.M.; Mugavero, M.J.; Saag, M.; et al. Incidence and timing of cancer in HIV-infected individuals following initiation of combination antiretroviral therapy. Clin. Infect. Dis. 2013, 57, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.F.; Scriven, J.; Meintjes, G.; Wilkinson, R.J. Immune reconstitution inflammatory syndrome in HIV-infected patients. HIV/AIDS 2015, 7, 49–64. [Google Scholar] [PubMed]
- Hosseinipour, M.C.; Sweet, K.M.; Xiong, J.; Namarika, D.; Mwafongo, A.; Nyirenda, M.; Chiwoko, L.; Kamwendo, D.; Hoffman, I.; Lee, J.; et al. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Glesby, M.J.; Hoover, D.R.; Weng, S.; Graham, N.M.; Phair, J.P.; Detels, R.; Ho, M.; Saah, A.J. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: Data from the multicenter AIDS cohort study. J. Infect. Dis. 1996, 173, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Mazzi, R.; Parisi, S.G.; Sarmati, L.; Uccella, I.; Nicastri, E.; Carolo, G.; Gatti, F.; Concia, E.; Andreoni, M. Efficacy of cidofovir on human herpesvirus 8 viraemia and Kaposi’s sarcoma progression in two patients with AIDS. AIDS 2001, 15, 2061–2062. [Google Scholar] [CrossRef] [PubMed]
- Bossini, N.; Sandrini, S.; Setti, G.; Luppi, M.; Maiorca, P.; Maffei, C.; Cancarini, G. Successful treatment with liposomal doxorubicin and foscarnet in a patient with widespread Kaposi’s sarcoma and human herpes virus 8-related, serious hemophagocytic syndrome, after renal transplantation. G. Ital. Nefrol. 2005, 22, 281–286. [Google Scholar] [PubMed]
- Sergerie, Y.; Boivin, G. Evaluation of susceptibility of human herpesvirus 8 to antiviral drugs by quantitative real-time PCR. J. Clin. Microbiol. 2003, 41, 3897–3900. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takahashi-Makise, N.; Suzu, S.; Hiyoshi, M.; Ohsugi, T.; Katano, H.; Umezawa, K.; Okada, S. Biscoclaurine alkaloid cepharanthine inhibits the growth of primary effusion lymphoma in vitro and in vivo and induces apoptosis via suppression of the NF-κB pathway. Int. J. Cancer 2009, 125, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, T.; Kariya, R.; Yano, S.; Morino-Koga, S.; Taura, M.; Suico, M.A.; Shimauchi, Y.; Matsuyama, S.; Okamoto, Y.; Shuto, T.; et al. Diethyldithiocarbamate induces apoptosis in HHV-8-infected primary effusion lymphoma cells via inhibition of the NF-κB pathway. Int. J. Oncol. 2012, 40, 1071–1078. [Google Scholar] [PubMed]
- Uldrick, T.S.; Goncalves, P.H.; Wyvill, K.M.; Peer, C.J.; Bernstein, W.; Aleman, K.; Polizzotto, M.N.; Venzon, D.; Steinberg, S.M.; Marshall, V.; et al. A phase Ib study of sorafenib (BAY 43-9006) in patients with Kaposi sarcoma. Oncologist 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Krown, S.E.; Lee, J.Y.; Honda, K.; Rapisuwon, S.; Wang, Z.; Aboulafia, D.; Reid, E.G.; Rudek, M.A.; Dezube, B.J.; et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS malignancy consortium protocol 042. J. Clin. Oncol. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. TLR-mediated innate immune recognition. Semin. Immunol. 2007, 19, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Fujiwara, N.; Ido, A.; Oono, M.; Takeuchi, Y.; Tateno, M.; Suzuki, K.; Takahashi, R.; Tooyama, I.; Taniguchi, N.; et al. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Pierce, S.K. How location governs toll-like receptor signaling. Traffic 2009, 10, 621–628. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G.; Freeze, H.H. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 2009, 11, 615–628. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Ogino, S.; Qian, Z.R. Toll-like receptor signaling in colorectal cancer: Carcinogenesis to cancer therapy. World J. Gastroenterol. 2014, 20, 17699–17708. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Amatya, R.; Jung, J.U. Multi-step regulation of innate immune signaling by Kaposi’s sarcoma-associated herpesvirus. Virus Res. 2015, 209, 39–44. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Damania, B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J. Virol. 2008, 82, 5440–5449. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Stopford, C.M.; West, J.A.; Bennett, C.L.; Giffin, L.; Damania, B. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J. Virol. 2015, 89, 11572–11583. [Google Scholar] [CrossRef] [PubMed]
- Mutocheluh, M.; Hindle, L.; Areste, C.; Chanas, S.A.; Butler, L.M.; Lowry, K.; Shah, K.; Evans, D.J.; Blackbourn, D.J. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J. Gen. Virol. 2011, 92, 2394–2398. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Vart, R.J.; Gratrix, F.; Westrop, S.J.; Emuss, V.; Wong, P.-P.; Robey, R.; Imami, N.; Bower, M.; Gotch, F.; et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 2008, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.A.; Reimer, E.; Todt, H.; Denker, B.; Gallo, A.; Konrad, A.; Ottinger, M.; Adler, H.; Stürzl, M.; Brune, W.; et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the toll-like receptor-induced proinflammatory cytokine response. J. Virol. 2014, 88, 9245–9259. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Gregory, S.M.; Sivaraman, V.; Su, L.; Damania, B. Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2011, 85, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.; Gubbels, R.; Ehlers, E.; Meyer, F.; Waterbury, T.; Lin, R.; Zhang, L. Kaposi sarcoma-associated herpesvirus degrades cellular toll-interleukin-1 receptor domain-containing adaptor-inducing β-interferon (TRIF). J. Boil. Chem. 2011, 286, 7865–7872. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liang, D.; Sun, R.; Jia, B.; Xia, T.; Xiao, H.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded replication and transcription activator impairs innate immunity via ubiquitin-mediated degradation of myeloid differentiation factor 88. J. Virol. 2015, 89, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Ramalingam, D.; Kieffer-Kwon, P.; Uldrick, T.S.; Yarchoan, R.; Ziegelbauer, J.M. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MyD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J. Virol. 2012, 86, 11663–11674. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, W.; Xiong, J.; Sherrod, C.J.; Henry, D.H.; Dittmer, D.P. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for Kaposi sarcoma herpesvirus lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E4762–E4768. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Monie, T.P. NLR activation takes a direct route. Trends Biochem. Sci. 2013, 38, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Ve, T.; Bentham, A.; Stacey, K.J.; Kobe, B. The molecular mechanisms of signaling by cooperative assembly formation in innate immunity pathways. Mol. Immunol. 2017, 86, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory nod-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Kanneganti, T.D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 2013, 255, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hopcraft, S.E.; Yang, F.; Petrucelli, A.; Guo, H.; Ting, J.P.; Dittmer, D.P.; Damania, B. NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. 2017, 13, e1006350. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008, 29, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ramanathan, A.; Miller, M.T.; Tang, G.Q.; Gale, M., Jr.; Patel, S.S.; Marcotrigiano, J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 2011, 479, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M., Jr.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.; Han, K.; Li, L.; Zhai, Z.; Shu, H. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Berke, I.C.; Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012, 31, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.S.; Randall, R.E.; Goodbourn, S. LGP2 plays a critical role in sensitizing MDA-5 to activation by double-stranded RNA. PLoS ONE 2013, 8, e64202. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbial. 2010, 13, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Lilue, J.; Stavrou, S.; Moran, E.A.; Ross, S.R. AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, Y.; Wang, H.; Wang, R. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates sting. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Vance, R.E. Sting and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E. Cytosolic DNA sensing: The field narrows. Immunity 2016, 45, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Paludan, S.R. Viral evasion of DNA-stimulated innate immune responses. Cell. Mol. Immunol. 2017, 14, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Cristea, I.M. The emerging role of nuclear viral DNA sensors. J. Boil. Chem. 2015, 290, 26412–26421. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Kerur, N.; Bottero, V.; Dutta, S.; Chakraborty, S.; Ansari, M.A.; Paudel, N.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 2013, 87, 4417–4431. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): Role of IFI16 in maintenance of KSHV latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-sting DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern recognition receptors in cancer progression and metastasis. Cancer Growth Metastasis 2015, 8, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Heinecke, J.L.; Ambs, S.; Glynn, S.; Young, H.A.; Trinchieri, G.; Wink, D.A. Molecular pathways: Toll-like receptors in the tumor microenvironment—Poor prognosis or new therapeutic opportunity. Clin. Cancer Res. 2013, 19, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Conroy, H.; Marshall, N.A.; Mills, K.H. TLR ligand suppression or enhancement of Treg cells? A double-edged sword in immunity to tumours. Oncogene 2008, 27, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Goto, Y.; Narita, N.; Hoon, D.S. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. 2009, 2 (Suppl. S1), 205–214. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Cataisson, C.; Hasan, U.; Yuspa, S.H.; Trinchieri, G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol. 2013, 34, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Harasymczuk, M.; Boyiadzis, M.; Kruk-Zagajewska, A.; Szyfter, W.; Zeromski, J.; Whiteside, T.L. Triggering of toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009, 69, 3105–3113. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.G.; Alvero, A.B.; Chen, R.; Silasi, D.A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Liu, G.-H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.L.; Crother, T.R.; Rabizadeh, S.; Hu, B.; Wang, H.; Chen, S.; Shimada, K.; Wong, M.H.; Michelsen, K.S.; Arditi, M. Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PLoS ONE 2010, 5, e13027. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.S.; Bondurant, K.L.; Wolff, R.K. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int. J. Cancer 2012, 130, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Isaza-Correa, J.M.; Liang, Z.; van den Berg, A.; Diepstra, A.; Visser, L. Toll-like receptors in the pathogenesis of human B cell malignancies. J. Hematol. Oncol. 2014, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007, 44, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.R.; Conejo-Garcia, J.R. TLR5 signaling, commensal microbiota and systemic tumor promoting inflammation: The three parcae of malignant progression. Oncoimmunology 2015, 4, e1021542. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhu, S.; Qiao, Y.; Liu, Y.; Chen, W.; Zhao, G.; Chen, J. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell 2014, 5, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukocyte Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Luddy, K.A.; Robertson-Tessi, M.; Tafreshi, N.K.; Soliman, H.; Morse, D.L. The role of toll-like receptors in colorectal cancer progression: Evidence for epithelial to leucocytic transition. Front. Immunol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Sandholm, J.; Selander, K.S. Toll-like receptor 9 in breast cancer. Front. Immunol. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, E.; Ntoufa, S.; Papakonstantinou, N.; Touloumenidou, T.; Laoutaris, N.; Anagnostopoulos, A.; Lamnissou, K.; Caligaris-Cappio, F.; Stamatopoulos, K.; Ghia, P.; et al. Toll-like receptor signaling pathway in chronic lymphocytic leukemia: Distinct gene expression profiles of potential pathogenic significance in specific subsets of patients. Haematologica 2011, 96, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Beller, E.; Kirchleitner, S.V.; Adunka, T.; Bourhis, H.; Siveke, J.; Mayr, D.; Kobold, S.; Endres, S.; Schnurr, M. Targeted activation of melanoma differentiation-associated protein 5 (MDA5) for immunotherapy of pancreatic carcinoma. Oncoimmunology 2015, 4, e1029698. [Google Scholar] [CrossRef] [PubMed]
- Sallets, A.; Kardosh, A.; Robinson, S.; Levy, R. In-situ vaccination using sting agonists combined with immune-modulating antibodies to treat lymphoma. Blood 2017, 130 (Suppl. S1), 4102. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Markers | Viral Markers/Proteins | Cellular Oncogenes | Growth Factors/Inflammatory Cytokines |
|---|---|---|---|
| CD4 Count [41,61,62] | KSHV Virus [63,64,65] | bcl-2 [49,66,67] | CD31 [57,64,68,69] |
| LANA/ORF73 [64,65,66,70] | c-kit [71,72] | CD34 [57,68,69] | |
| vCyclin/ORF72 [67,73,74] | p53 [46,75,76] | CD36 [51,77,78] | |
| vFLIP/ORF71 [49,74,79,80] | pRb [47,81,82] | CD138/Syndecan-1 [83,84] | |
| K12/Kaposins [49,85,86] | FLI1 [57,87,88] | ||
| miRNAs [48,85,86,89] | D2-40 [57,90,91] | ||
| vIRFs [48,92,93,94] | Podoplanin [54,55,95] | ||
| vIL-6 [49,50,59] | VEGFR3 [39,56,95,96] | ||
| vGPCR/ORF74 [48,97] | LYVE-1 [39,54,95,96] | ||
| TNF-α [58,59] | |||
| bFGF [39,58,98,99] | |||
| Oncostatin M [58,60,100] | |||
| vIL-6 [49,50,59] | |||
| IL-I [58,59,101] |
| PRRs | Cellular Localization | PAMPs | Associated Tumors |
|---|---|---|---|
| TLR-1/2 | Plasma membrane | Lipoprotein | Colon cancer [191] |
| TLR-3 | Endosome | Double stranded (ds)RNA | Melanoma, Colorectal adenoma, low-grade B-cell Lymphoma, Solid tumors [192,193] |
| TLR-4 | Plasma membrane | Lipopolysaccharides | Lung cancer, Non-Hodgkin’s lymphoma [194] |
| TLR-5 | Plasma membrane | Flagellin | Advanced/Metastatic solid tumors [195] |
| TLR-7/8 | Endosome | Single stranded (ss)RNA | Ovarian cancer, Solid tumors [196,197,198] |
| TLR-9 | Endosome | CpG-DNA | Colorectal cancer, Breast cancer, Chronic Lymphocytic Leukemia [199,200,201] |
| MDA5 | Cytoplasm | Long dsRNA | Solid tumors [202] |
| STING | Cytoplasm | dsDNA | Advanced/Metastatic solid tumors [203] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uppal, T.; Sarkar, R.; Dhelaria, R.; Verma, S.C. Role of Pattern Recognition Receptors in KSHV Infection. Cancers 2018, 10, 85. https://doi.org/10.3390/cancers10030085
Uppal T, Sarkar R, Dhelaria R, Verma SC. Role of Pattern Recognition Receptors in KSHV Infection. Cancers. 2018; 10(3):85. https://doi.org/10.3390/cancers10030085
Chicago/Turabian StyleUppal, Timsy, Roni Sarkar, Ranjit Dhelaria, and Subhash C. Verma. 2018. "Role of Pattern Recognition Receptors in KSHV Infection" Cancers 10, no. 3: 85. https://doi.org/10.3390/cancers10030085
APA StyleUppal, T., Sarkar, R., Dhelaria, R., & Verma, S. C. (2018). Role of Pattern Recognition Receptors in KSHV Infection. Cancers, 10(3), 85. https://doi.org/10.3390/cancers10030085