Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
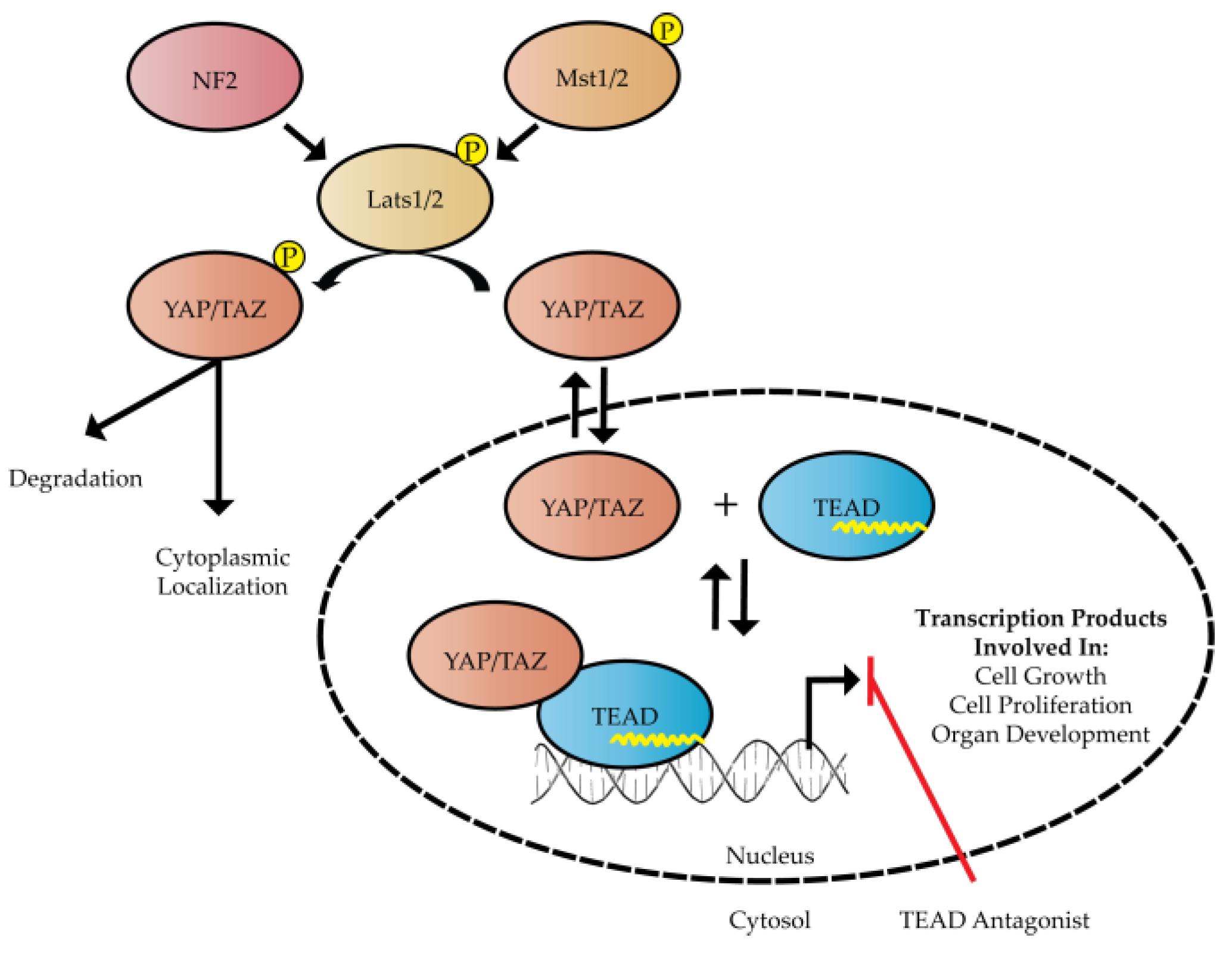
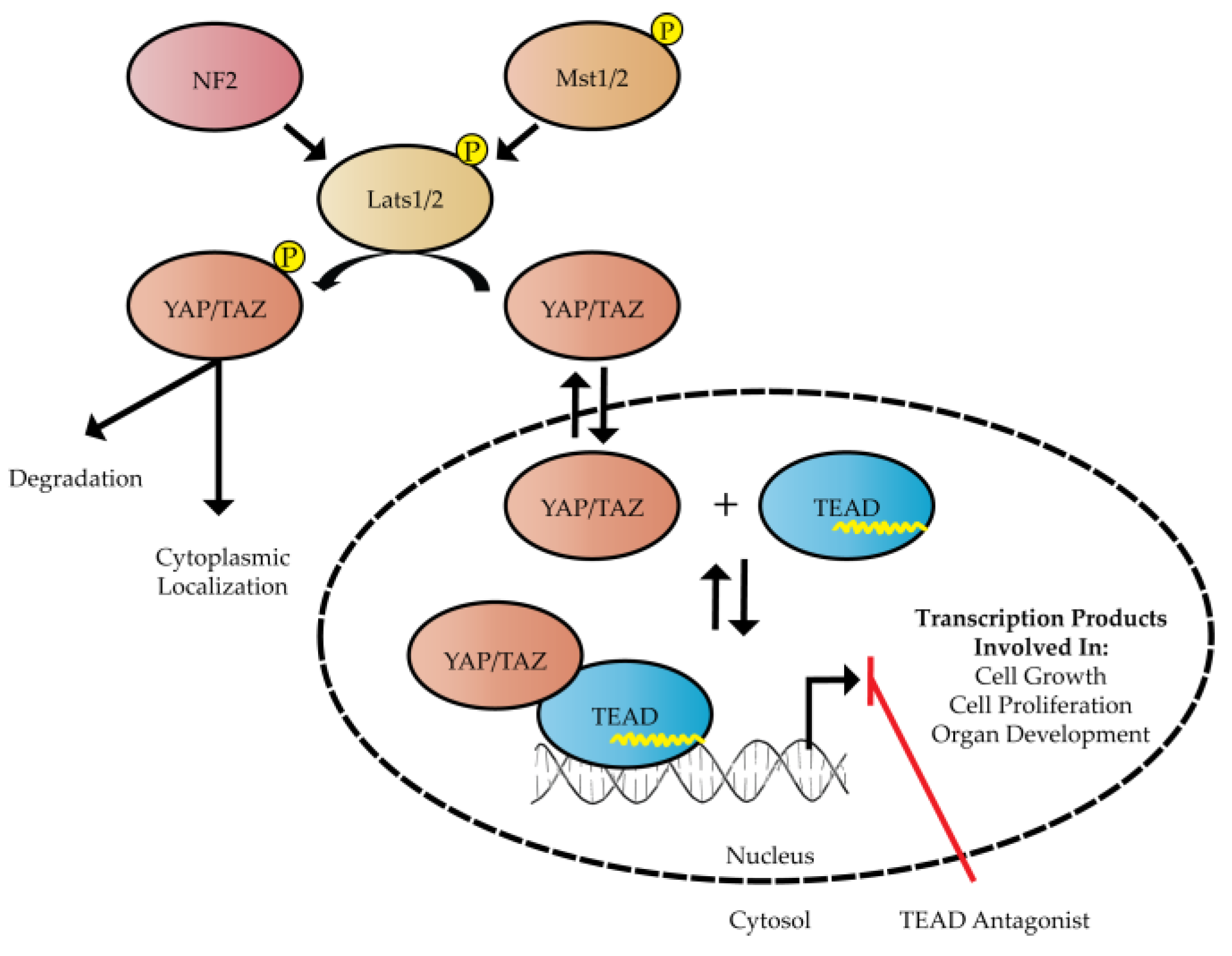
:1. Introduction
2. Structure and Function of Hippo Transcription Factors
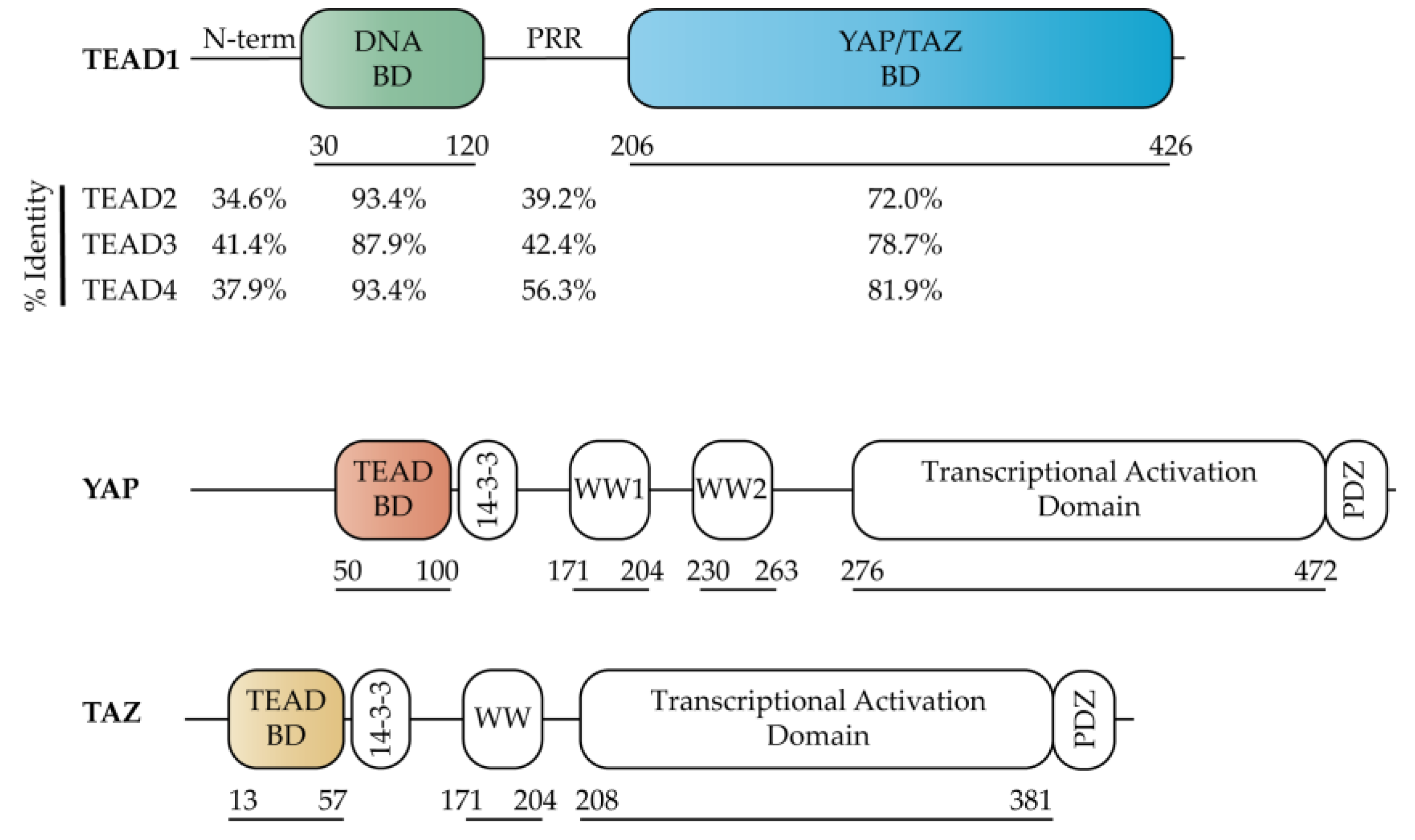
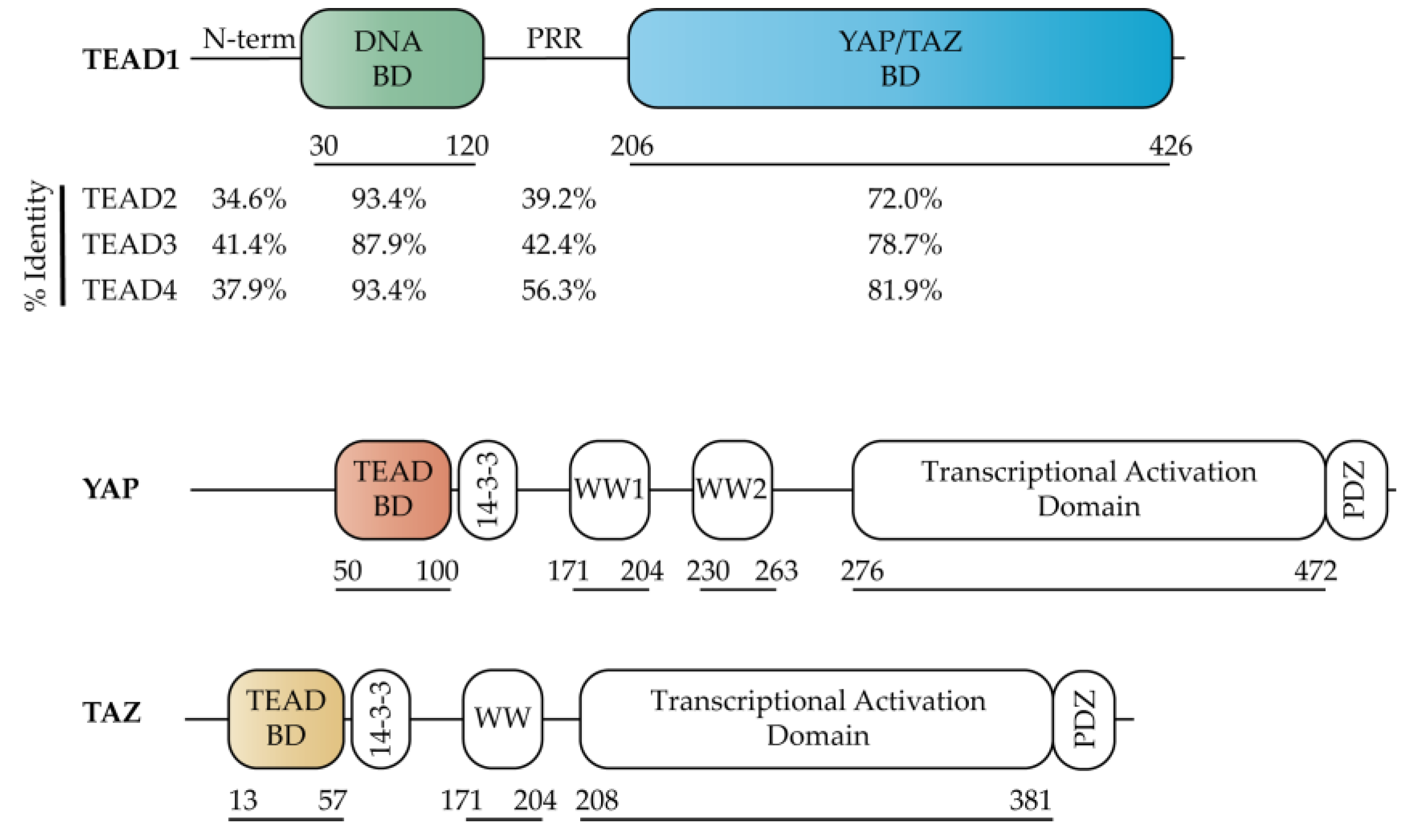
2.1. TEAD Co-Activators
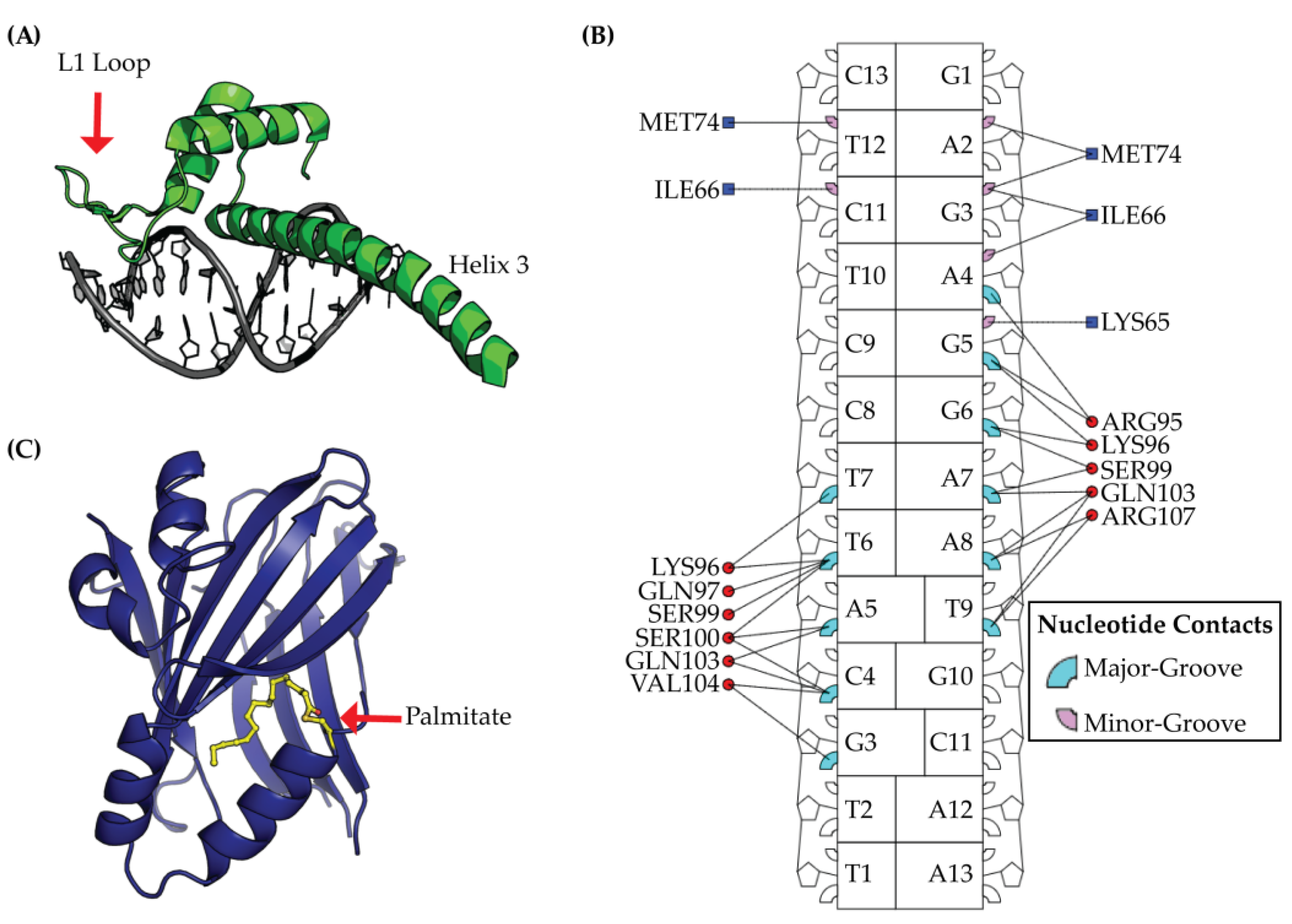
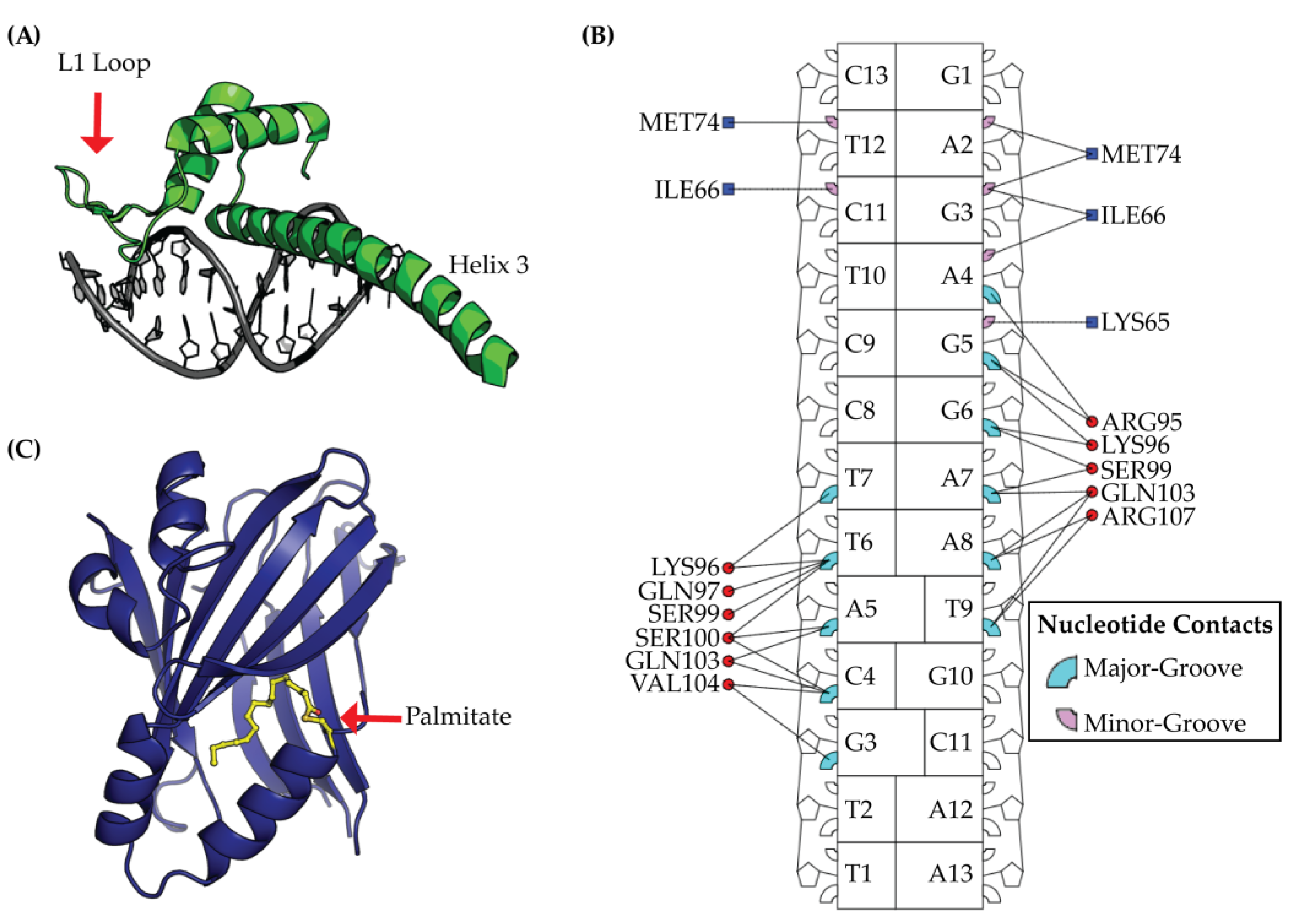
2.2. TEAD DNA Binding Domain (DBD)
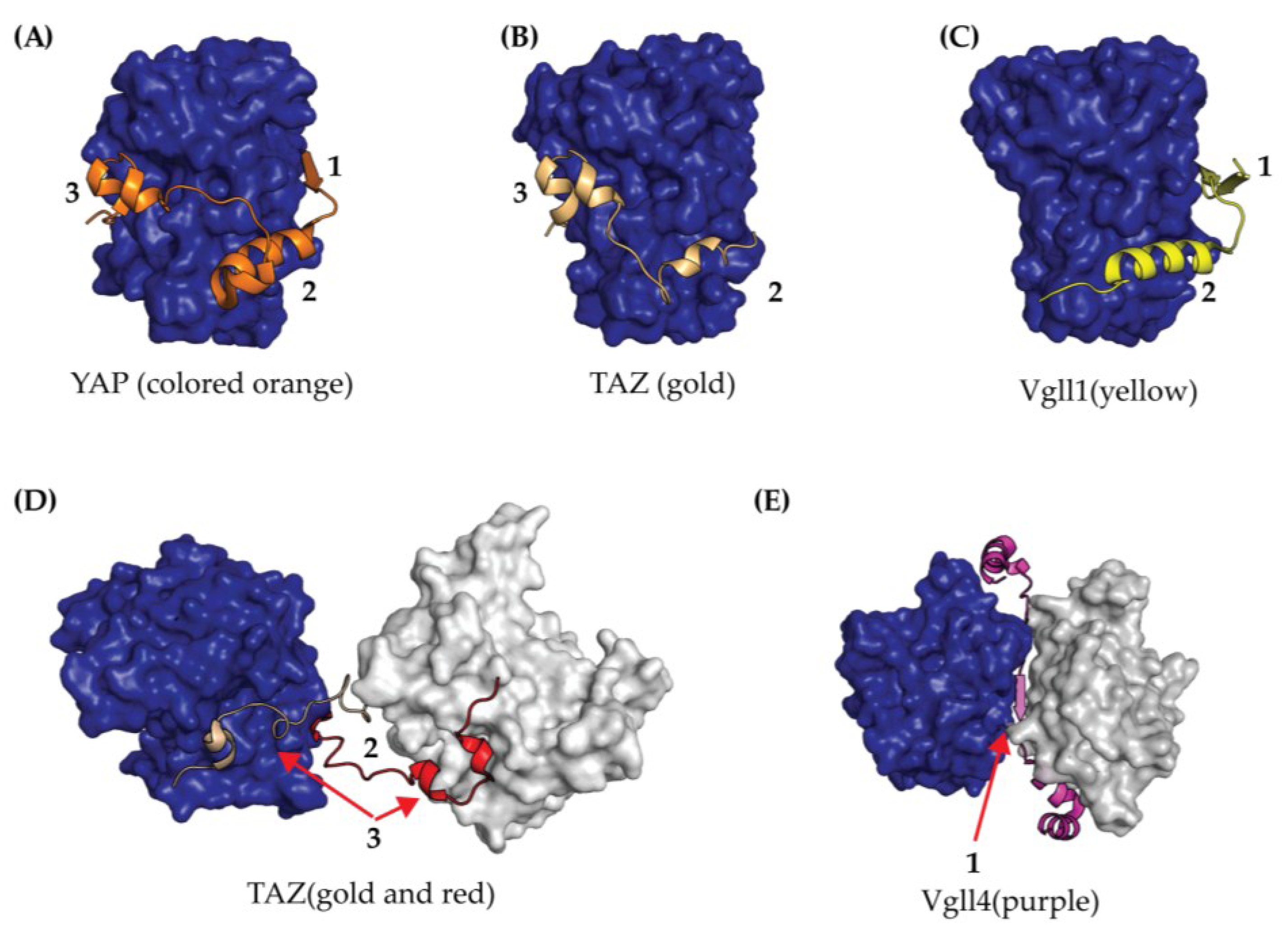
2.3. TEAD YAP Binding Domain (YBD)
3. Physiological Roles of TEAD
3.1. TEAD in Early Development
3.2. Oncogenic Function of TEAD
3.3. TEAD is Required for YAP/TAZ Oncogenesis
3.4. TEAD Modulates YAP Oncogenesis
4. TEAD as a Therapeutic Target
4.1. Targeting the DNA Binding Domain (DBD)
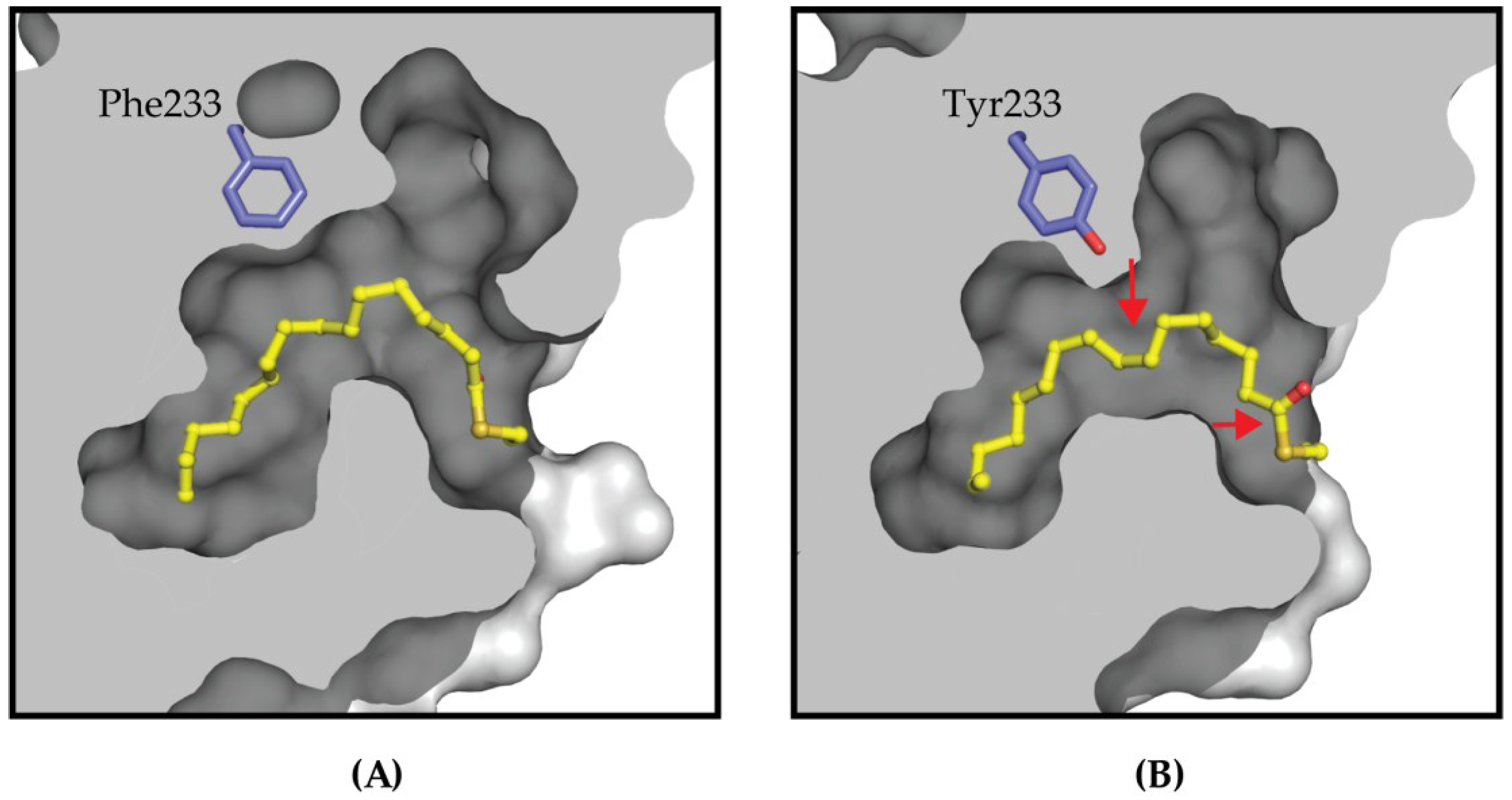
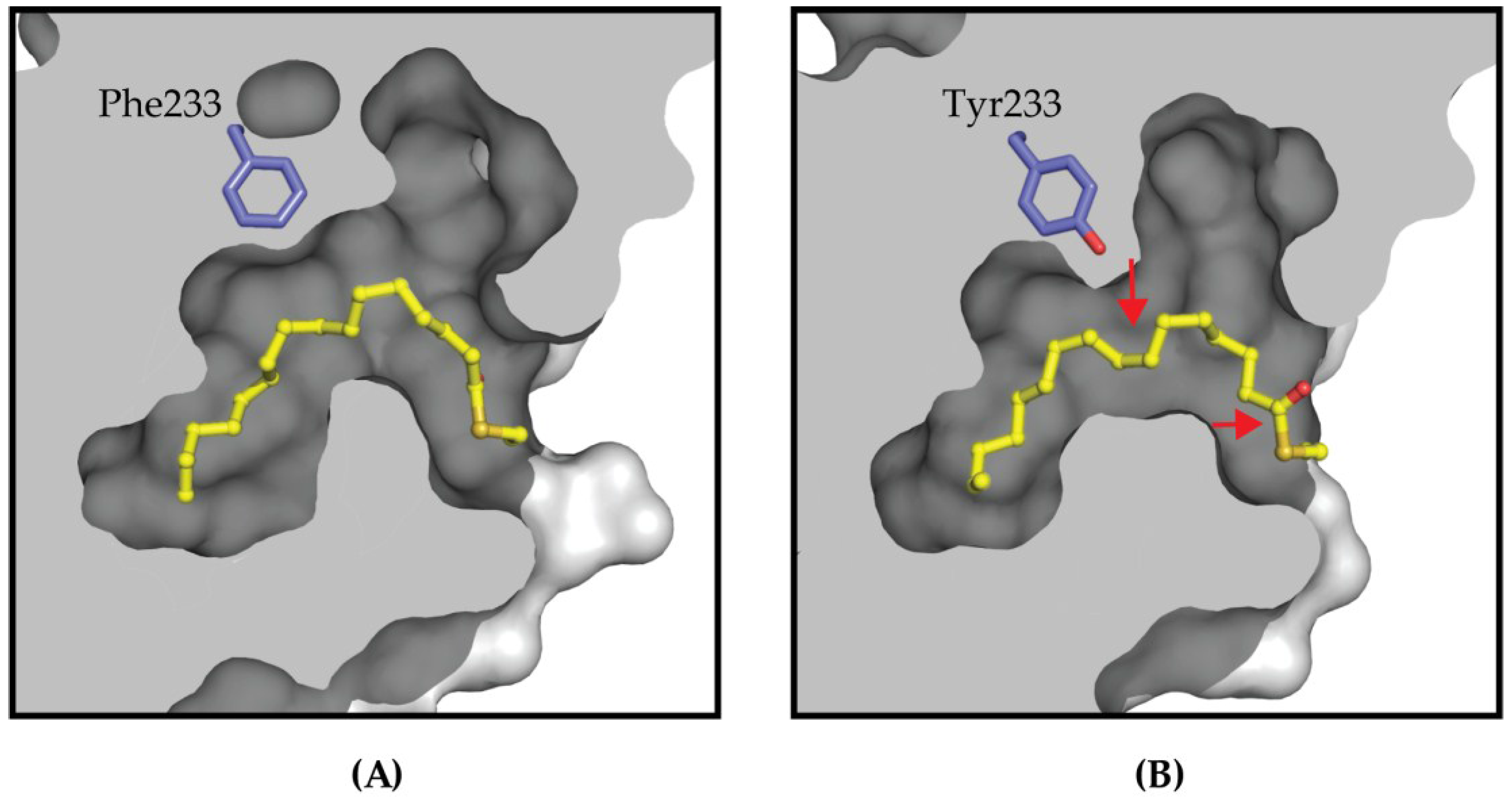
4.2. Targeting the Lipid Pocket
4.3. Targeting the TEAD-YAP Interface
5. Challenges and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by lats and CK1 regulates YAP stability through scf(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Slattery, M.; Ma, L.; Crofts, A.; White, K.P.; Mann, R.S.; Irvine, K.D. Genome-wide association of yorkie with chromatin and chromatin-remodeling complexes. Cell Rep. 2013, 3, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Smolen, G.A.; Haber, D.A. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila hippo pathway. Cancer Res. 2008, 68, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell. Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [PubMed]
- Kaneko, K.J.; DePamphilis, M.L. Regulation of gene expression at the beginning of mammalian development and the tead family of transcription factors. Dev. Genet. 1998, 22, 43–55. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Friedrich, G.A.; Soriano, P. Transcriptional enhancer factor 1 disruption by a retroviral gene TRAP leads to heart defects and embryonic lethality in mice. Genes Dev. 1994, 8, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.J.; Cullinan, E.B.; Latham, K.E.; DePamphilis, M.L. Transcription factor mTEAD-2 is selectively expressed at the beginning of zygotic gene expression in the mouse. Development 1997, 124, 1963–1973. [Google Scholar] [PubMed]
- Jacquemin, P.; Hwang, J.J.; Martial, J.A.; Dolle, P.; Davidson, I. A novel family of developmentally regulated mammalian transcription factors containing the TEA/ATTS DNA binding domain. J. Biol. Chem. 1996, 271, 21775–21785. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, P.; Sapin, V.; Alsat, E.; Evain-Brion, D.; Dolle, P.; Davidson, I. Differential expression of the TEF family of transcription factors in the murine placenta and during differentiation of primary human trophoblasts in vitro. Dev. Dyn. 1998, 212, 423–436. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.; Upadhyay, A.; Giuntini, F.; Eggleston, I.; Furutani-Seiki, M.; Ishima, R.; Bagby, S. Structural features and ligand binding properties of tandem ww domains from YAP and TAZ, nuclear effectors of the hippo pathway. Biochemistry 2011, 50, 3300–3309. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.I.; Sudol, M. The WW domain of yes-associated protein binds a proline-rich ligand that differs from the consensus established for SRC homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, C.J.; Oka, T.; Mazack, V.; Hilman, D.; Gat, U.; Muramatsu, T.; Inazawa, J.; Golden, A.; Carey, D.J.; Farooq, A.; et al. Identification, basic characterization and evolutionary analysis of differentially spliced mrna isoforms of human YAP1 gene. Gene 2012, 509, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999, 18, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, T.; Miyamura, N.; Hata, S.; Miura, R.; Hirayama, J.; Nishina, H. The pdz-binding motif of yes-associated protein is required for its co-activation of TEAD-mediated CTGF transcription and oncogenic cell transforming activity. Biochem. Biophys. Res. Commun. 2014, 443, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Anbanandam, A.; Albarado, D.C.; Nguyen, C.T.; Halder, G.; Gao, X.; Veeraraghavan, S. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc. Natl. Acad. Sci. USA 2006, 103, 17225–17230. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Polaczyk, P.; Kraus, M.E.; Hudson, A.; Kim, J.; Laughon, A.; Carroll, S. The vestigial and scalloped proteins act together to directly regulate wing-specific gene expression in drosophila. Genes Dev. 1998, 12, 3900–3909. [Google Scholar] [CrossRef] [PubMed]
- Benhaddou, A.; Keime, C.; Ye, T.; Morlon, A.; Michel, I.; Jost, B.; Mengus, G.; Davidson, I. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ. 2012, 19, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, E.; Taipale, J. Structural perspective of cooperative transcription factor binding. Curr. Opin. Struct. Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.W.; Desai, D.; Khan, S.; Eberhardt, N.L. Cooperative binding of TEF-1 to repeated ggaatg-related consensus elements with restricted spatial separation and orientation. DNA Cell Biol. 2000, 19, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Carroll, S.B. Binding of the vestigial co-factor switches the DNA-target selectivity of the scalloped selector protein. Development 2001, 128, 3295–3305. [Google Scholar] [PubMed]
- Lee, D.S.; Vonrhein, C.; Albarado, D.; Raman, C.S.; Veeraraghavan, S. A potential structural switch for regulating DNA-binding by tead transcription factors. J. Mol. Biol. 2016, 428, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; He, F.; Chen, M.; Hua, L.; Wang, W.; Jiao, S.; Zhou, Z. DNA-binding mechanism of the hippo pathway transcription factor TEAD4. Oncogene 2017, 36, 4362–4369. [Google Scholar] [CrossRef] [PubMed]
- Sagendorf, J.M.; Berman, H.M.; Rohs, R. Dnaprodb: An interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2017, 45, W89–W97. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural basis of yap recognition by TEAD4 in the hippo pathway. Genes Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD transcription factors is required for their stability and function in hippo pathway signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of tead proteins regulates transcriptional output of the hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Mesrouze, Y.; Meyerhofer, M.; Bokhovchuk, F.; Fontana, P.; Zimmermann, C.; Martin, T.; Delaunay, C.; Izaac, A.; Kallen, J.; Schmelzle, T.; et al. Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ. Protein Sci. 2017, 26, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Hannoush, R.N.; Sun, J. The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat. Chem. Biol. 2010, 6, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Yu, J.; Tomchick, D.R.; Pan, D.; Luo, X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. USA 2010, 107, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, M.; Sara, T.; Platzer, G.; Mateos, B.; Bokhovchuk, F.; Chene, P.; Konrat, R. (1)h, (13)c, (15)n resonance assignment of human YAP 50-171 fragment. Biomol. NMR Assign. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cairns, L.; Tran, T.; Kavran, J.M. Structural insights into the regulation of hippo signaling. ACS Chem. Biol. 2017, 12, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the VGLL1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Sawada, A.; Kiyonari, H.; Ukita, K.; Nishioka, N.; Imuta, Y.; Sasaki, H. Redundant roles of TEAD1 and TEAD2 in notochord development and the regulation of cell proliferation and survival. Mol. Cell. Biol. 2008, 28, 3177–3189. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.J.; Kohn, M.J.; Liu, C.; DePamphilis, M.L. Transcription factor TEAD2 is involved in neural tube closure. Genesis 2007, 45, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Kohn, M.J.; Karavanova, I.; Kaneko, K.J.; Vullhorst, D.; DePamphilis, M.L.; Buonanno, A. Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 2007, 134, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Hogg, K.; Robinson, W.P.; Beristain, A.G. Activation of endocrine-related gene expression in placental choriocarcinoma cell lines following DNA methylation knock-down. Mol. Hum. Reprod. 2014, 20, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.X.; Li, X.Y.; Zhang, Q.; Zhao, K.; Zhang, C.P.; Xue, C.H.; Yang, K.; Tian, Z.B. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5199–5205. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Park, J.L.; Kim, H.J.; Park, Y.K.; Kim, J.H.; Sohn, H.A.; Noh, S.M.; Song, K.S.; Kim, W.H.; Kim, Y.S.; et al. Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis 2014, 35, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.H.; Zhang, W. Tead1 enhances proliferation via activating SP1 in colorectal cancer. Biomed. Pharmacother. 2016, 83, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, G.; Yang, Y.; Mei, Z.; Liang, Z.; Cui, A.; Wu, T.; Liu, C.Y.; Cui, L. Increased tead4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene 2016, 35, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Nowee, M.E.; Snijders, A.M.; Rockx, D.A.; de Wit, R.M.; Kosma, V.M.; Hamalainen, K.; Schouten, J.P.; Verheijen, R.H.; van Diest, P.J.; Albertson, D.G.; et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J. Pathol. 2007, 213, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Adelaide, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67, 11565–11575. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.F.; Shepherd, C.J.; Rizzo, S.; Brewer, D.; Jhavar, S.; Dodson, A.R.; Cooper, C.S.; Eeles, R.; Falconer, A.; Kovacs, G.; et al. TEAD1 and c-Cbl are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br. J. Cancer 2008, 99, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Skotheim, R.I.; Autio, R.; Lind, G.E.; Kraggerud, S.M.; Andrews, P.W.; Monni, O.; Kallioniemi, O.; Lothe, R.A. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006, 28, 315–326. [Google Scholar] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.R.; Chaerkady, R.; Hu, S.; Wan, J.; Qian, J.; Zhu, H.; Pandey, A.; Kern, S.E. Unbiased discovery of interactions at a control locus driving expression of the cancer-specific therapeutic and diagnostic target, mesothelin. J. Proteome Res. 2012, 11, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.; Lim, C.J.; Guo, K.; Ng, C.P.; Lee, I.; Hunziker, W.; Zeng, Q.; Hong, W. A role for taz in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008, 68, 2592–2598. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Yao, T.J.; Lee, N.P.; Ng, I.O.; Chan, Y.T.; Zender, L.; Lowe, S.W.; Poon, R.T.; Luk, J.M. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009, 115, 4576–4585. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a SRC/yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Berube, J.; Fromont, A.; Bell, J.B. Ability of scalloped deletion constructs to rescue SD mutant wing phenotypes in drosophila melanogaster. Genome 2004, 47, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.H.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of tead4 attenuates the hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting transcriptional enhanced associate domains (TEADS). J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.Y.; Kang, C.; Chia, C.S.; Luo, X.; Hong, W.; et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Michels, S.; Schmidt-Erfurth, U. Photodynamic therapy with verteporfin: A new treatment in ophthalmology. Semin. Ophthalmol. 2001, 16, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Sim, A.Y.L.; Tan, S.K.J.; Verma, C.; Song, H. Targeting YAP/TAZ-TEAD protein-protein interactions using fragment-based and computational modeling approaches. PLoS ONE 2017, 12, e0178381. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, G.; Thurston, D.E. Targeting protein-protein interactions for therapeutic intervention: A challenge for the future. Future Med. Chem. 2009, 1, 65–93. [Google Scholar] [CrossRef] [PubMed]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. https://doi.org/10.3390/cancers10030081
Holden JK, Cunningham CN. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers. 2018; 10(3):81. https://doi.org/10.3390/cancers10030081
Chicago/Turabian StyleHolden, Jeffrey K., and Christian N. Cunningham. 2018. "Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors" Cancers 10, no. 3: 81. https://doi.org/10.3390/cancers10030081
APA StyleHolden, J. K., & Cunningham, C. N. (2018). Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers, 10(3), 81. https://doi.org/10.3390/cancers10030081