New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response



Abstract
1. Introduction
2. Localized Regulation of Akt Activation and Activity
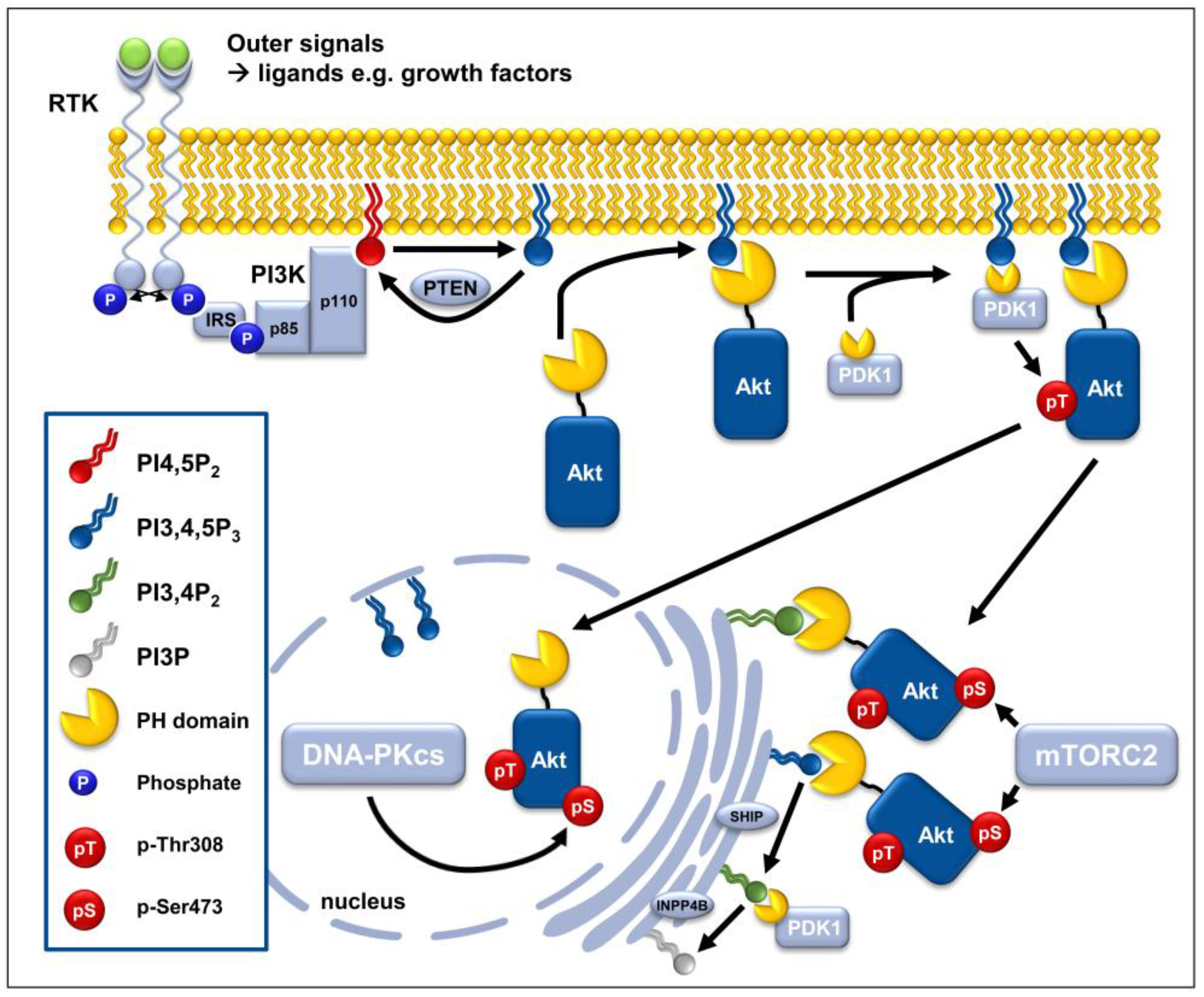
2.1. Phosphorylation and Activation of Akt
2.1.1. PI3K-Dependent Phosphorylation of Akt at the Cytoplasmic Membrane
2.1.2. mTORC2-Dependent Activation of AKT at Subcellular Membrane Compartments
2.1.3. DNA-PKcs-Dependent Phosphorylation of Akt in the Nuclear Compartment
2.1.4. Additional Phosphorylation Sites of Akt
Perspective
2.2. Termination of Akt Activity by Dephosphorylation
2.2.1. Lipid Phosphatases PTEN and INPP4B
2.2.2. Protein Phosphatases PHLPP1/2, PP2A
Perspective
2.3. Further Factors Regulating Akt Activation
2.3.1. 5′-Lipid Phosphatases
2.3.2. PH-Domain Containing Proteins
2.3.3. AIM2 and HSP90
2.3.4. Ubiquitin-Modifying Enzymes
2.3.5. miRNA
Perspective
3. Subcellular Network of Akt Target Proteins
3.1. General Aspects of Target Protein Selection
3.1.1. Importance of the Akt Isoform for Target Protein Selection
3.1.2. Importance of Akt’s Phosphorylation State for Target Protein Selection
3.1.3. Importance of Subcellular Localization for Target Protein Selection
Perspective
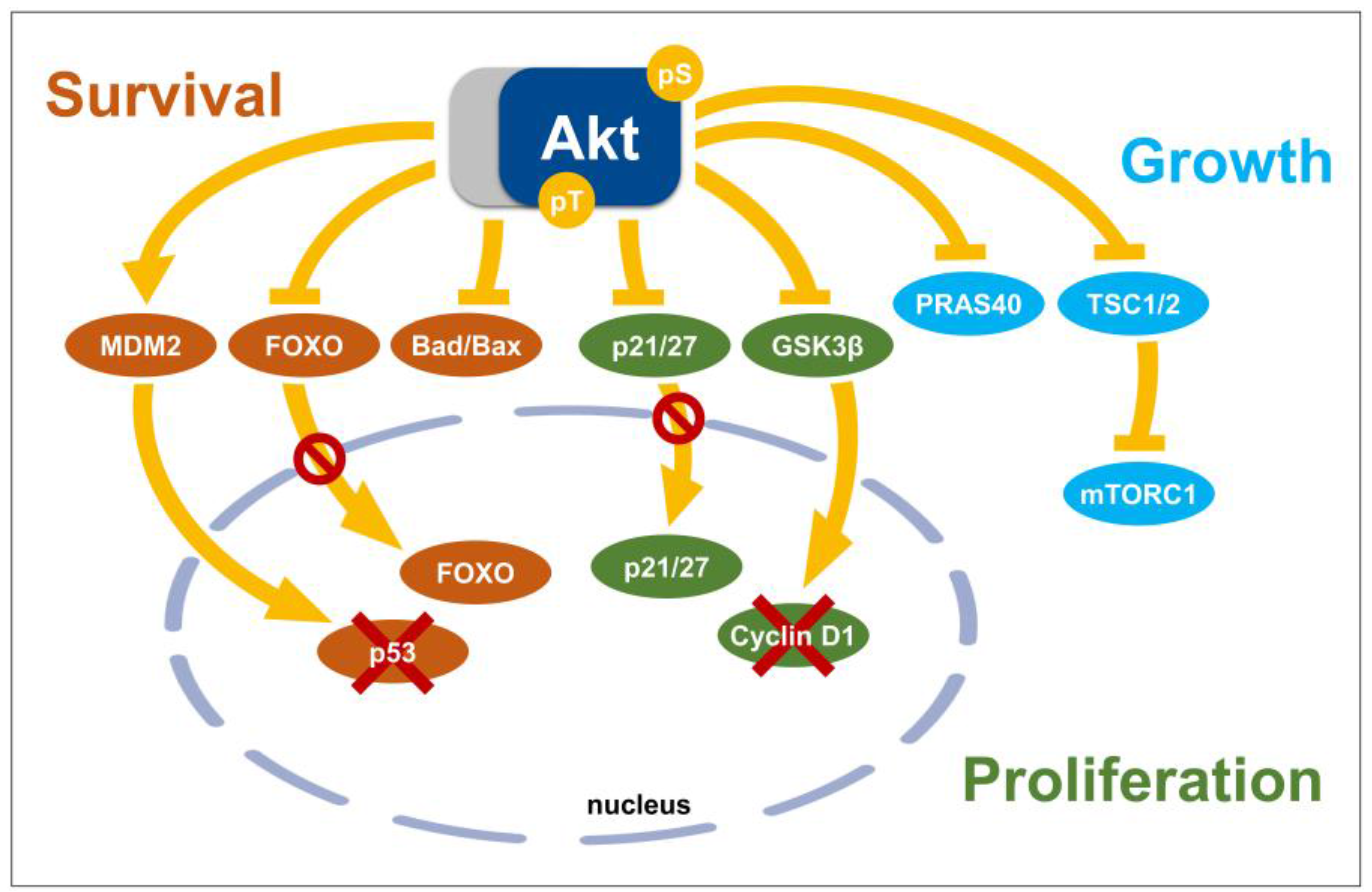
3.2. Akt Substrates with Functions as Key Regulators of Cell Signaling Networks
3.2.1. Glycogen Synthase Kinase 3, GSK3
3.2.2. FOXO Transcription Factors
3.2.3. TSC2, mTORC1, and PRAS40
3.2.4. Mdm2
3.3. Direct Effector Proteins of Akt with Roles in Cell Cycle Regulation and Cell Death
3.3.1. The Inhibitors of Cyclin-Dependent Kinase, p21 and p27
3.3.2. Pro-Apoptotic Members of the Bcl-2 Family, Bad and Bax
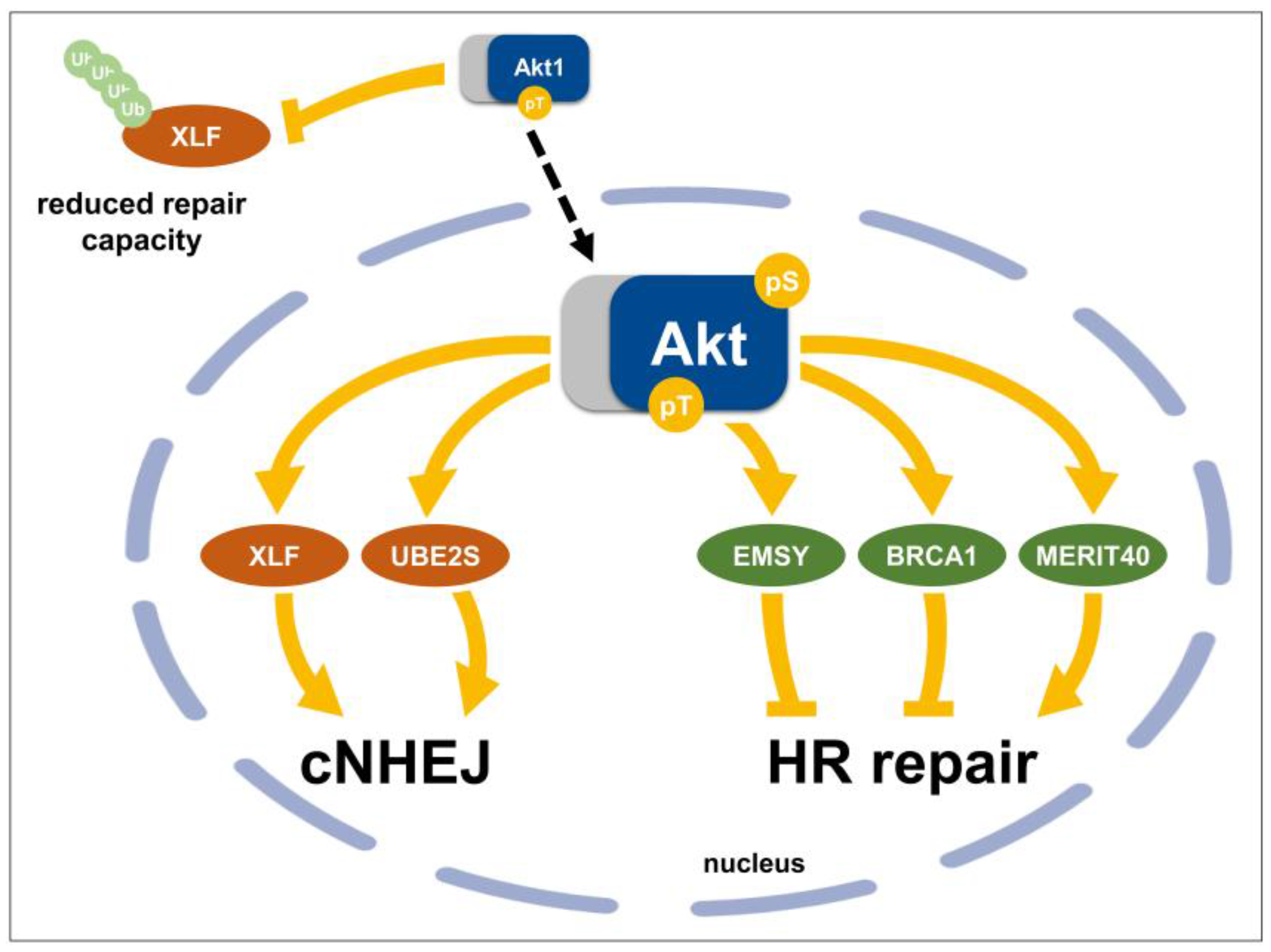
3.4. Direct Nuclear Akt Target Proteins with Roles in DSB Repair
3.4.1. Chk1 (Checkpoint Kinase 1)
3.4.2. BRCA1 (Breast Cancer 1)
3.4.3. MERIT40 (Mediator of Rap80 Interactions and Targeting 40 kDa)
3.4.4. EMSY (BRCA2-Interacting Transcriptional Repressor)
3.4.5. XLF (Cernunnos/XRCC4-Like Factor)
3.4.6. UBE2S (Ubiquitin-Conjugating Enzyme E2S)
Perspective
4. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. Pi3k and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase b is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the Pi3k/Akt pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase akt pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the pten tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of Akt1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase b isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/pkbalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Tschopp, O.; Di-Poi, N.; Bruder, E.; Baudry, A.; Dummler, B.; Wahli, W.; Hemmings, B.A. Dosage-dependent effects of Akt1/protein kinase balpha (pkbalpha) and Akt3/pkbgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol. Cell. Biol. 2005, 25, 10407–10418. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, W.N.; Chen, X.; Peng, X.D.; Jeon, S.M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Henderson, V.; Smith, B.; Burton, L.J.; Randle, D.; Morris, M.; Odero-Marah, V.A. Snail promotes cell migration through Pi3K/Akt-dependent rac1 activation as well as Pi3K/Akt-independent pathways during prostate cancer progression. Cell Adhes. Migr. 2015, 9, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Wagner, A.J.; Conzen, S.D.; Jordan, J.; Bellacosa, A.; Tsichlis, P.N.; Hay, N. The Pi 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997, 11, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the Akt signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 42700. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 acts downstream of DNA-pk in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. Akt/Pkb signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific Pi3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef]
- Kim, D.; Chung, J. Akt: Versatile mediator of cell survival and beyond. J. Biochem. Mol. Biol. 2002, 35, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, Y.; McCoull, D.; Davidson, L.; Leslie, N.R.; Fairservice, A.; Gray, A.; Lucocq, J.; Downes, C.P. Localization of agonist-sensitive Ptdins(3,4,5)p3 reveals a nuclear pool that is insensitive to PTEN expression. J. Cell Sci. 2006, 119, 5160–5168. [Google Scholar] [CrossRef] [PubMed]
- Watt, S.A.; Kular, G.; Fleming, I.N.; Downes, C.P.; Lucocq, J.M. Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem. J. 2002, 363, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. Emerging evidence of signalling roles for Pi(3,4)p2 in class I and II Pi3k-regulated pathways. Biochem. Soc. Trans. 2016, 44, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; Hemmings, B.A.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase b define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Laguerre, M.; Parker, P.J.; Larijani, B. Role of a novel Ph-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, e17. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Kitani, T.; Matsuzaki, H.; Konishi, H.; Kikkawa, U.; Fujisawa, H. Studies on the phosphorylation of protein kinase b by Ca2+/calmodulin-dependent protein kinases. J. Biochem. 2000, 127, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/Pkb by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a Pkb/Akt hydrophobic motif ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Caudwell, F.B.; Andjelkovic, M.; Hemmings, B.A.; Cohen, P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996, 399, 333–338. [Google Scholar] [CrossRef]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and amp-pnp. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. Sin1/Mip1 maintains rictor-mTOR complex integrity and regulates akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to akt-foxo and pkcalpha, but not s6k1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between akt and mTORC2 via sin1 phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of akt1 enhances radiation toxicity of human tumor cells by inhibiting DNA-pkcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Persad, S.; Attwell, S.; Gray, V.; Mawji, N.; Deng, J.T.; Leung, D.; Yan, J.; Sanghera, J.; Walsh, M.P.; Dedhar, S. Regulation of protein kinase b/akt-serine 473 phosphorylation by integrin-linked kinase: Critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 2001, 276, 27462–27469. [Google Scholar] [CrossRef] [PubMed]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-oh kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase b/akt by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The tsc1-tsc2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. Ptdins(3,4,5)p3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Sinkovics, B.; Szczygiel, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Walther, T.C. Torc2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.A.; Buck, M.; Cloonan, N.; Hancock, J.F.; Suhrbier, A.; Sculley, T.; Bushell, G. Human Sin1 contains Ras-binding and Pleckstrin homology domains and suppresses ras signalling. Cell. Signal. 2007, 19, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, N.; Chung, G.H.; Lete, M.G.; Alonso, A.; Byrne, R.D.; Calleja, V.; Larijani, B. Endomembrane ptdins(3,4,5)p3 activates the Pi3K-Akt pathway. J. Cell Sci. 2015, 128, 3456–3465. [Google Scholar] [CrossRef] [PubMed]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. Pi3K-C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of akt phosphorylation at ser473 and thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Shpiro, N.; Alessi, D.R. Akt is efficiently activated by pif-pocket- and Ptdins(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem. J. 2012, 448, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Feng, J.; Li, Y.; Hammarsten, O.; Brazil, D.P.; Hemmings, B.A. DNA-dependent protein kinase-mediated phosphorylation of protein kinase b requires a specific recognition sequence in the c-terminal hydrophobic motif. J. Biol. Chem. 2009, 284, 6169–6174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Yue, P.; Tao, H.; Ramalingam, S.S.; Owonikoko, T.K.; Deng, X.; Wang, Y.; Fu, H.; Khuri, F.R.; et al. Protein phosphatase 2A and DNA-dependent protein kinase are involved in mediating rapamycin-induced Akt phosphorylation. J. Biol. Chem. 2013, 288, 13215–13224. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Huang, J.; Basu, A. Protein kinase cepsilon activates protein kinase B/Akt via DNA-pk to protect against tumor necrosis factor-alpha-induced cell death. J. Biol. Chem. 2006, 281, 22799–22807. [Google Scholar] [CrossRef] [PubMed]
- Surucu, B.; Bozulic, L.; Hynx, D.; Parcellier, A.; Hemmings, B.A. In vivo analysis of protein kinase b (PKB)/Akt regulation in DNA-PKCS-null mice reveals a role for Pkb/Akt in DNA damage response and tumorigenesis. J. Biol. Chem. 2008, 283, 30025–30033. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-pk mediates Akt activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-pkcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, M.; Lu, Y.; Liang, J.; Lorenzi, P.L.; Bai, S.; Hawke, D.H.; Li, J.; Dogruluk, T.; Scott, K.L.; et al. Coordinate phosphorylation of multiple residues on single Akt1 and Akt2 molecules. Oncogene 2014, 33, 3463–3472. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase Ck2 phosphorylates and upregulates Akt/Pkb. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in Pkc and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Bulek, K.; Xiao, H.; Yu, M.; Gao, J.; Sun, L.; Beurel, E.; Kaidanovich-Beilin, O.; Fox, P.L.; DiCorleto, P.E.; et al. Inactivation of the enzyme gsk3alpha by the kinase IKKI promotes akt-mTOR signaling pathway that mediates interleukin-1-induced th17 cell maintenance. Immunity 2012, 37, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.F. Nuclear inositide signalling—Expansion, structures and clarification. Biochim. Biophys. Acta 2006, 1761, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Lavi, S.; Iwai, K.; Reiss, Y.; Ciechanover, A.; et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-cbl/sli-1. Mol. Cell 1999, 4, 1029–1040. [Google Scholar] [CrossRef]
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Redondo-Munoz, J.; Perez-Garcia, V.; Cortes, I.; Chagoyen, M.; Carrera, A.C. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Mol. Cell. Biol. 2011, 31, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.J.; Lehmann, P.Z.; Li, W. Nuclear pi3k signaling in cell growth and tumorigenesis. Front. Cell. Dev. Biol. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between Akt and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim. Biophys. Acta 2014, 1846, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.D.; Lum, M.A.; Xu, C.; Black, J.D.; Wang, X. Ubiquitin-dependent regulation of phospho-Akt dynamics by the ubiquitin E3 ligase, NEDD4-1, in the insulin-like growth factor-1 response. J. Biol. Chem. 2013, 288, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, S.J.; Ferguson, D.T.; Mitchell, C.A.; Ooms, L.M. Regulation of Pi3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci. Rep. 2017, 37, BSR20160432. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, pten/mmac1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Ittmann, M.M. Chromosome 10 alterations in prostate adenocarcinoma (review). Oncol. Rep. 1998, 5, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Jendrossek, V.; Henkel, M.; Hennenlotter, J.; Vogel, U.; Ganswindt, U.; Muller, I.; Handrick, R.; Anastasiadis, A.G.; Kuczyk, M.; Stenzl, A.; et al. Analysis of complex protein kinase b signalling pathways in human prostate cancer samples. BJU Int. 2008, 102, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the Pi3K/Akt pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Helman, L.J. Levels of pten protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene 2003, 22, 8205–8211. [Google Scholar] [CrossRef] [PubMed]
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Simmonet Gouy, V.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Recher, C. The level of Akt phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Maiti, S.; Mandal, C. PTEN negatively regulates mTORC2 formation and signaling in grade IV glioma via Rictor hyperphosphorylation at Thr1135 and direct the mode of action of an mTORC1/2 inhibitor. Oncogenesis 2016, 5, e227. [Google Scholar] [CrossRef] [PubMed]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits Pi3K signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, I.U.; Hodgson, M.C.; Bowden, W.A.; Ittmann, M.M. Inpp4b: The new kid on the Pi3K block. Oncotarget 2011, 2, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Furnari, F.; Newton, A.C. Phlpp: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, A.K.; Niederst, M.J.; Newton, A.C. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2013, 280, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Pratt, C.P.; Zeeman, M.E.; Schultz, N.; Taylor, B.S.; O’Neill, A.; Castillo-Martin, M.; Nowak, D.G.; Naguib, A.; Grace, D.M.; et al. Identification of phlpp1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 2011, 20, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, X.; Wang, J.; Li, T.; Jin, T.; Lei, D.; Pan, X. Aberrant expression of PHLPP1 and PHLPP2 correlates with poor prognosis in patients with hypopharyngeal squamous cell carcinoma. PLoS ONE 2015, 10, e0119405. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Yang, H.; Wang, W.; Xie, Y.; Hu, W.; Ye, M.; Chen, X. High PHLPP expression is associated with better prognosis in patients with resected lung adenocarcinoma. BMC Cancer 2015, 15, 687. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Deng, J.; Zhang, L.; Xie, X.; Guo, X.; Sun, C.; Zhang, R.; Liang, H. Lower expression of PH domain leucine-rich repeat protein phosphatase 1 (PHLPP1) association with poor prognosis of gastric cancer. Int. J. Clin. Exp. Med. 2015, 8, 20481–20489. [Google Scholar] [PubMed]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.C. A new role of protein phosphatase 2A in adenoviral E1A protein-mediated sensitization to anticancer drug-induced apoptosis in human breast cancer cells. Cancer Res. 2004, 64, 5938–5942. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of thr-308 of Akt, cell proliferation, and survival by the b55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Koch, U.; Hehlgans, S.; Sandfort, V.; Stanchi, F.; Zips, D.; Baumann, M.; Shevchenko, A.; Pilarsky, C.; Haase, M.; et al. Pinch1 regulates Akt1 activation and enhances radioresistance by inhibiting PP1alpha. J. Clin. Investig. 2010, 120, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Thayyullathil, F.; Chathoth, S.; Shahin, A.; Kizhakkayil, J.; Hago, A.; Patel, M.; Galadari, S. Protein phosphatase 1-dependent dephosphorylation of Akt is the prime signaling event in sphingosine-induced apoptosis in jurkat cells. J. Cell Biochem. 2011, 112, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-independent role of aim2 in suppressing colon tumorigenesis via DNA-pk and akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. Ph domain-only protein phlda3 is a p53-regulated repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O'Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via Akt. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Hendrickson, A.E.W.; Yun, S.S.; Han, J.J.; Schneider, P.A.; Koh, B.D.; Stenson, M.J.; Wellik, L.E.; Shing, J.C.; Peterson, K.L.; et al. Dual mTORC1/mTORC2 inhibition diminishes akt activation and induces puma-dependent apoptosis in lymphoid malignancies. Blood 2012, 119, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Su, C.H.; Wang, C.Y.; Lan, K.H.; Li, C.P.; Chao, Y.; Lin, H.C.; Lee, S.D.; Lee, W.P. Akt phosphorylation at thr308 and ser473 is required for chip-mediated ubiquitination of the kinase. Cell Signal. 2011, 23, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kim, S.Y.; Jung, J.H.; Yoon, Y.; Cha, H.J.; Lee, H.; Kim, K.; Kim, J.; An, I.S.; Um, H.D.; et al. Akt is negatively regulated by the mulan e3 ligase. Cell Res. 2012, 22, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Koren, J.; Zhang, Y.J.; Xu, Y.F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and chip coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The skp2-scf e3 ligase regulates akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The e3 ligase traf6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gao, Y.; Li, L.; Jin, G.; Cai, Z.; Chao, J.I.; Lin, H.K. K63-linked ubiquitination in kinase activation and cancer. Front. Oncol. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.F.; He, K.; Wang, N.; Sang, Z.H.; Qiu, X.; Xu, G.; Jian, Z.; Liang, B.; Li, T.; Li, H.Y.; et al. Nedd4 ubiquitinates traf3 to promote cd40-mediated Akt activation. Nat. Commun. 2014, 5, 4513. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Josse, C.; Bouznad, N.; Geurts, P.; Irrthum, A.; Huynh-Thu, V.A.; Servais, L.; Hego, A.; Delvenne, P.; Bours, V.; Oury, C. Identification of a microRNA landscape targeting the pi3k/akt signaling pathway in inflammation-induced colorectal carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G229–G243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Ding, Y.; Li, Q.; Wang, R.; Liu, T.; Sun, Q.; Yang, H.; Peng, S.; Wang, W.; et al. Mir-20a induces cell radioresistance by activating the PTEN/Pi3K/Akt signaling pathway in hepatocellular carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Salajegheh, A.; Smith, R.A.; Lam, A.K. MicroRNA-126 suppresses proliferation of undifferentiated (braf(v600e) and braf(wt)) thyroid carcinoma through targeting pik3r2 gene and repressing pi3k-akt proliferation-survival signalling pathway. Exp. Cell Res. 2015, 339, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.Q.; Yi, H.M.; Ye, X.; Zhu, J.F.; Yi, H.; Li, L.N.; Xiao, T.; Yuan, L.; Li, J.Y.; Wang, Y.Y.; et al. MiRNA-203 reduces nasopharyngeal carcinoma radioresistance by targeting IL8/Akt signaling. Mol. Cancer Ther. 2015, 14, 2653–2664. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Ahn, J.; Guo, D.; Votaw, J.R.; Shim, H. MicroRNA-302 replacement therapy sensitizes breast cancer cells to ionizing radiation. Pharm. Res. 2013, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Hwang, Y.H.; Lee, D.J.; Kim, D.H.; Park, J.M.; Wu, H.G.; Kim, I.A. MicroRNA-203 modulates the radiation sensitivity of human malignant glioma cells. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Sun, B.; Jiang, Y.; Zheng, J.; Yang, N.; Ji, C.; Liang, Z.; Shi, J.; Zhang, R.; Liu, Y.; et al. MicroRNA-31 inhibits lung adenocarcinoma stem-like cells via down-regulation of MET-PI3K-AKT signaling pathway. Anti-Cancer Agents Med. Chem. 2016, 16, 501–518. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Hu, L.; He, Y.; Shi, Z.; Tang, S.; Chen, Y. Abnormal expression of mir-21 and mir-95 in cancer stem-like cells is associated with radioresistance of lung cancer. Cancer Investig. 2015, 33, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Abreu, C.; Prieto, D.; Morande, P.; Ruiz, S.; Fernandez-Calero, T.; Naya, H.; Libisch, G.; Robello, C.; Landoni, A.I.; et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia 2015, 29, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, I.; Tapia, O.; Leal, P.; Sandoval, A.; Varga, M.G.; Letelier, P.; Buchegger, K.; Bizama, C.; Espinoza, J.A.; Peek, R.M.; et al. miR-101-2, miR-125b-2 and miR-451a act as potential tumor suppressors in gastric cancer through regulation of the PI3K/AKT/mTOR pathway. Cell. Oncol. 2016, 39, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.; Liu, Z.; Yao, B.; Han, S.; Yang, W. MicroRNA-519a promotes tumor growth by targeting PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Int. J. Oncol. 2016, 48, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Saji, M.; Vasko, V.; Kada, F.; Allbritton, E.H.; Burman, K.D.; Ringel, M.D. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem. Biophys. Res. Commun. 2005, 332, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Rabinovitz, I.; Sun, J.F.; Prendergast, G.C.; Benjamin, L.E. Rhob controls akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003, 17, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Akt/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed]
- Alt, J.R.; Cleveland, J.L.; Hannink, M.; Diehl, J.A. Phosphorylation-dependent regulation of Cyclin d1 nuclear export and Cyclin d1-dependent cellular transformation. Genes Dev. 2000, 14, 3102–3114. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Bartkowiak, K.; Grupp, K.; Brandt, B.; Pantel, K.; Jucker, M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell Signal. 2011, 23, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Gustafsson, A.S.; Pendekanti, P.N.; Glimelius, B.; Stenerlow, B. The influence of Akt isoforms on radiation sensitivity and DNA repair in colon cancer cell lines. Tumour biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Balzano, D.; Fawal, M.A.; Velazquez, J.V.; Santiveri, C.M.; Yang, J.; Pastor, J.; Campos-Olivas, R.; Djouder, N.; Lietha, D. Alternative activation mechanisms of protein kinase b trigger distinct downstream signaling responses. J. Biol. Chem. 2015, 290, 24975–24985. [Google Scholar] [CrossRef] [PubMed]
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavare, J.M. Akt phosphorylation on Thr308 but not on ser473 correlates with akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Ye, Q.; Liu, S.; She, Q.B. Akt inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E-BP1 axis. Oncotarget 2015, 6, 13962–13977. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.F.; Hunter, R.W.; Hers, I. mTORC2 protein complex-mediated Akt (protein kinase B) serine 473 phosphorylation is not required for Akt1 activity in human platelets (corrected]. J. Biol. Chem. 2011, 286, 24553–24560. [Google Scholar] [CrossRef] [PubMed]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Hemmings, B.A. Pikking on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Drozdz, M.M.; Vaux, D.J. Shared mechanisms in physiological and pathological nucleoplasmic reticulum formation. Nucleus 2017, 8, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Maiti, D.; Bhattacharyya, A.; Basu, J. Lipoarabinomannan from mycobacterium tuberculosis promotes macrophage survival by phosphorylating bad through a phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 2001, 276, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Polzien, L.; Benz, R.; Rapp, U.R. Can bad pores be good? New insights from examining bad as a target of raf kinases. Adv. Enzyme Regul. 2010, 50, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Altiok, S.; Batt, D.; Altiok, N.; Papautsky, A.; Downward, J.; Roberts, T.M.; Avraham, H. Heregulin induces phosphorylation of brca1 through phosphatidylinositol 3-kinase/Akt in breast cancer cells. J. Biol. Chem. 1999, 274, 32274–32278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. Akt regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in BRCA1-deficient cells by blocking the chk1-rad51 pathway. Oncogene 2013, 32, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- King, F.W.; Skeen, J.; Hay, N.; Shtivelman, E. Inhibition of chk1 by activated PKB/Akt. Cell Cycle 2004, 3, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Hegarat, N.; Black, E.J.; Scott, M.T.; Hochegger, H.; Gillespie, D.A. Akt/PKB suppresses DNA damage processing and checkpoint activation in late g2. J. Cell Biol. 2010, 190, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.P.; Mollenauer, M.N.; Xu, Z.; Turck, C.W.; Weiss, A. Akt-dependent phosphorylation specifically regulates cot induction of NF-κB-dependent transcription. Mol. Cell. Biol. 2002, 22, 5962–5974. [Google Scholar] [CrossRef] [PubMed]
- Ezell, S.A.; Polytarchou, C.; Hatziapostolou, M.; Guo, A.; Sanidas, I.; Bihani, T.; Comb, M.J.; Sourvinos, G.; Tsichlis, P.N. The protein kinase Akt1 regulates the interferon response through phosphorylation of the transcriptional repressor emsy. Proc. Natl. Acad. Sci. USA 2012, 109, E613–E621. [Google Scholar] [CrossRef] [PubMed]
- Ezell, S.A.; Tsichlis, P.N. Akt1, EMSY, BRCA2 and type I IFN signaling: A novel arm of the IFN response. Transcription 2012, 3, 305–309. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ibanez, A.; Rio, P.; Casado, J.A.; Bueren, J.A.; Fernandez-Luna, J.L.; Pipaon, C. Elevated levels of IL-1beta in Fanconi anaemia group a patients due to a constitutively active phosphoinositide 3-kinase-akt pathway are capable of promoting tumour cell proliferation. Biochem. J. 2009, 422, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, T.; Nagashima, T.; Komatsu, N.; Kirito, K.; Furukawa, Y.; Kobayashi Si, S.; Liu, J.M.; Ozawa, K. Phosphorylation of fanconi anemia protein, FANCA, is regulated by Akt kinase. Biochem. Biophys. Res. Commun. 2002, 291, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Erdamar, S.; Dai, H.; Wheeler, T.M.; Frolov, A.; Scardino, P.T.; Thompson, T.C.; Ayala, G.E. Forkhead protein FKHR and its phosphorylated form p-FKHR in human prostate cancer. Hum. Pathol. 2007, 38, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase b. J. Biol. Chem. 1999, 274, 17179–17183. [Google Scholar] [CrossRef] [PubMed]
- Yip, W.K.; Leong, V.C.; Abdullah, M.A.; Yusoff, S.; Seow, H.F. Overexpression of phospho-akt correlates with phosphorylation of EGF receptor, FKHR and bad in nasopharyngeal carcinoma. Oncol. Rep. 2008, 19, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Kashii, Y.; Uchida, M.; Kirito, K.; Tanaka, M.; Nishijima, K.; Toshima, M.; Ando, T.; Koizumi, K.; Endoh, T.; Sawada, K.; et al. A member of forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3-kinase-akt activation pathway in erythropoietin signal transduction. Blood 2000, 96, 941–949. [Google Scholar] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in pc12 cells. J. Biol. Chem. 2000, 275, 39152–39158. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Ichino, A.; Hayashi, T.; Yamamoto, T.; Kikkawa, U. Regulation of intracellular localization and transcriptional activity of FOXO4 by protein kinase B through phosphorylation at the motif sites conserved among the foxo family. J. Biochem. 2005, 138, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; de Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin a degradation and LMNA gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Bertacchini, J.; Beretti, F.; Lattanzi, G.; Bavelloni, A.; Riccio, M.; Ruzzene, M.; Marin, O.; Arrigoni, G.; Parnaik, V.; et al. Lamin a ser404 is a nuclear target of akt phosphorylation in C2C12 cells. J. Proteome Res. 2008, 7, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.C. HER-2/neu induces p53 ubiquitination via akt-mediated Mdm2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Davids, M.S.; Rodon, J.; Abrisqueta, P.; Kasar, S.N.; Lager, J.; Jiang, J.; Egile, C.; Awan, F.T. Phase i trial of the pan-PI3K inhibitor pilaralisib (SAR245408/XL147) in patients with chronic lymphocytic leukemia (CLL) or relapsed/refractory lymphoma. Clin. Cancer Res. 2015, 21, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dowbenko, D.; Lasky, L.A. Akt/pkb phosphorylation of p21cip/waf1 enhances protein stability of p21cip/waf1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Rossig, L.; Badorff, C.; Holzmann, Y.; Zeiher, A.M.; Dimmeler, S. Glycogen synthase kinase-3 couples akt-dependent signaling to the regulation of p21cip1 degradation. J. Biol. Chem. 2002, 277, 9684–9689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.H.; Hung, M.C. Cytoplasmic localization of p21cip1/waf1 by akt-induced phosphorylation in her-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sato, S.; Katayama, K.; Tsuruo, T. Akt-dependent phosphorylation of p27kip1 promotes binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 2002, 277, 28706–28713. [Google Scholar] [CrossRef] [PubMed]
- Larrea, M.D.; Liang, J.; Da Silva, T.; Hong, F.; Shao, S.H.; Han, K.; Dumont, D.; Slingerland, J.M. Phosphorylation of p27kip1 regulates assembly and activation of Cyclin d1-cdk4. Mol. Cell. Biol. 2008, 28, 6462–6472. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. Pkb/akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated g1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the akt/pkb substrate pras40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich akt substrate as a 14-3-3 binding partner. J. Biol. Chem. 2003, 278, 10189–10194. [Google Scholar] [CrossRef] [PubMed]
- Urano-Tashiro, Y.; Sasaki, H.; Sugawara-Kawasaki, M.; Yamada, T.; Sugiyama, A.; Akiyama, H.; Kawasaki, Y.; Tashiro, F. Implication of akt-dependent prp19 alpha/14-3-3beta/cdc5l complex formation in neuronal differentiation. J. Neurosci. Res. 2010, 88, 2787–2797. [Google Scholar] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal s6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Hu, Z.; He, S.; Wang, F.; Shen, H.; Zhang, H. Tsc1/Tsc2 inactivation inhibits Akt through mTORC1-dependent up-regulation of STAT3-pten cascade. Cancer Lett. 2011, 313, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, X.; Liu, Q.; Xu, J.; Ge, H.; Wang, Z.; Wang, H.; Wang, Z.; Shi, C.; Xu, X.; et al. Ube2s, a novel substrate of akt1, associates with ku70 and regulates DNA repair and glioblastoma multiforme resistance to chemotherapy. Oncogene 2017, 36, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Wang, B. RNF8- and Ube2S-dependent ubiquitin lysine 11-linkage modification in response to DNA damage. Mol. Cell 2017, 66, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/protein kinase b-dependent phosphorylation and inactivation of wee1hu promote cell cycle progression at g2/m transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of xlf impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase b. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Frame, S. The renaissance of gsk3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates Cyclin d1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Kakuda, S.; Ochiai, Y.; Nakagawa, H.; Kuwahara, Y.; Takai, Y.; Kobayashi, J.; Komatsu, K.; Fukumoto, M. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3β-mediated Cyclin D1 overexpression. Oncogene 2010, 29, 4826–4837. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; He, X.; Hsu, J.M.; Xia, W.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Liu, J.C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple ras-dependent phosphorylation pathways regulate myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.V.; Jangamreddy, J.R.; Grabarek, J.; Schweizer, F.; Klonisch, T.; Cieslar-Pobuda, A.; Los, M.J. Nuclear localized akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell Cycle 2015, 14, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. Foxo transcription factors; regulation by akt and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Ma, Q.; Chen, L.; Li, P.; Zhang, M.; Ramamoorthy, S.; Nawaz, Z.; Shimojima, T.; Wang, H.; Yang, Y.; et al. Mdm2 acts downstream of p53 as an e3 ligase to promote foxo ubiquitination and degradation. J. Biol. Chem. 2009, 284, 13987–14000. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Hergert, P. Foxo3a and disease progression. World J. Biol. Chem. 2014, 5, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. Foxo transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Coffer, P.J. The extending network of foxo transcriptional target genes. Antioxid. Redox Signal. 2011, 14, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhou, R.; Kong, Y.; Wang, J.; Xia, W.; Guo, J.; Liu, J.; Sun, H.; Liu, K.; Yang, J.; et al. S-equol, a secondary metabolite of natural anticancer isoflavone daidzein, inhibits prostate cancer growth in vitro and in vivo, though activating the akt/foxo3a pathway. Curr. Cancer Drug Targets 2016, 16, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Rimmele, P.; Bigarella, C.L.; Yalcin, S.; Ghaffari, S. Evidence for akt-independent regulation of foxo1 and foxo3 in haematopoietic stem and progenitor cells. Cell Cycle 2016, 15, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. Pras40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. Pras40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian tor complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. Msin1 is necessary for akt/pkb phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Roy, D. Activation of phosphorylation of plasma membrane insulin-like growth factor-i receptors in the kidney of syrian hamsters by diethylstilbestrol. Carcinogenesis 1995, 16, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex Integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Ricoult, S.J.; Ben-Sahra, I.; Manning, B.D. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at ser-2448 is mediated by p70s6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. Tbc1d7 is a third subunit of the tsc1-tsc2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb gtpase is a direct target of tsc2 gap activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Arrazola, P.; Hino, O.; Kobayashi, T.; Yeung, R.S.; Ru, B.; Pan, D. Tsc tumour suppressor proteins antagonize amino-acid-tor signalling. Nat. Cell Biol. 2002, 4, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Rheb fills a gap between tsc and tor. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 s6 kinase and 4e-bp1 through their tor signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and s6k1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. Srebp activity is regulated by mTORC1 and contributes to akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by redd1 and the tsc1/tsc2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The Mdm2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Jackson, S.P. DNA-pk, atm and atr as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell Biol. 2001, 13, 225–231. [Google Scholar] [CrossRef]
- Kastan, M.B.; Lim, D.S.; Kim, S.T.; Xu, B.; Canman, C. Multiple signaling pathways involving atm. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of atm, atr and DNA-pkcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.C. Physiological regulation of akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar] [PubMed]
- Terasima, T.; Tolmach, L.J. Variations in several responses of hela cells to x-irradiation during the division cycle. Biophys. J. 1963, 3, 11–33. [Google Scholar] [CrossRef]
- Sherr, C.J. Growth factor-regulated g1 Cyclins. Stem Cell 1994, 12 (Suppl. 1), 47–55; discussion 55–47. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. Cdk inhibitors: Positive and negative regulators of g1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Canepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. Ink4 proteins, a family of mammalian cdk inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Cucciolla, V.; Oliva, A.; Zappia, V.; Della Ragione, F. P27kip1 metabolism: A fascinating labyrinth. Cell Cycle 2007, 6, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Child, E.S.; Mann, D.J. The intricacies of p21 phosphorylation: Protein/protein interactions, subcellular localization and stability. Cell Cycle 2006, 5, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. Cdk inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Kench, J.G.; Morey, A.L.; Lee, C.S.; Biankin, S.A.; Head, D.R.; Hugh, T.B.; Henshall, S.M.; Sutherland, R.L. Overexpression of p21(waf1/cip1) is an early event in the development of pancreatic intraepithelial neoplasia. Cancer Res. 2001, 61, 8830–8837. [Google Scholar] [PubMed]
- Blagosklonny, M.V. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle 2002, 1, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Gurian-West, M.; Schmidt, A.; Hall, A.; Roberts, J.M. P27kip1 modulates cell migration through the regulation of rhoa activation. Genes Dev. 2004, 18, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.C.; Komata, T.; Kondo, Y.; Kanzawa, T.; Kondo, S.; Germano, I.M. Molecular response of human glioblastoma multiforme cells to ionizing radiation: Cell cycle arrest, modulation of the expression of Cyclin-dependent kinase inhibitors, and autophagy. J. Neurosurg. 2003, 98, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Kawai, Y.; Komori, H.; Rokutanda, S.; Eguchi, Y.; Tsujimoto, Y.; Asahina, I.; Komori, T. Bcl2 deficiency activates foxo through akt inactivation and accelerates osteoblast differentiation. PLoS ONE 2014, 9, e86629. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Myers, C.J.; Jung, D.K.; Lu, B. Nvp-bez-235 enhances radiosensitization via blockade of the Pi3K/mTOR pathway in cisplatin-resistant non-small cell lung carcinoma. Genes Cancer 2014, 5, 293–302. [Google Scholar] [PubMed]
- Yamaguchi, H.; Wang, H.G. The protein kinase pkb/akt regulates cell survival and apoptosis by inhibiting bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of bax ser184 by akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Jendrossek, V. The intrinsic apoptosis pathways as a target in anticancer therapy. Curr. Pharm. Biotechnol. 2012, 13, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical nhej share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Montaser-Kouhsari, L.; Beck, A.H.; Toker, A. Merit40 is an akt substrate that promotes resolution of DNA damage induced by chemotherapy. Cell Rep. 2015, 11, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. Akt1 inhibits homologous recombination by inducing cytoplasmic retention of brca1 and rad51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Yin, Y.; Shen, Q.; Chen, Y.; Li, R.; Chang, W.; Bai, J.; Liu, W.; Shi, L.; Zhang, P. Akt inhibitor mk-2206 enhances the effect of cisplatin in gastric cancer cells. Biomed. Rep. 2016, 4, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Handrick, R.; Rubel, A.; Faltin, H.; Eibl, H.; Belka, C.; Jendrossek, V. Increased cytotoxicity of ionizing radiation in combination with membrane-targeted apoptosis modulators involves downregulation of protein kinase b/akt-mediated survival-signaling. Radiother. Oncol. 2006, 80, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Rudner, J.; Ruiner, C.E.; Handrick, R.; Eibl, H.J.; Belka, C.; Jendrossek, V. The akt-inhibitor erufosine induces apoptotic cell death in prostate cancer cells and increases the short term effects of ionizing radiation. Radiat. Oncol. 2010, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Spiegelberg, D.; Glimelius, B.; Stenerlow, B.; Nestor, M. Evaluation of cancer stem cell markers cd133, cd44, cd24: Association with akt isoforms and radiation resistance in colon cancer cells. PLoS ONE 2014, 9, e94621. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Eccles, L.A.; Tseng, J.; Cybulska, P.; Wielgos, M.; Powell, S.N.; Levine, D.A. The emsy threonine 207 phospho-site is required for emsydriven suppression of DNA damage repair. Oncotarget 2017, 8, 13792–13804. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Besnard, E.; Fisher, D.; Piette, J.; Dulic, V. Chk1 is dispensable for g2 arrest in response to sustained DNA damage when the atm/p53/p21 pathway is functional. Oncogene 2011, 30, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Kyrieleis, O.J.; McIntosh, P.B.; Webb, S.R.; Calder, L.J.; Lloyd, J.; Patel, N.A.; Martin, S.R.; Robinson, C.V.; Rosenthal, P.B.; Smerdon, S.J. Three-dimensional architecture of the human brca1-a histone deubiquitinase core complex. Cell Rep. 2016, 17, 3099–3106. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.L.; Isaacs, C. Brca mutation testing in determining breast cancer therapy. Cancer J. 2011, 17, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Soo Lee, N.; Kim, Y.; Kim, H. Factors forming the brca1-a complex orchestrate brca1 recruitment to the sites of DNA damage. Acta Biochim. Biophys. Sin. 2016, 48, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Paramasivam, M.; Aressy, B.; Wu, J.; Bellani, M.; Tong, W.; Seidman, M.M.; Greenberg, R.A. Merit40 cooperates with brca2 to resolve DNA interstrand cross-links. Genes Dev. 2015, 29, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Huang, J.; Chen, J. Merit40 facilitates brca1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Patterson-Fortin, J.; Messick, T.E.; Feng, D.; Shanbhag, N.; Wang, Y.; Greenberg, R.A. Merit40 controls brca1-rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009, 23, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.F.; Milner, J.; Brown, L.A.; Hsu, F.; Gilks, B.; et al. Emsy links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef]
- Hou, J.; Wang, Z.; Yang, L.; Guo, X.; Yang, G. The function of emsy in cancer development. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 5061–5066. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. Xlf interacts with the xrcc4-DNA ligase iv complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Length-dependent binding of human xlf to DNA and stimulation of xrcc4.DNA ligase iv activity. J. Biol. Chem. 2007, 282, 11155–11162. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Yu, Y.; Fang, S.; Lees-Miller, S.P.; Tainer, J.A. Xlf regulates filament architecture of the XRCC4.Ligase iv complex. Structure 2010, 18, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Vergnes, A.; Ristic, D.; Wyman, C.; Modesti, M.; Junop, M. A human xrcc4-xlf complex bridges DNA. Nucleic Acids Res. 2012, 40, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mahaney, B.L.; Yano, K.; Ye, R.; Fang, S.; Douglas, P.; Chen, D.J.; Lees-Miller, S.P. DNA-pk and atm phosphorylation sites in XLF/cernunnos are not required for repair of DNA double strand breaks. DNA Repair 2008, 7, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Fan, Y.-Z. Ubiquitin-conjugating enzyme E2S (UBE2S), a potential tumor therapeutic target. Clin. Oncol. 2016, 1, 1132. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| miRNA Name | Function | Reference |
|---|---|---|
| miR-20a | Activation of mir-20a correlated with decreased activation of PTEN leading to higher radioresistance. Inhibition of miR-20a by anti-miR-20a led to cell radiosensitization. PI3K inhibitor LY294002 could radiosensitize hepatocellular carcinoma cells. | [110] |
| miR-21 miR-95 | Upregulation of miR-21 and miR-95 expression in lung cancer cells correlating with poor prognosis. Radiosensitization by applying anti-miR-21 and anti-miR-95. | [116] |
| miR-22 | miR-22 activates PI3K/Akt pathway inducing enhanced proliferation of chronic lymphocytic leukemia (CLL) B-cells. | [117] |
| miR-31 | Inhibiting effects of miR-31 evoked downregulation of PI3K/Akt pathway in lung adenocarcinoma cells. | [115] |
| miR-101-2 miR-125b-2 miR-451a | Exogenous expression of miR-101-2, miR-125b-2, and miR-451a suppressed tumor growth in gastric cancer through decreasing the expression of PI3K/Akt pathway. | [118] |
| miR-126 | miR-126 suppressed proliferation of undifferentiated thyroid carcinoma through repressing PI3K/Akt pathway. | [111] |
| miR-203 | miR-203 is critical factor in radioresistance of nasopharyngeal carcinoma cells by targeting IL8/AKT signaling. This effect could be abolished by an agomir. Overexpression of miR-203 could achieve radiosensitization by affecting DNA repair as well as the PI3K/Akt pathway in malignant glioma cells. | [112] |
| miR-205 | Enhanced expression of miR-205 evokes downregulation of PTEN resulting in enhanced Akt phosphorylation and radioresistance. | [119] |
| miR-302 | Downregulation of miR-302 evokes elevated level of phosphorylated Akt. Restoration of miR-302 expression returned this effect and sensitized cells to radiotherapy. | [113] |
| miR-519a | miR-519a can promote tumor growth in hepatocellular carcinoma targeting PTEN/PI3K/Akt pathway. | [120] |
| Target Name | Function | References |
|---|---|---|
| BAD | Pro-apoptotic protein. Phosphorylation by Akt inhibits its function and promotes cell survival. | [136,137] |
| BRCA1 | Breast cancer susceptibility gene product and tumor suppressor. Phosphorylation by Akt alters its function, perhaps by preventing nuclear localization. | [138,139] |
| CHK1 | DNA damage effector that regulates G2/M transition during DNA damage. Phosphorylation by Akt inhibits its function by preventing phosphorylation by ATM/ATR. | [140,141,142] |
| Cot | Oncogene. Phosphorylation by Akt induces NF-kB-dependent transcription. | [143] |
| EMSY | Oncogenic interacting partner of BRCA2. EMSY overexpression disrupts the BRCA2/RAD51 interaction. | [144,145] |
| FANCA | ATPase involved in DNA repair. Phosphorylation by Akt negatively regulates its function. | [146,147] |
| FOXO1A | Transcription factor involved in cell cycle arrest, apoptosis, and glucose metabolism. Phosphorylation by Akt causes export from the nucleus and inhibits its activity. | [148,149,150] |
| FOXO3A | Transcription factor involved in cell cycle arrest and apoptosis. Phosphorylation by Akt causes export from the nucleus and inhibits its activity. | [151,152,153] |
| FOXO4 | Transcription factor involved in cell cycle arrest, apoptosis, and insulin signaling. Phosphorylation by Akt causes export from the nucleus and inhibits its activity. | [154] |
| Lamin A/C | Plays a role in nuclear assembly, chromatin organization, nuclear membrane and telomere dynamics. Phosphorylation by Akt promotes its degradation. | [155,156] |
| Mdm2 | As an E3 ubiquitin-protein ligase Mdm2 mediates ubiquitination of p53/TP53. | [124,157,158] |
| Merit40 | Component of the BRCA1-A complex that contributes to DNA repair. Phosphorylation by Akt leads to enhanced DNA repair and survival. | [159] |
| p21 | p21 as a CDK-inhibitor regulates cell cycle and survival. Phosphorylation by Akt leads to release from PCNA that results in elevated progression. | [160,161,162] |
| p27 | Cyclin-dependent kinase inhibitor that mediates G1 arrest. Phosphorylation by Akt promotes 14-3-3 binding and cytoplasmic localization resulting in enhanced proliferative effect. | [163,164,165] |
| PRAS40 | Binds to and inhibits mTOR. Phosphorylation causes 14-3-3 binding and facilitates its phosphorylation by mTORC1. | [166,167] |
| PRP19 | PRP19is a member of the spliceosome that also functions in DNA double strand break repair. Phosphorylation by Akt allows 14-3-3 binding and promotes its degradation. | [168] |
| TSC2 | Tumor suppressor that can inhibit mTOR. Phosphorylation by Akt inhibits its function. | [169,170] |
| UBE2S | Ubiquitin-conjugating enzyme E2S plays a role in NHEJ complex. Phosphorylation by Akt enhances its stability by inhibiting proteasomal degradation. | [171,172] |
| Wee1 | Inhibits cell cycle progression. Phosphorylation by Akt inhibits its function associated with changed localization of Wee1 from nuclear to cytoplasmic. | [173] |
| XLF | Involved in NHEJ and promotes the Ligase IV recruitment to the DNA damage site. XLF interacts with XRCC4 to create long filaments promoting ligation of DNA broken ends. Phosphorylation by Akt negatively affects DNA repair. | [174] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. https://doi.org/10.3390/cancers10030078
Szymonowicz K, Oeck S, Malewicz NM, Jendrossek V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers. 2018; 10(3):78. https://doi.org/10.3390/cancers10030078
Chicago/Turabian StyleSzymonowicz, Klaudia, Sebastian Oeck, Nathalie M. Malewicz, and Verena Jendrossek. 2018. "New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response" Cancers 10, no. 3: 78. https://doi.org/10.3390/cancers10030078
APA StyleSzymonowicz, K., Oeck, S., Malewicz, N. M., & Jendrossek, V. (2018). New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers, 10(3), 78. https://doi.org/10.3390/cancers10030078