Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer

 , ,
, , 


Abstract
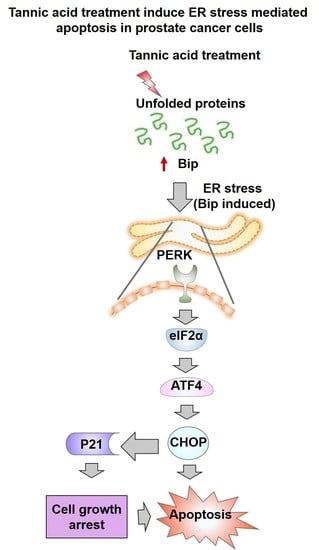
:
1. Introduction
2. Results
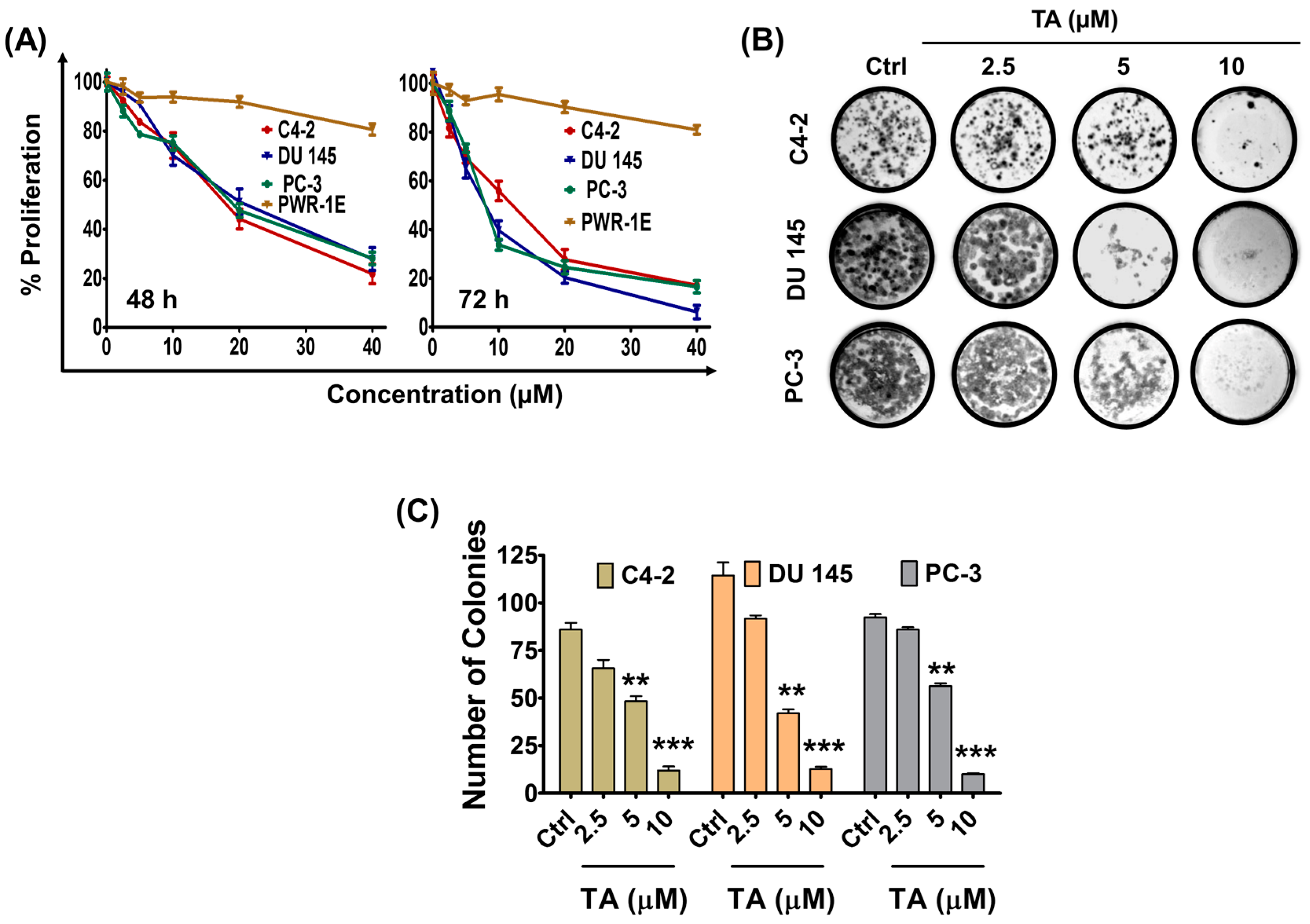
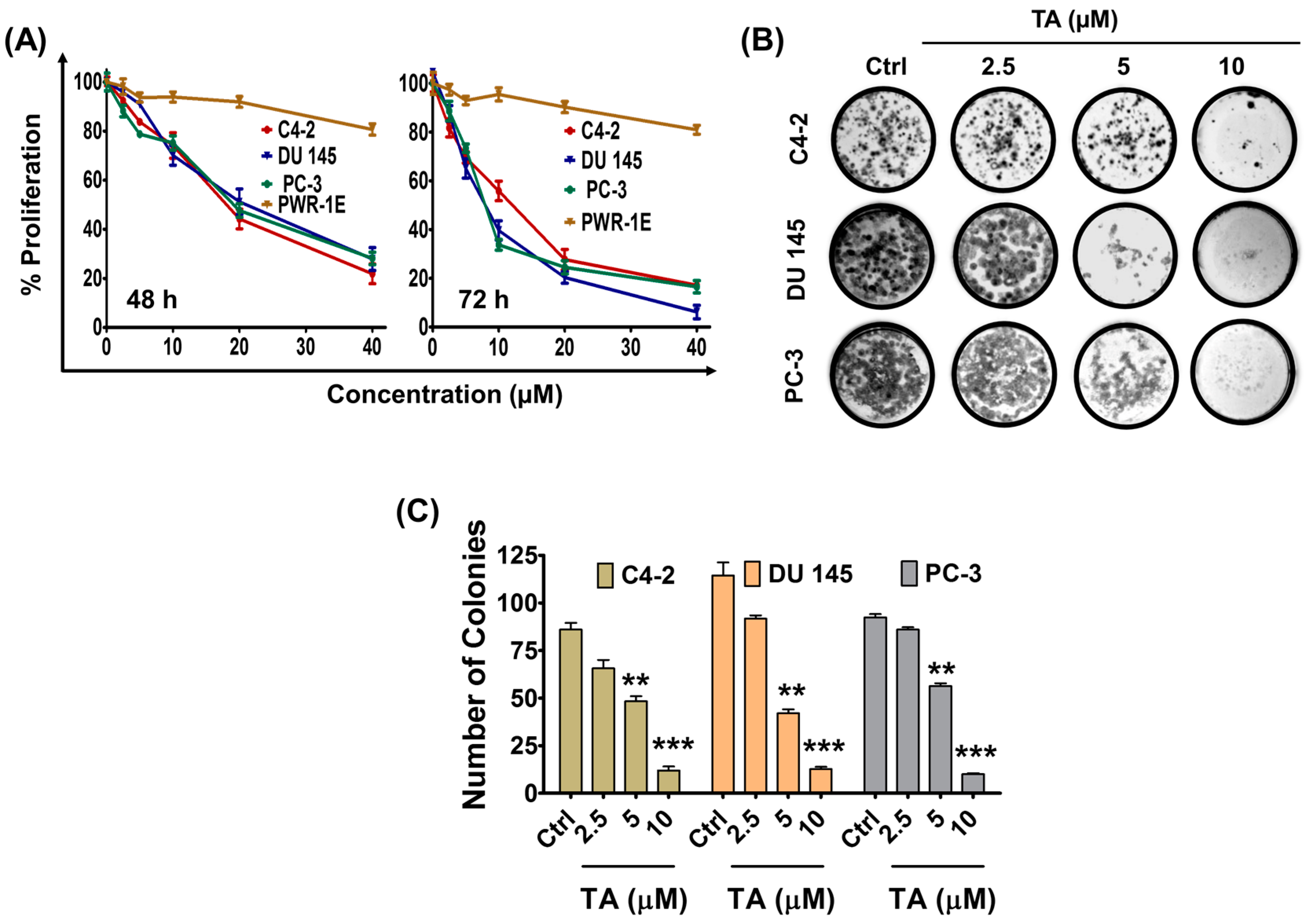
2.1. Tannic Acid Treatment Suppresses Proliferation and Clonogelnicity of Prostate Cancer Cells
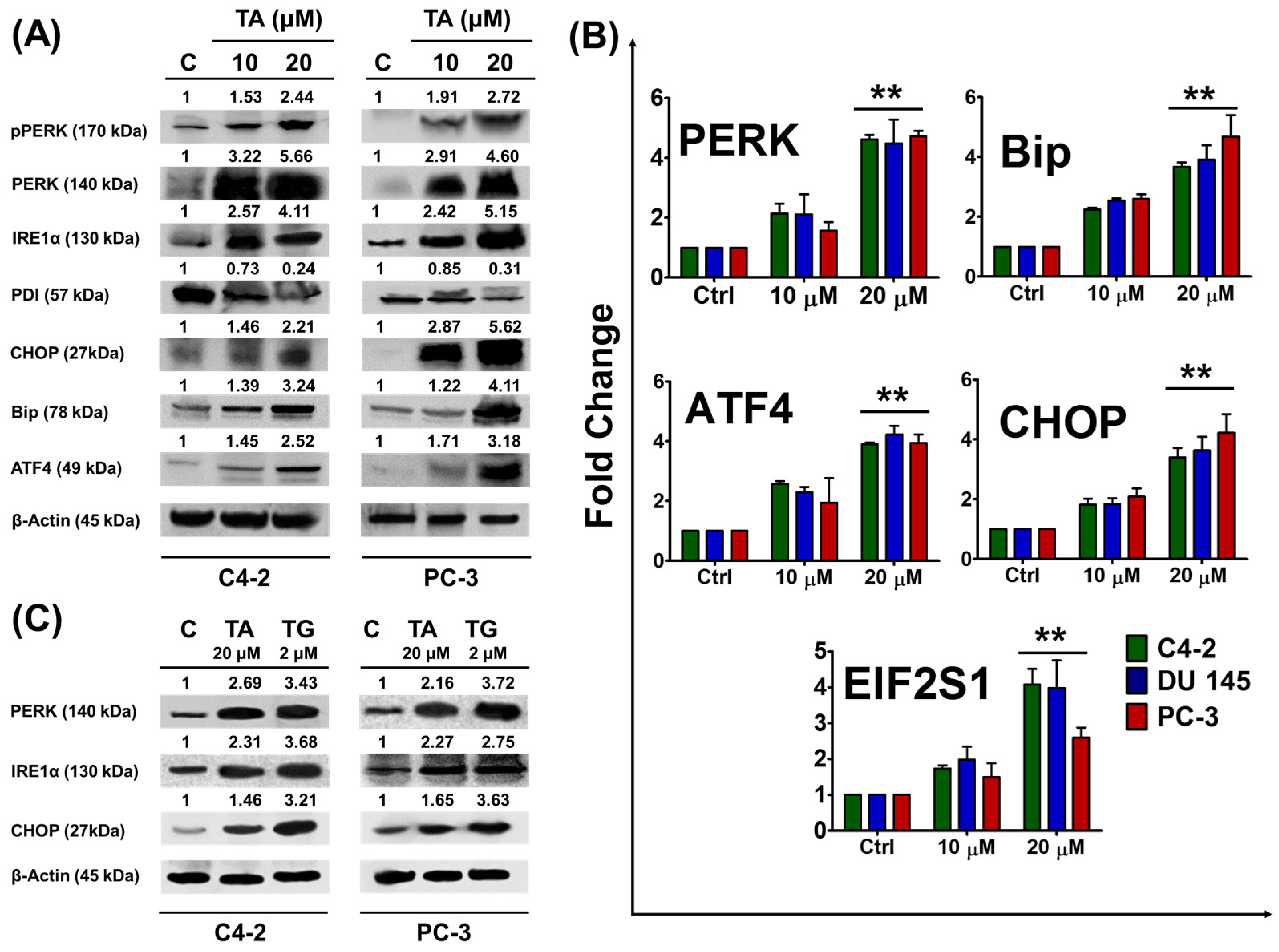
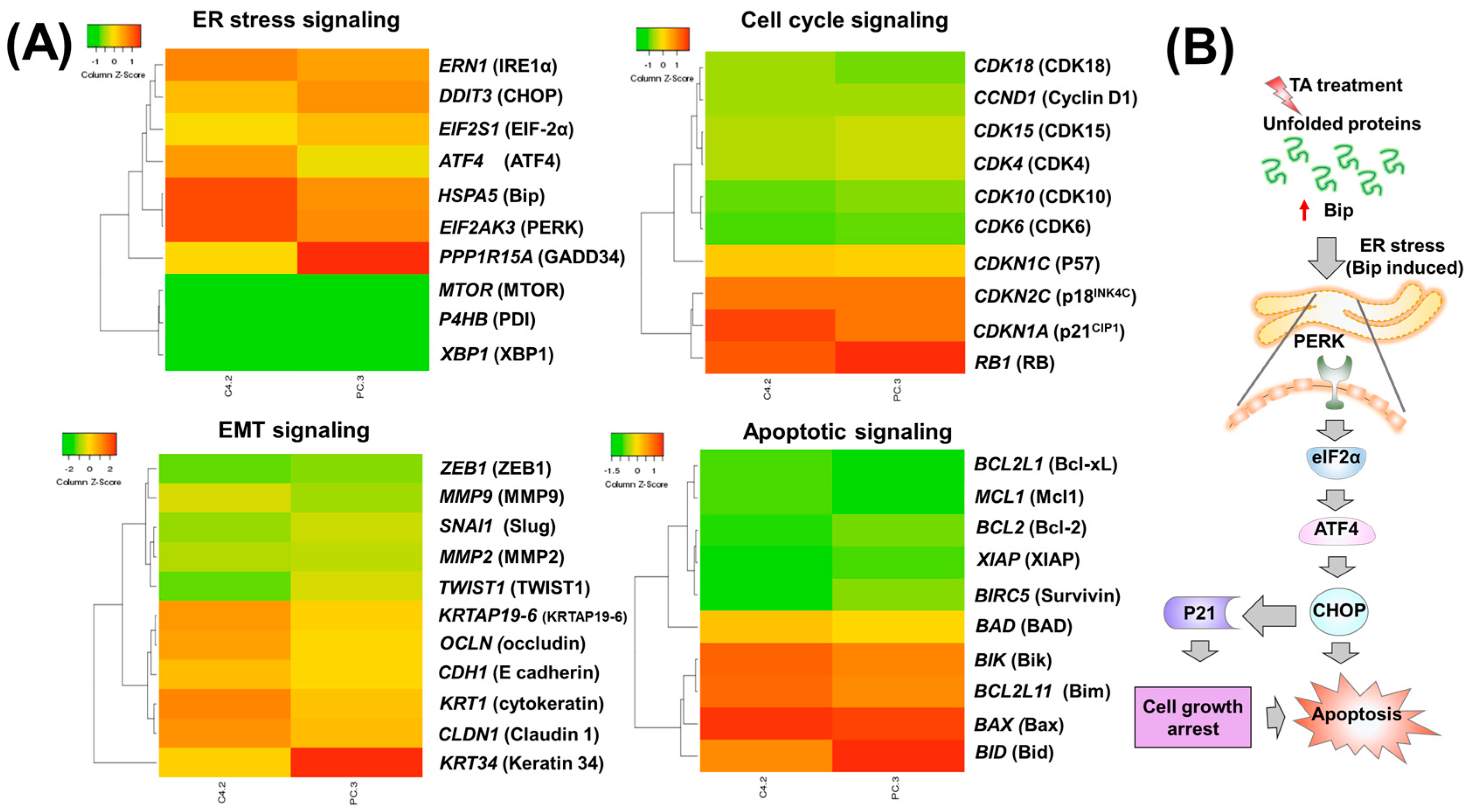
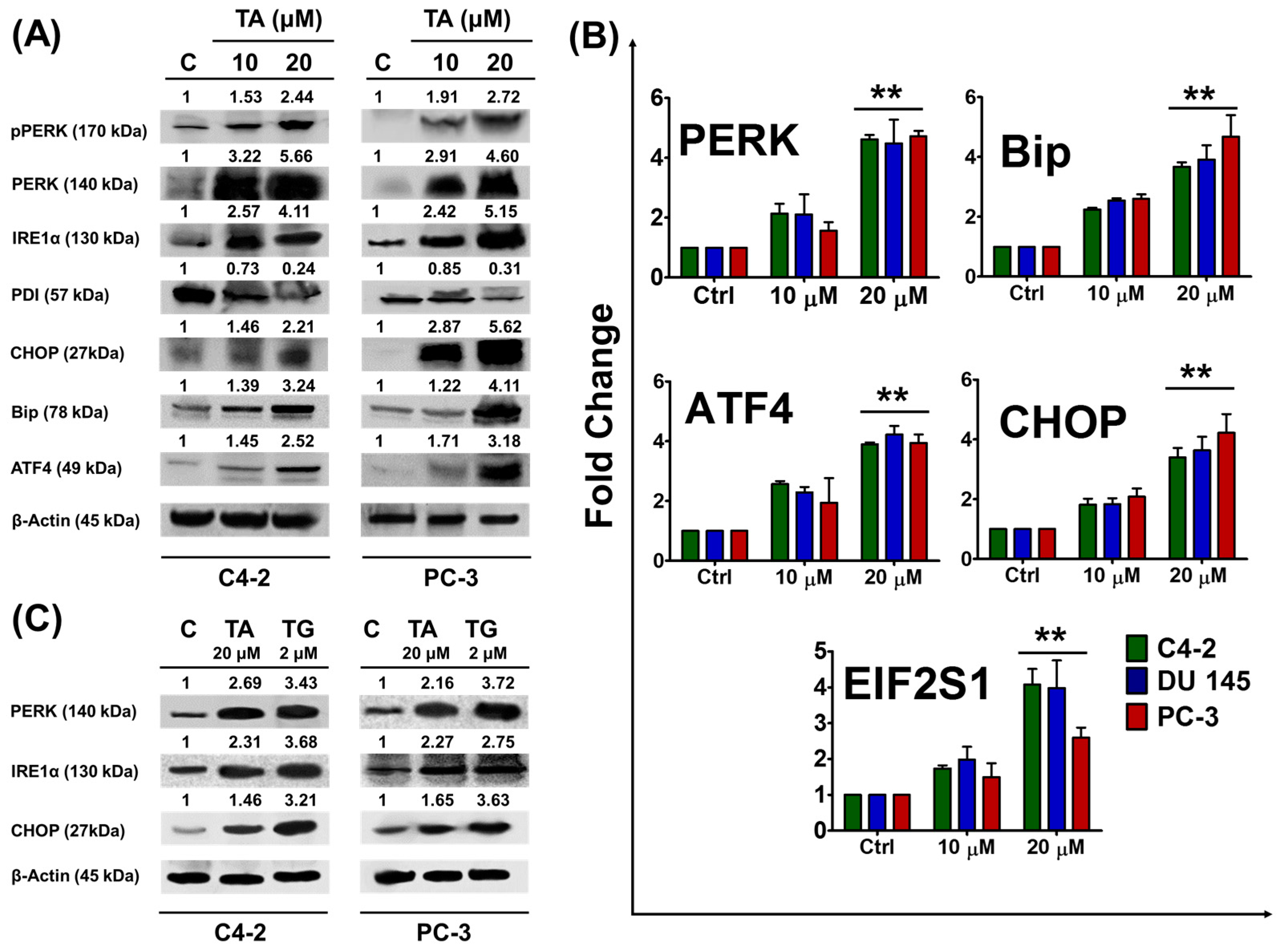
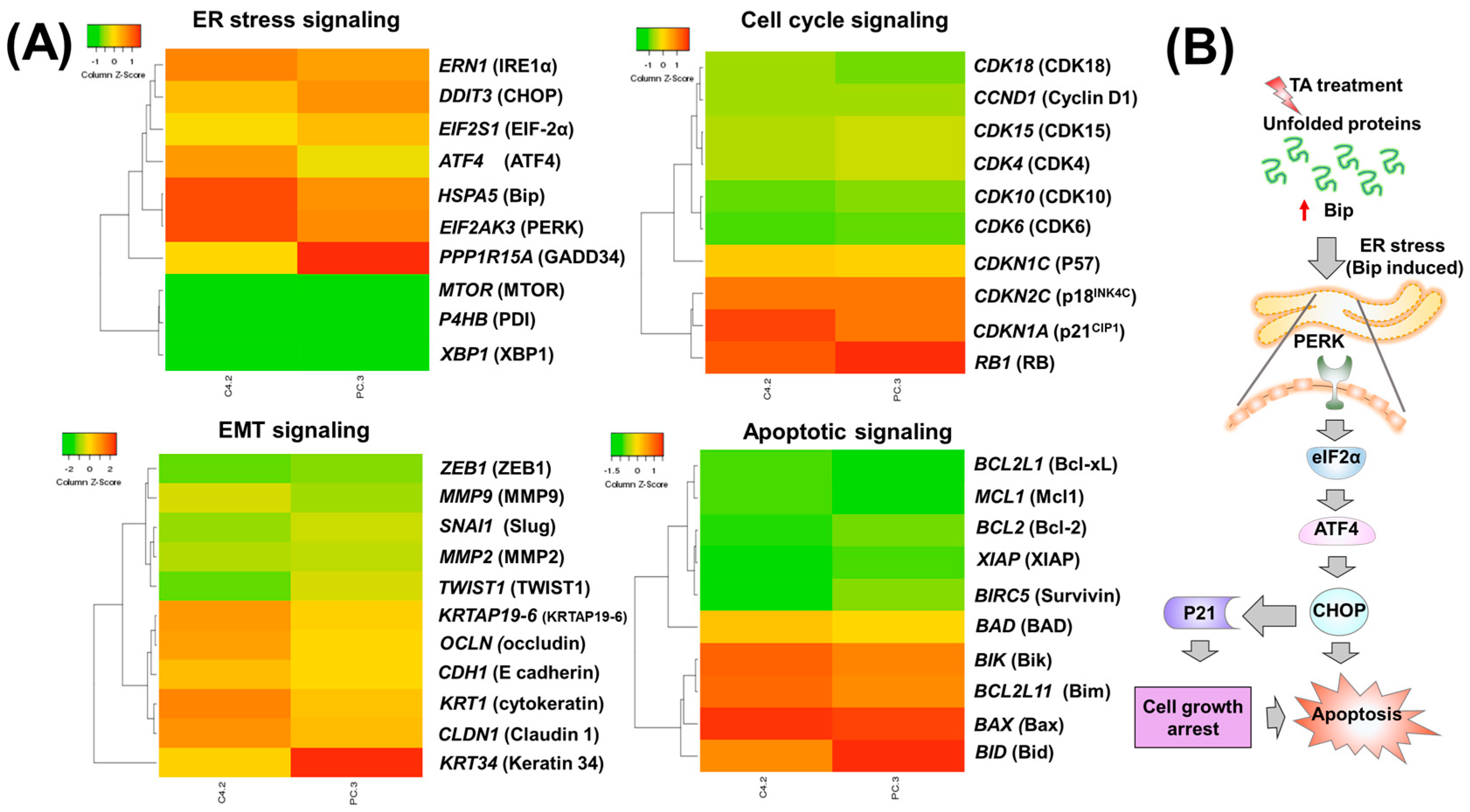
2.2. Tannic Acid Treatment Upregulates the Expression of ER Stress Regulatory Proteins
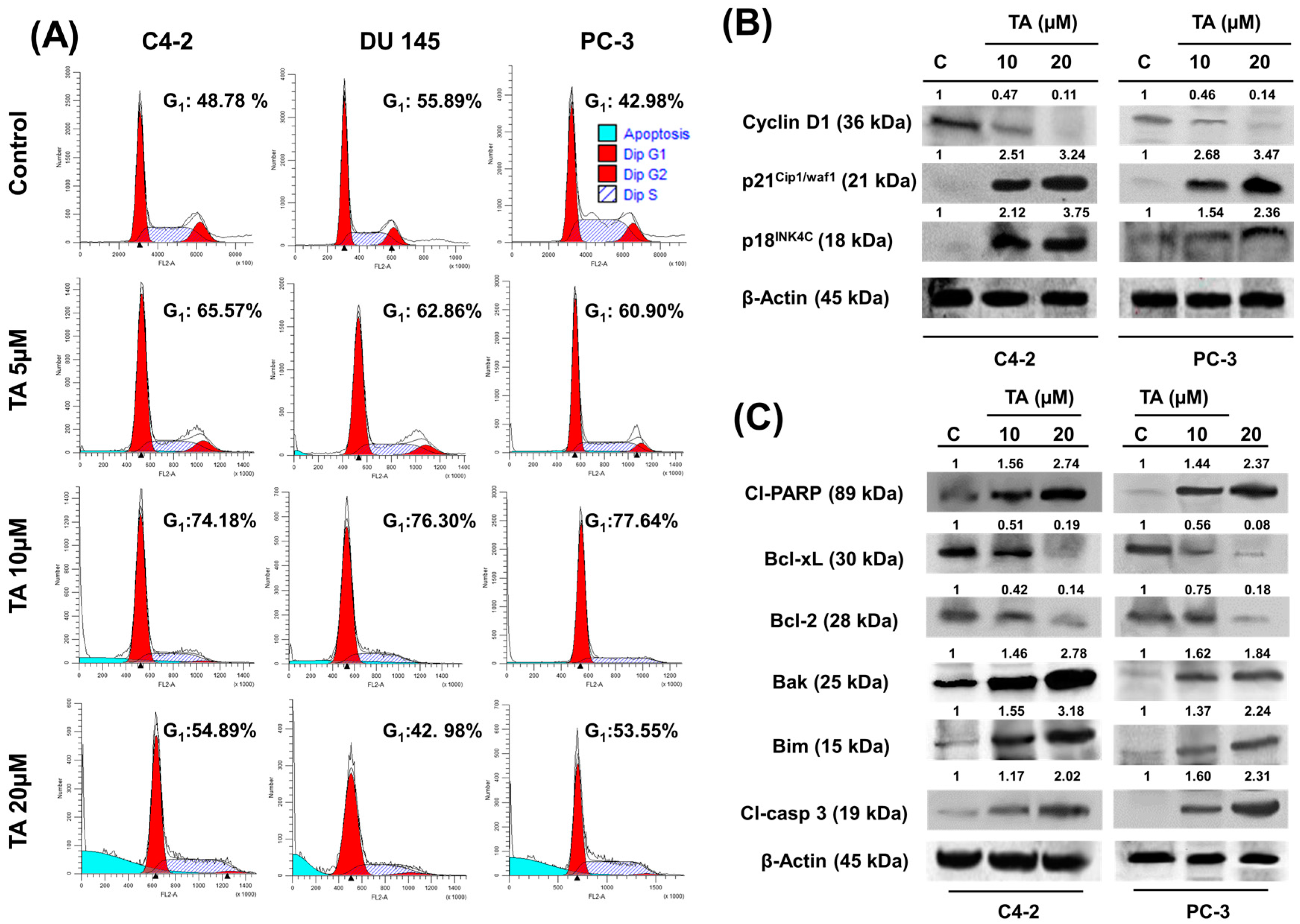
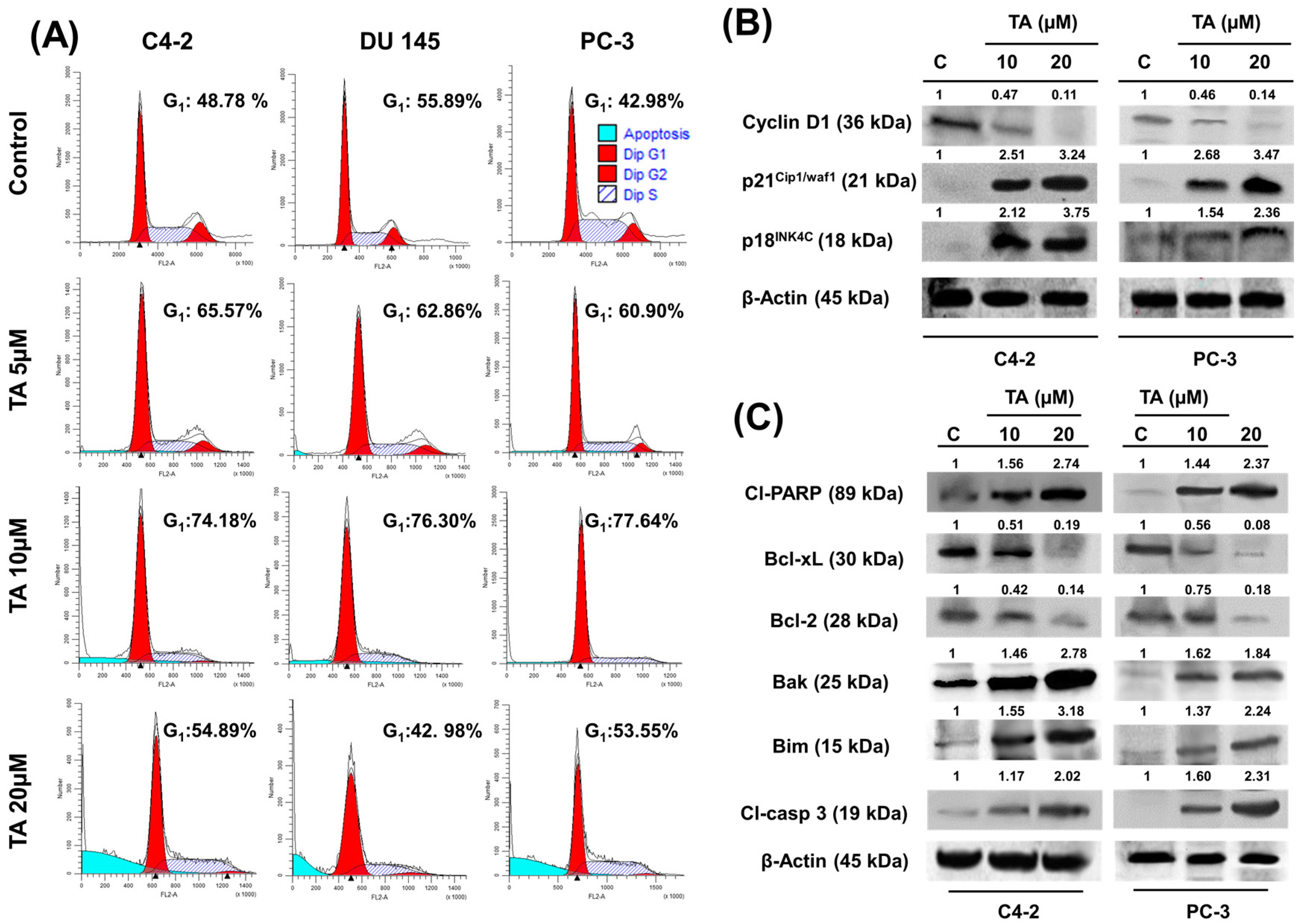
2.3. TA Treatment Effectively Arrests the Cell Cycle and Modulates Cell Cycle Regulatory Proteins
2.4. Tannic Acid Treatment Induced Apoptosis via ER Stress Mediated Signaling
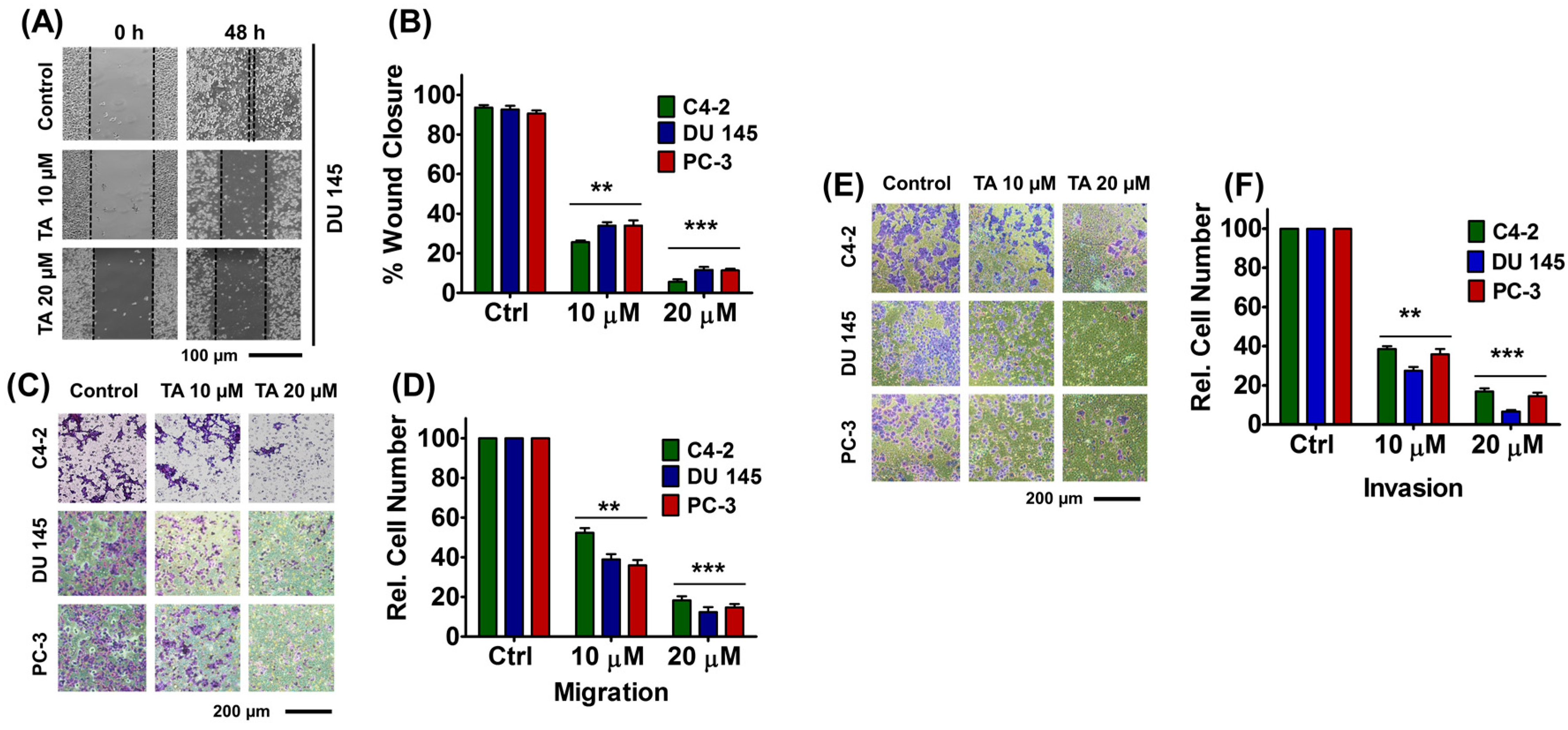
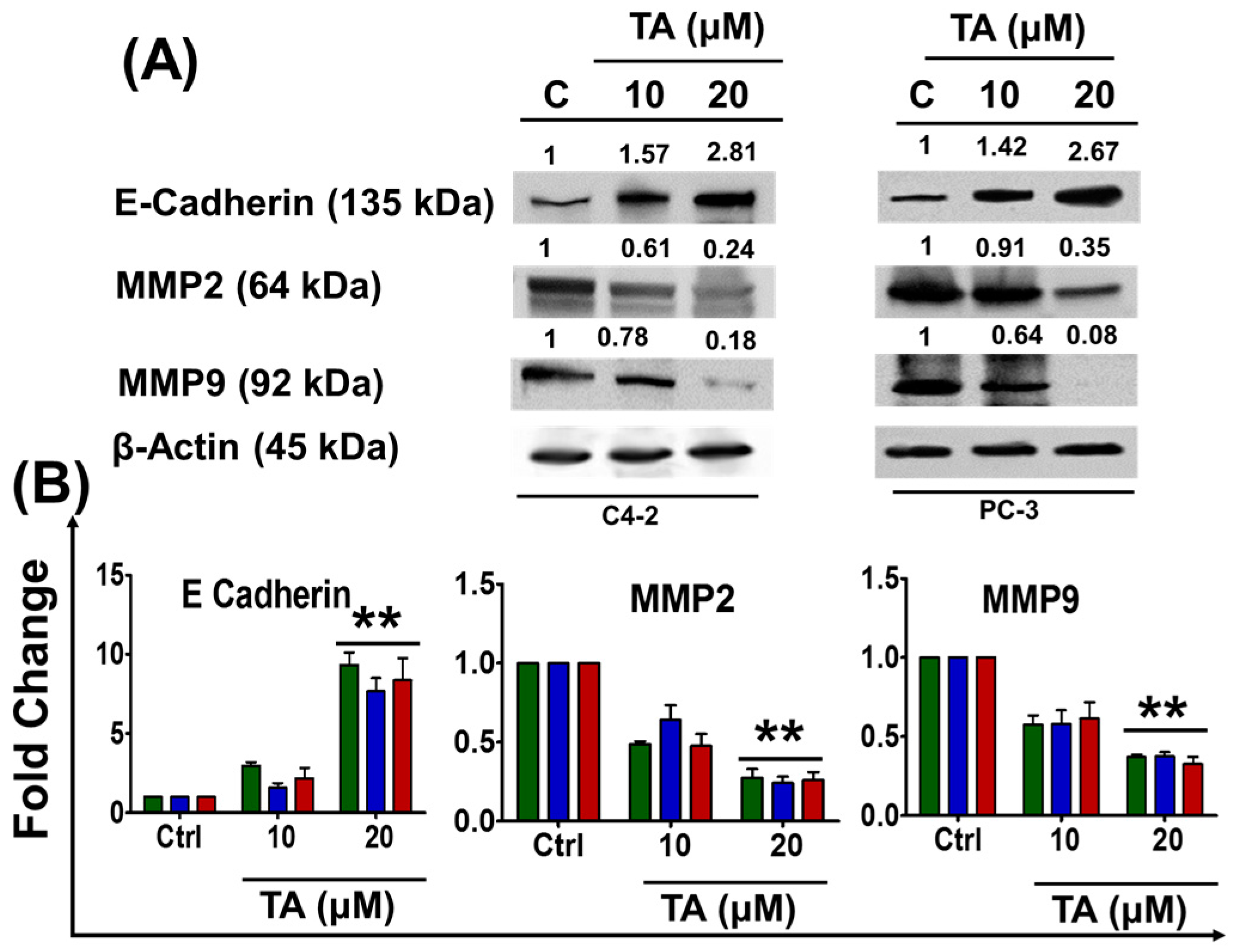
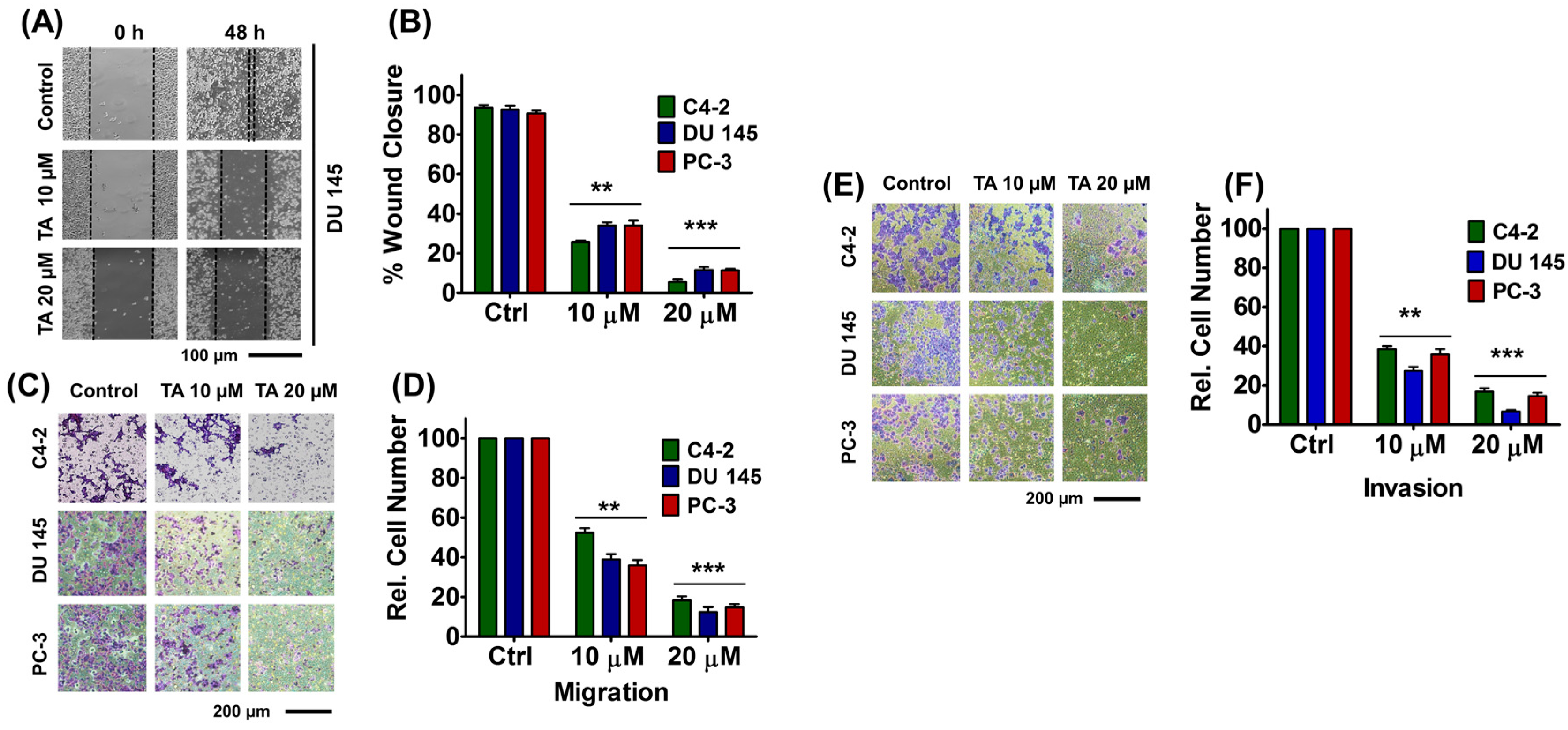
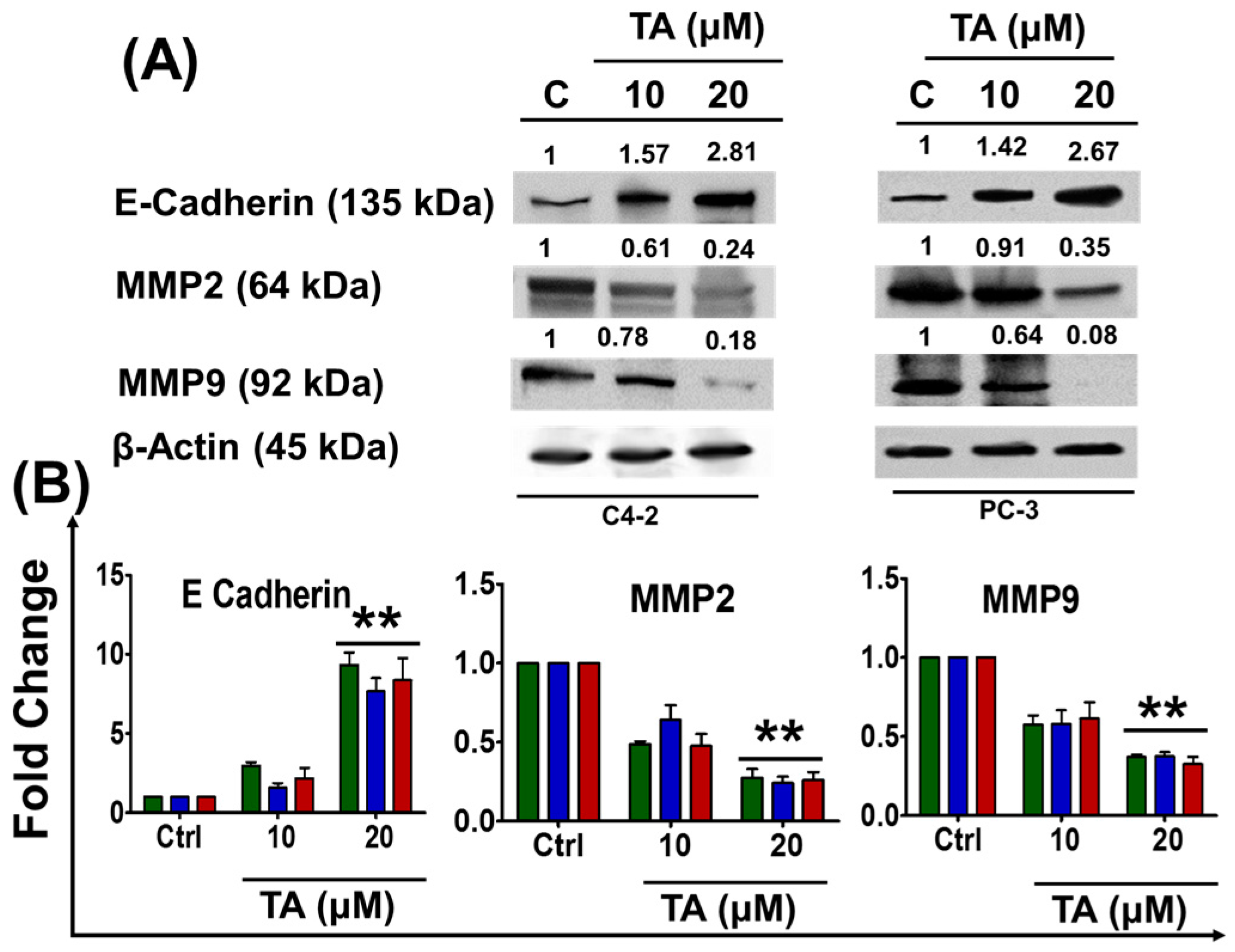
2.5. Tannic Acid Treatment Inhibited Migratory and Invasive Potentials of Prostate Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Proliferation Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. Wound Healing Assay
4.6. Cell Migration Assay
4.7. Cell Invasion Assay
4.8. cDNA Synthesis and Quantitative mRNA Expression by Real-Time PCR
4.9. mRNA Purification, Ethanol Precipitation and Micro Array Studies
4.10. Western Blot Analysis
4.11. Statistical Analysis
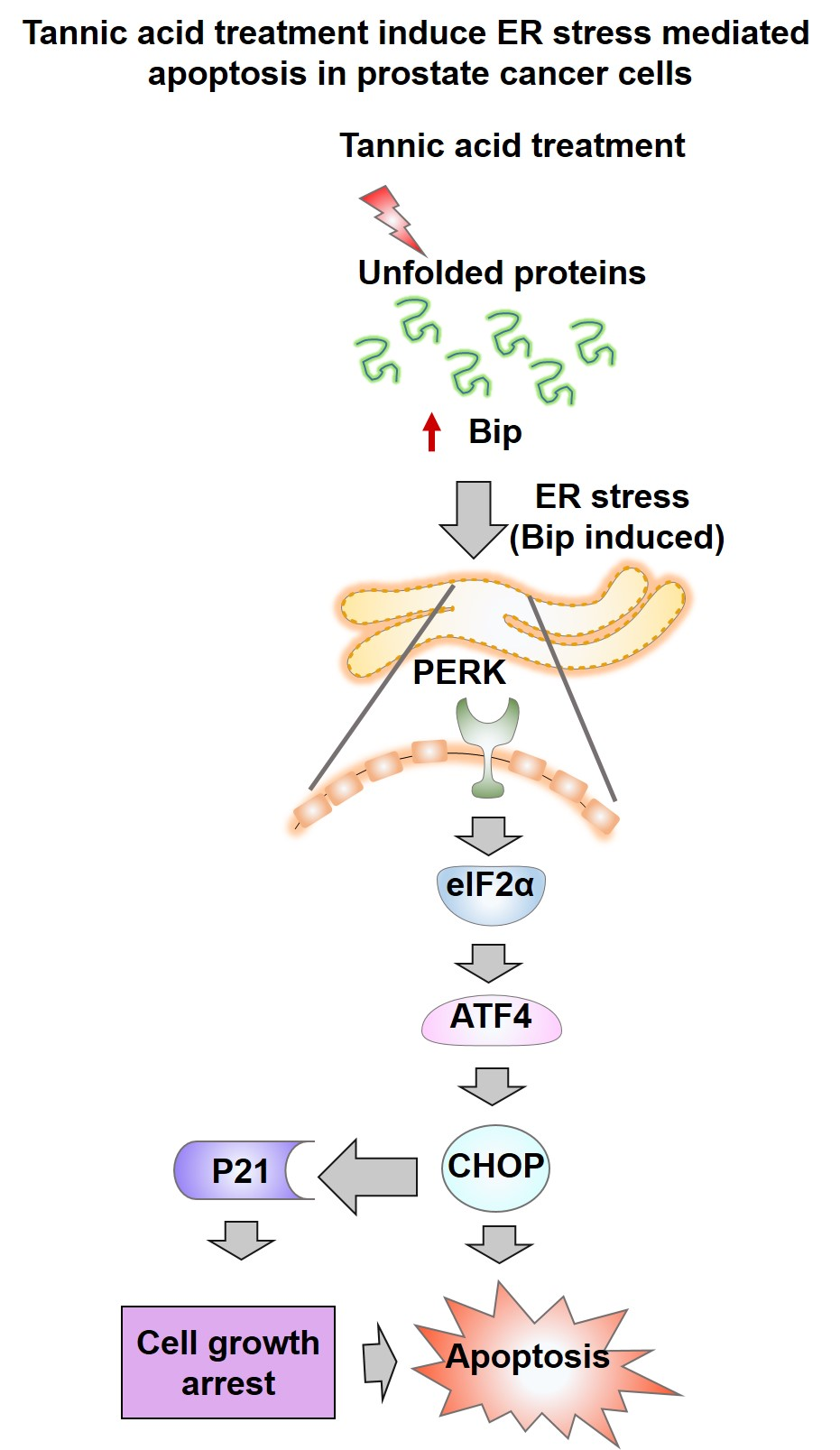
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; von Minckwitz, G.; Seidman, A.D. Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 2002, 7 (Suppl. 6), 13–19. [Google Scholar] [CrossRef] [PubMed]
- Mohapatro, S.K.; Dandapat, M.C.; Padhi, N.C. Toxicity and side-effects of combination chemohormonal therapy of advanced breast cancer. J. Indian Med. Assoc. 1992, 90, 39–42. [Google Scholar] [PubMed]
- Storm, M.; Sheng, X.; Arnoldussen, Y.J.; Saatcioglu, F. Prostate cancer and the unfolded protein response. Oncotarget 2016, 7, 54051–54066. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lee, A.S. Stress induction of grp78/bip and its role in cancer. Curr. Mol. Med. 2006, 6, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Hou, Y.-F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.-M.; Yin, X.-M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. Er stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.J.; Shin, S.H.; Lee, H.J.; Lee, K.W. Polyphenols as small molecular inhibitors of signaling cascades in carcinogenesis. Pharmacol. Ther. 2011, 130, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.H.; Kristal, A.R.; Stanford, J.L. Fruit and vegetable intakes and prostate cancer risk. J. Natl. Cancer Inst. 2000, 92, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.J.; Yen, G.C. Chemopreventive effects of dietary phytochemicals against cancer invasion and metastasis: Phenolic acids, monophenol, polyphenol, and their derivatives. Cancer Treat. Rev. 2012, 38, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Ferruelo, A.; Romero, I.; Cabrera, P.M.; Arance, I.; Andres, G.; Angulo, J.C. Effects of resveratrol and other wine polyphenols on the proliferation, apoptosis and androgen receptor expression in lncap cells. Actas Urol. Esp. 2014, 38, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.R.; Jones, P.; Benghuzzi, H.; Tucci, M. A comparison of the morphological changes associated with conventional and sustained treatment with pigallocatechin3gallate, thymoquinone, and tannic acid on lncap cells. Biomed. Sci. Instrum. 2008, 44, 465–470. [Google Scholar] [PubMed]
- Vanella, L.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Cardile, V.; Kim, D.H.; Abraham, N.G.; Sorrenti, V. Apoptotic markers in a prostate cancer cell line: Effect of ellagic acid. Oncol. Rep. 2013, 30, 2804–2810. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.T.; Wong, T.Y.; Wei, C.I.; Huang, Y.W.; Lin, Y. Tannins and human health: A review. Crit. Rev. Food Sci. Nutr. 1998, 38, 421–464. [Google Scholar] [CrossRef] [PubMed]
- Lublin, A.; Isoda, F.; Patel, H.; Yen, K.; Nguyen, L.; Hajje, D.; Schwartz, M.; Mobbs, C. FDA-approved drugs that protect mammalian neurons from glucose toxicity slow aging dependent on cbp and protect against proteotoxicity. PLoS ONE 2011, 6, e27762. [Google Scholar] [CrossRef] [PubMed]
- Beniwal, V.; Kumar, A.; Sharma, J.; Chhokar, V. Recent advances in industrial application of tannases: A review. Recent Pat. Biotechnol. 2013, 7, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Salako, N.O. Antifungal properties of tannic acid and aqueous extracts of serindea warneckei. Odontostomatol. Trop. 1990, 13, 117–120. [Google Scholar] [PubMed]
- Dong, G.; Liu, H.; Yu, X.; Zhang, X.; Lu, H.; Zhou, T.; Cao, J. Antimicrobial and anti-biofilm activity of tannic acid against staphylococcus aureus. Nat. Prod. Res. 2017, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Booth, B.W.; Inskeep, B.D.; Shah, H.; Park, J.P.; Hay, E.J.; Burg, K.J.L. Tannic acid preferentially targets estrogen receptor-positive breast cancer. Int. J. Breast Cancer 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Sane, M.S.; Gupta, C. Tannic acid ameliorates doxorubicin-induced cardiotoxicity and potentiates its anti-cancer activity: Potential role of tannins in cancer chemotherapy. Toxicol. Appl. Pharmacol. 2011, 251, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Perchellet, J.-P.; Gali, H.U.; Perchellet, E.M.; Klish, D.S.; Armbrust, A.D. Antitumor-promoting activities of tannic acid, ellagic acid, and several gallic acid derivatives in mouse skin. In Plant Polyphenols: Synthesis, Properties, Significance; Hemingway, R.W., Laks, P.E., Eds.; Springer: Boston, MA, USA, 1992; pp. 783–801. [Google Scholar]
- Sun, Y.; Zhang, T.; Wang, B.; Li, H.; Li, P. Tannic acid, an inhibitor of poly(adp-ribose) glycohydrolase, sensitizes ovarian carcinoma cells to cisplatin. Anti-Cancer Drugs 2012, 23, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Joung, Y.H.; Kang, D.Y.; Sp, N.; Byun, H.J.; Hwang, T.S.; Sasidharakurup, H.; Lee, C.H.; Cho, K.H.; Park, K.D.; et al. Tannic acid inhibits egfr/stat1/3 and enhances p38/stat1 signalling axis in breast cancer cells. J. Cell. Mol. Med. 2017, 21, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Smith, D.M.; Dou, Q.P. Tannic acid potently inhibits tumor cell proteasome activity, increases p27 and bax expression, and induces g1 arrest and apoptosis. Cancer Epidemiol. Biomarkers Prev. 2001, 10, 1083–1088. [Google Scholar] [PubMed]
- Pomp, J.; Wike, J.L.; Ouwerkerk, I.J.; Hoogstraten, C.; Davelaar, J.; Schrier, P.I.; Leer, J.W.; Thames, H.D.; Brock, W.A. Cell density dependent plating efficiency affects outcome and interpretation of colony forming assays. Radiother. Oncol. 1996, 40, 121–125. [Google Scholar] [CrossRef]
- Zhu, G.Y.; Wong, B.C.; Lu, A.; Bian, Z.X.; Zhang, G.; Chen, H.B.; Wong, Y.F.; Fong, W.F.; Yang, Z. Alkylphenols from the roots of ardisia brevicaulis induce G1 arrest and apoptosis through endoplasmic reticulum stress pathway in human non-small-cell lung cancer cells. Chem. Pharm. Bull. 2012, 60, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.J.; Bailey, H.H.; Mukhtar, H. Green tea polyphenols for prostate cancer chemoprevention: A translational perspective. Phytomedicine 2010, 17, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.P.; Sarkar, S.; Das, S.; Azab, B.; Santhekadur, P.K.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B.; et al. Targeted apoptotic effects of thymoquinone and tamoxifen on xiap mediated akt regulation in breast cancer. PLoS ONE 2013, 8, e61342. [Google Scholar] [CrossRef] [PubMed]
- Voulgari, A.; Pintzas, A. Epithelial–mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Morgia, G.; Falsaperla, M.; Malaponte, G.; Madonia, M.; Indelicato, M.; Travali, S.; Mazzarino, M.C. Matrix metalloproteinases as diagnostic (MMP-13) and prognostic (MMP-2, MMP-9) markers of prostate cancer. Urol. Res. 2005, 33, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Al-Hijazeen, M.; Lee, E.J.; Mendonca, A.; Ahn, D.U. Effects of tannic acid on lipid and protein oxidation, color, and volatiles of raw and cooked chicken breast meat during storage. Antioxidants 2016, 5, E19. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.H., Jr.; Mazzio, E.; Badisa, R.B.; Zhu, Z.P.; Agharahimi, M.; Millington, D.J.; Goodman, C.B. Differential cytotoxicity of triphala and its phenolic constituent gallic acid on human prostate cancer lncap and normal cells. Anticancer Res. 2011, 31, 3739–3745. [Google Scholar] [PubMed]
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013, 33, 4683–4694. [Google Scholar] [PubMed]
- Maas, N.L.; Diehl, J.A. Molecular pathways: The perks and pitfalls of targeting the unfolded protein response in cancer. Clin. Cancer Res. 2015, 21, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of nf-y (cbf) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 2015, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the chop promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce c/ebp-homologous protein (chop/gadd153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. Chop is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Minami, M.; Takeda, K.; Sakao, Y.; Akira, S. Ectopic expression of chop (gadd153) induces apoptosis in m1 myeloblastic leukemia cells. FEBS Lett. 1996, 395, 143–147. [Google Scholar] [CrossRef]
- Barone, M.V.; Crozat, A.; Tabaee, A.; Philipson, L.; Ron, D. Chop (gadd153) and its oncogenic variant, tls-chop, have opposing effects on the induction of g1/s arrest. Genes Dev. 1994, 8, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, M.; Chen, N.; Ma, A.; Zhu, C.; Zhao, R.; Jiang, M.; Zhou, J.; Ye, L.; Fu, H.; et al. Glycyrrhetinic acid induces g1phase cell cycle arrest in human nonsmall cell lung cancer cells through endoplasmic reticulum stress pathway. Int. J. Oncol. 2015, 46, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Kolar, Z.; Murray, P.G.; Scott, K.; Harrison, A.; Vojtesek, B.; Dusek, J. Relation of bcl-2 expression to androgen receptor, p21waf1/cip1, and cyclin d1 status in prostate cancer. Mol. Pathol. 2000, 53, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Yeh, R.D.; Chen, J.C.; Lai, T.Y.; Yang, J.S.; Yu, C.S.; Chiang, J.H.; Lu, C.C.; Yang, S.T.; Yu, C.C.; Chang, S.J.; et al. Gallic acid induces g(0)/g(1) phase arrest and apoptosis in human leukemia hl-60 cells through inhibiting cyclin d and e, and activating mitochondria-dependent pathway. Anticancer Res. 2011, 31, 2821–2832. [Google Scholar] [PubMed]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. Chop favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [PubMed]
- Karakurt, S.; Adali, O. Tannic acid inhibits proliferation, migration, invasion of prostate cancer and modulates drug metabolizing and antioxidant enzymes. Anticancer Agents Med. Chem. 2016, 16, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ebeling, M.C.; Chauhan, N.; Thompson, P.A.; Gara, R.K.; Ganju, A.; Yallapu, M.M.; Behrman, S.W.; Zhao, H.; Zafar, N.; et al. Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res. 2015, 75, 2292–2304. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Dey, G.; Banerjee, I.; Dey, K.K.; Parida, S.; Kumar, B.N.P.; Das, C.K.; Pal, I.; Mukherjee, M.; Misra, M.; et al. Somatostatin receptor targeted liposomes with diacerein inhibit il-6 for breast cancer therapy. Cancer Lett. 2017, 388, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Fabrication of curcumin encapsulated plga nanoparticles for improved therapeutic effects in metastatic cancer cells. J. Colloid Interface Sci. 2010, 351, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Puvvada, N.; Rajput, S.; Kumar, B.N.P.; Mandal, M.; Pathak, A. Exploring the fluorescence switching phenomenon of curcumin encapsulated niosomes: In vitro real time monitoring of curcumin release to cancer cells. RSC Adv. 2013, 3, 2553–2557. [Google Scholar] [CrossRef]
- Nagesh, P.K.; Johnson, N.R.; Boya, V.K.; Chowdhury, P.; Othman, S.F.; Khalilzad-Sharghi, V.; Hafeez, B.B.; Ganju, A.; Khan, S.; Behrman, S.W.; et al. Psma targeted docetaxel-loaded superparamagnetic iron oxide nanoparticles for prostate cancer. Colloids Surf. B Biointerfaces 2016, 144, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Puvvada, N.; Kumar, B.N.; Sarkar, S.; Konar, S.; Bharti, R.; Dey, G.; Mazumdar, A.; Pathak, A.; Fisher, P.B.; et al. Overcoming akt induced therapeutic resistance in breast cancer through sirna and thymoquinone encapsulated multilamellar gold niosomes. Mol. Pharm. 2015, 12, 4214–4225. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.N.P.; Rajput, S.; Dey, K.K.; Parekh, A.; Das, S.; Mazumdar, A.; Mandal, M. Celecoxib alleviates tamoxifen-instigated angiogenic effects by ros-dependent vegf/vegfr2 autocrine signaling. BMC Cancer 2013, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.N.P.; Puvvada, N.; Rajput, S.; Sarkar, S.; Das, S.K.; Emdad, L.; Sarkar, D.; Venkatesan, P.; Pal, I.; Dey, G.; et al. Sequential release of drugs from hollow manganese ferrite nanocarriers for breast cancer therapy. J. Mater. Chem. B 2015, 3, 90–101. [Google Scholar] [CrossRef]
- Rajput, S.; Kumar, B.N.; Banik, P.; Parida, S.; Mandal, M. Thymoquinone restores radiation-induced tgf-beta expression and abrogates EMT in chemoradiotherapy of breast cancer cells. J. Cell. Physiol. 2015, 230, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Sikander, M.; Hafeez, B.B.; Malik, S.; Alsayari, A.; Halaweish, F.T.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Cucurbitacin D exhibits potent anti-cancer activity in cervical cancer. Sci. Rep. 2016, 6, 36594. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.; Lin, J.; Hu, X.; Li, B.; Xue, A.; Shen, Y.; Jiang, J.; Zhang, M.; Xie, J.; et al. Deregulation of rgs17 expression promotes breast cancer progression. J. Cancer 2015, 6, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.-P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Setua, S.; Khan, S.; Yallapu, M.M.; Behrman, S.W.; Sikander, M.; Khan, S.S.; Jaggi, M.; Chauhan, S.C. Restitution of tumor suppressor microRNA-145 using magnetic nanoformulation for pancreatic cancer therapy. J. Gastrointest. Surg. 2017, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Zhou, D.; Song, Y.; Pei, D.; Wang, J.; Guo, Z.; Yang, R. Bacterial mRNA purification by magnetic capture-hybridization method. Microbiol. Immunol. 2004, 48, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Dey, G.; Bharti, R.; Dhanarajan, G.; Das, S.; Dey, K.K.; Kumar, B.N.; Sen, R.; Mandal, M. Marine lipopeptide iturin a inhibits akt mediated gsk3beta and foxo3a signaling and triggers apoptosis in breast cancer. Sci. Rep. 2015, 5, 10316. [Google Scholar] [CrossRef] [PubMed]
- Prashanth Kumar, B.N.; Rajput, S.; Bharti, R.; Parida, S.; Mandal, M. Bi2536—A plk inhibitor augments paclitaxel efficacy in suppressing tamoxifen induced senescence and resistance in breast cancer cells. Biomed. Pharmacother. 2015, 74, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Rico-Bautista, E.; Zhu, W.; Kitada, S.; Ganapathy, S.; Lau, E.; Krajewski, S.; Ramirez, J.; Bush, J.A.; Yuan, Z.; Wolf, D.A. Small molecule-induced mitochondrial disruption directs prostate cancer inhibition via UPR signaling. Oncotarget 2013, 4, 1212–1229. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primers | Reverse Primers |
|---|---|---|
| BiP | 5′-CATGGTTCTCACTAAAATGAAAGG-3′ | 5′-GCTGGTACAGTAACAACTG-3′ |
| CHOP/GADD153 | 5′-GCCTTTCTCCTTTGGGACACTGTCCAGC-3′ | 5′-CTCGGCGAGTCGCCTCTACTTCCC-3′ |
| ATF4 | 5′-GCATGCTCTGTTTCGAATGGA-3′ | 5′-CCAACGTGGTCAAGAGCTCAT-3′ |
| PERK | 5′-TCTTGGTTGGGTCTGATGAAT-3′ | 5′-GATGTTCTTGCTGTAGTGGGGG-3′ |
| EIF2S1 | 5′-ACGACAACCCTGGAGAGAAC-3′ | 5′-TATGACATTCCAAGGCCGACA-3′ |
| E-Cadherin | 5′-TGGAGGAATTCTTGCTTTGC-3′ | 5′-CGTACATGTCAGCCAGCTTC-3′ |
| MMP2 | 5′-TCTCCTGACATTGACCTTGGC-3′ | 5′-CAAGGTGCTGGCTGAGTAGATC-3′ |
| MMP9 | 5′-TTGACAGCGACAAGAAGTGG-3′ | 5′-GCCATTCACGTCGTCCTTAT-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagesh, P.K.B.; Hatami, E.; Chowdhury, P.; Kashyap, V.K.; Khan, S.; Hafeez, B.B.; Chauhan, S.C.; Jaggi, M.; Yallapu, M.M. Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer. Cancers 2018, 10, 68. https://doi.org/10.3390/cancers10030068
Nagesh PKB, Hatami E, Chowdhury P, Kashyap VK, Khan S, Hafeez BB, Chauhan SC, Jaggi M, Yallapu MM. Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer. Cancers. 2018; 10(3):68. https://doi.org/10.3390/cancers10030068
Chicago/Turabian StyleNagesh, Prashanth K.B., Elham Hatami, Pallabita Chowdhury, Vivek K. Kashyap, Sheema Khan, Bilal B. Hafeez, Subhash C. Chauhan, Meena Jaggi, and Murali M. Yallapu. 2018. "Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer" Cancers 10, no. 3: 68. https://doi.org/10.3390/cancers10030068
APA StyleNagesh, P. K. B., Hatami, E., Chowdhury, P., Kashyap, V. K., Khan, S., Hafeez, B. B., Chauhan, S. C., Jaggi, M., & Yallapu, M. M. (2018). Tannic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Prostate Cancer. Cancers, 10(3), 68. https://doi.org/10.3390/cancers10030068