Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts

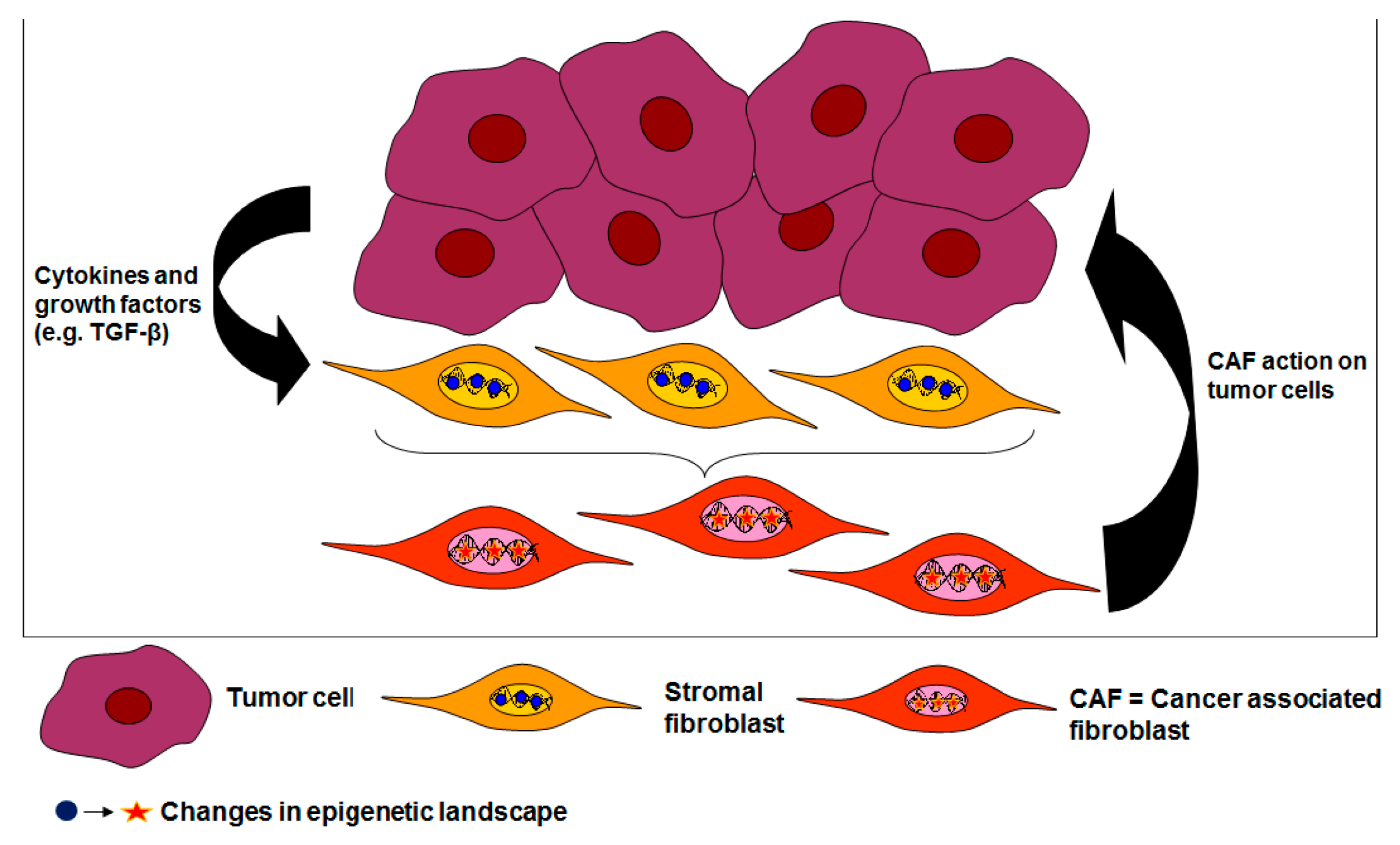{kind=link}
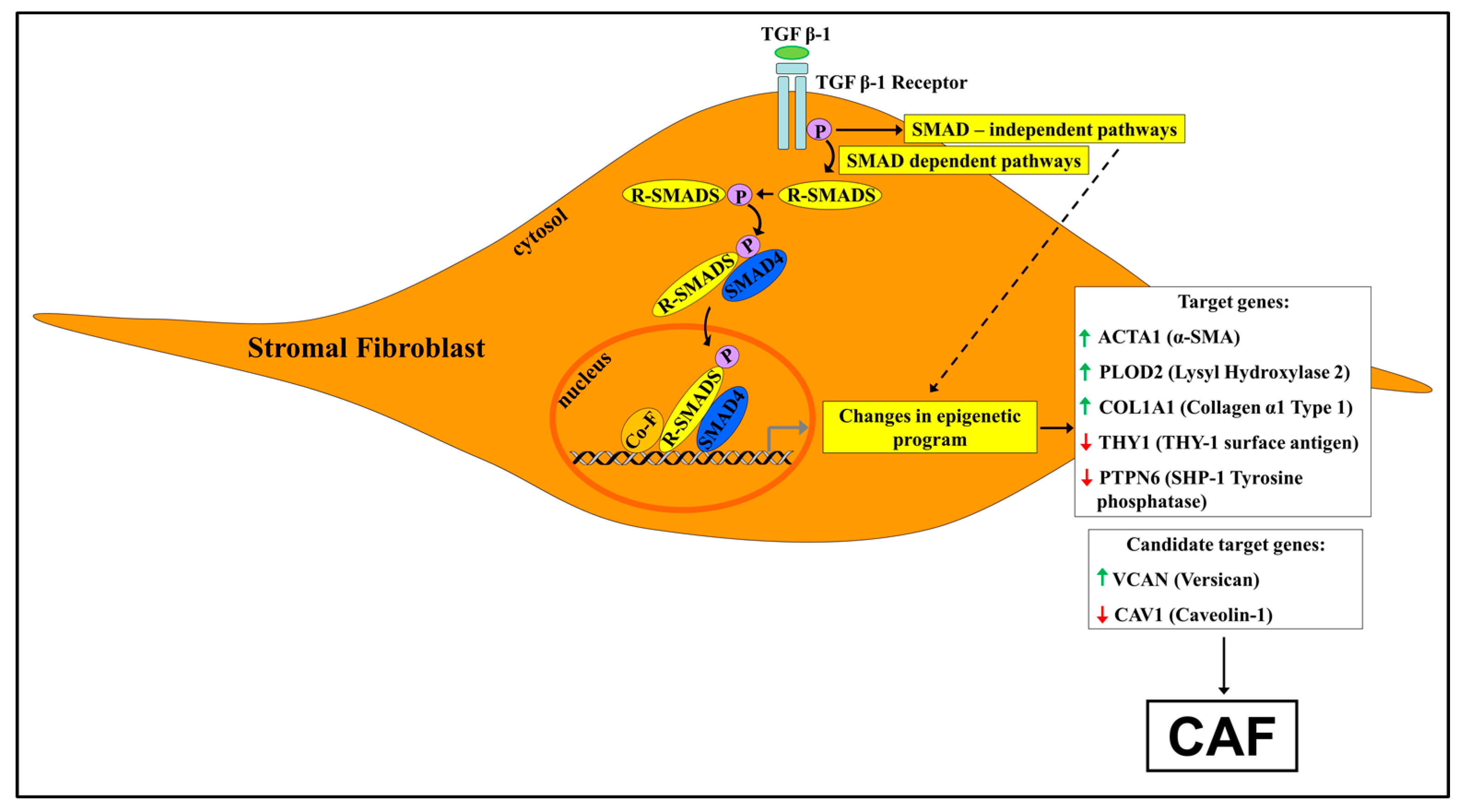{kind=link}
Abstract
1. Introduction
2. The Durable Gene Signature of Cancer-Associated Fibroblasts: Clonal Somatic Mutations or Epigenetic Changes?
3. Epigenetic Changes Underlie Trans-Differentiation of Stromal Fibroblasts to CAFs
3.1. Cancer-Associated Fibroblasts and the DNA Methylome
3.2. Cancer-Associated Fibroblasts and Post-Translational Histone Modifications
4. The Involvement of TGF-β1 Signaling in Shaping the Epigenetic Landscape of Cancer-Associated Fibroblasts
4.1. A Synopsis of TGF-β1 Signaling
4.2. TGF-β1 Signaling and the Epigenetic Signature of Cancer-Associated Fibroblasts
4.2.1. The ACTA2 Gene
4.2.2. The PLOD2 Gene
4.2.3. The COL1A1 Gene
4.2.4. The Thy1/CD90 Gene
4.2.5. The PTPN6 Gene
4.2.6. The Bet Proteins
4.2.7. The CAV-1 Gene
4.2.8. The VCAN Gene
4.2.9. TGF-β Signaling and Metabolic Reprogramming of Cancer-Associated Fibroblasts
5. TGF-β1 Signaling, Cancer-Associated Fibroblasts and Neoplasia: Colon Cancer as a Paradigm for the Human Disease
6. Concluding Remarks
7. Few (Out of Many) Outstanding Questions
Author Contributions
Conflicts of Interest
Abbreviations
| CAFs | Cancer-associated fibroblasts |
| TGF-β1 | Transforming Growth Factor-β1 |
| CRC | Colorectal cancer |
| TME | Tumor microenvironment |
| EMT | Epithelial–mesenchymal transition |
| ECM | Extracellular matrix |
| LOH | Loss of heterozygosity |
| FFPE | Formalin-fixed paraffin-embedded |
| 5mCyt | 5-methylcytosine |
| DNMT | DNA methyl transferase |
| NSCLC | Non-small cell lung cancer |
| PDAC | Pancreatic ductal adenocarcinoma |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription protein |
| SOCS1 | Suppressor of cytokine signaling 1 |
| LIF | Leukemia-inducing factor |
| LAP | Latency associated peptide |
| LTBP | Large TGF-β binding protein |
| SMAD | Suppressor of mothers against decapentaplegic |
| SMURF | SMAD ubiquitin regulatory factor |
| a-SMA | α-smooth muscle actin |
| LHX | Lysyl hydroxylase |
| HDAC | Histone deacetylase |
| HAT | Histone acetyl transferase |
| CMS | Colorectal cancer mesenchymal subtype |
References
- Tape, C.J. The heterocellular emergence of colorectal cancer. Trends Cancer 2017, 3, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Battle, E. Targeting the microenvironment in advanced colorectal cancer. Trends Cancer 2016, 2, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell. Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer -associated fibroblasts’-more than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell. Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Delinasios, J.G.; Angeli, F.; Koumakis, G.; Kumar, S.; Kang, W.G.; Sica, G.; Iacopino, F.; Lama, G.; Lamprecht, S.; Sigal-Batikoff, I.; et al. Proliferating fibroblasts and HeLa cells co-cultured in vitro reciprocally influence growth patterns, protein expression, chromatin features and cell survival. Anticancer Res. 2015, 35, 1881–1916. [Google Scholar] [PubMed]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug. Deliv. 2016, 99, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.C.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersal, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Delinassios, J.G. Cytocidal effects of human fibroblasts on HeLa cells in vitro. Biol. Cell 1987, 59, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Angeli, F.; Koumakis, G.; Chen, M.C.; Kumar, S.; Delinassios, J.G. Role of stromal fibroblasts in cancer: Promoting or impeding? Tumour Biol. 2009, 30, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hine, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Rønnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 1993, 68, 696–707. [Google Scholar] [PubMed]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast–myofibroblast terminal differentiation—the role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2016, 142, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; Sayles, L.C.; Vaka, D.; Khatri, P.; Gevaert, O.; Chen, R.; Zheng, Y.; Gillespie, A.K.; Clarke, N.; Xu, Y.; et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res. 2012, 15, 5744–5756. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, I.; Lehtonen, S.; Sormunen, R.; Skarp, S.; Lehtilahti, E.; Pietilä, M.; Sequeiros, R.B.; Lehenkari, P.; Kuvaj, P. Breast cancer carcinoma-associated fibroblasts differ from breast fibroblasts in immunological and extracellular matrix regulating pathways. Exp. Cell Res. 2016, 344, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.T.; Brentani, H.; Roela, R.A.; Katayama, M.L.; Lima, L.; Rolim, C.F.; Milani, C.; Folgueira, M.A.; Brentani, M.M. Differences in transcriptional effects of 1α,25 dihydroxyvitamin D3 on fibroblasts associated to breast carcinomas and from paired normal breast tissues. J. Steroid Biochem. Mol. Biol. 2013, 133, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar] [PubMed]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002, 32, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Wernert, N.; Löcherbach, C.; Wellmann, A.; Behrens, P.; Hügel, A. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001, 21, 2259–2264. [Google Scholar] [PubMed]
- Fukino, K.; Shen, L.; Matsumoto, S.; Morrison, C.D.; Mutter, G.L.; Eng, G. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004, 64, 7231–7236. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, I.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Tuhkanen, H.; Anttila, M.; Kosma, V.M.; Ylä-Herttuala, S.; Heinonen, S.; Kuronen, A.; Juhola, M.; Tammi, R.; Tammi, M.; Mannermaa, A. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int. J. Cancer 2004, 109, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, N.R.; Morales, C.P.; Damle, S.; Boman, B.; Wright, W.E.; Kopelovich, L.; Shay, J.W. Spontaneous immortalization of clinically normal colon-derived fibroblasts from a familial adenomatous polyposis patient. Neoplasia 2004, 6, 258–265. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bischoff, F.Z.; Yim, S.O.; Pathak, S.; Grant, G.; Siciliano, M.J.; Giovanella, B.C.; Strong, L.C.; Tainsky, M.A. Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: Aneuploidy and immortalization. Cancer Res. 1990, 50, 7979–7984. [Google Scholar] [PubMed]
- Rogan, E.M.; Bryan, T.M.; Hukku, B.; Maclean, K.; Chang, A.C.; Moy, E.L.; Englezou, A.S.G.; Warneford, S.G.; Dalla-Pozza, L.; Reddel, R.R. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell Biol. 1995, 15, 4745–4753. [Google Scholar] [CrossRef] [PubMed]
- Etzold, A.; Galetzka, D.; Weis, E.; Bartsch, O.; Haaf, T.; Spix, C.; Itzel, T.; Schweiger, S.; Strand, D.; Strand, S.; et al. CAF-like state in primary skin fibroblasts with constitutional BRCA1 epimutation sheds new light on tumor suppressor deficiency-related changes in healthy tissue. Epigenetics 2016, 11, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Corver, W.E.; Ter Haar, N.T.; Fleuren, G.J.; Oosting, J. Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations. Cell Oncol. 2011, 34, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Hu, M.; Sridhar, A.; Opeskin, K.; Fox, S.; Shipitsin, M.; Trivett, M.; Thompson, E.E.; Ramakrishna, M.; Gorringe, K.L.; et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet. 2008, 40, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [PubMed]
- Akahane, T.; Hirasawa, A.; Tsuda, H.; Kataoka, F.; Nishimura, S.; Tanaka, H.; Tominaga, E.; Nomura, H.; Chiyoda, T.; Iguchi, Y.; et al. The origin of stroma surrounding epithelial ovarian cancer cells. Int. J. Gynecol. Pathol. 2013, 32, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Goggins, M. Pancreatic cancer-associated fibroblasts display normal allelotypes. Cancer Biol. Ther. 2008, 7, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Bianchi-Frias, D.; Basom, R.; Delrow, J.J.; Coleman, I.M.; Dakhova, O.; Qu, X.; Fang, M.; Franco, O.E.; Ericson, N.G.; Bielas, J.H.; et al. Cells comprising the prostate cancer microenvironment lack recurrent clonal somatic genomic aberrations. Mol. Cancer Res. 2016, 14, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.; Polyak, K.; Haviv, I. Clonal mutations in the cancer-associated fibroblasts: The case against genetic coevolution. Cancer Res. 2009, 69, 6765–6768. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.; Qiu, W.; Haviv, I. Genetic changes in tumour microenvironments. J. Pathol. 2011, 223, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Rummel, S.; Valente, A.L.; Kane, J.L.; Shriver, C.D.; Ellsworth, R.E. Genomic (in)stability of the breast tumor microenvironment. Mol. Cancer Res. 2012, 10, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed]
- DeClerk, Y.A.; Pienta, K.J.; Woodhouse, E.C.; Singer, D.S.; Mohla, S. The tumor microenvironment at a turning point knowledge gained over the last decade, and challenges and opportunities ahead: A white paper from the NCI TME Network. Cancer Res. 2017, 77, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Minarovits, J.; Banati, F.; Szenthe, K.; Niller, H.H. Epigenetic Regulation. Adv. Exp. Med. Biol. 2016, 879, 1–25. [Google Scholar] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Bannister, J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: Association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; van den Brûle, F.; Velculescu, V.; Polyak, K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.A.; Kim, Y.I.; Salas, M.C.; Gamble, M.V.; Shibata, W.; Muthupalani, S.; Sohn, K.J.; Abrams, J.A.; Fox, J.G.; Wang, T.C.; et al. Folic acid increases global DNA methylation and reduces inflammation to prevent Helicobacter-associated gastric cancer in mice. Gastroenterology 2012, 142, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, S.A.; Lipkin, M. Chemoprevention of colon cancer by calcium, vitamin D and folate: Molecular mechanisms. Nat. Rev. Cancer 2003, 8, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Adany, R.; Iozzo, R.V. Altered methylation of versican proteoglycan gene in human colon carcinoma. Biochem. Biophys. Res. Commun. 1990, 171, 1402–1413. [Google Scholar] [CrossRef]
- Carthy, J.M.; Meredith, A.J.; Boroomand, S.; Abraham, T.; Luo, Z.; Knight, D.; McManus, B.M. Versican VI overexpression induces a myofibroblast-likephenotype in cultured fibroblasts. PLoS ONE 2015, 10, e0133056. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Mrazek, A.A.; Carmical, J.R.; Wood, T.G.; Hellmich, M.R.; Eltorky, M.; Bohanon, F.J.; Chao, C. Colorectal cancer-associated fibroblasts are genotypically distinct. Curr. Cancer Ther. Rev. 2014, 10, 97–218. [Google Scholar] [CrossRef] [PubMed]
- Moyal-Atias, K.; Sigal-Batikoff, I.; Fich, A.; Lamprecht, S. Global DNA hypomethylation in stromal fibroblasts imposed by colon cancer cells. Unpublished work. 2017. [Google Scholar]
- Lamprecht, S.; Deliniasos, J.; Fich, A. Do cancer cells impose epigenetic changes on normal fibroblasts? A colon-oriented preliminary inquiry. Anticancer Res. 2014, 10, 6020. [Google Scholar]
- Sigal-Batikoff, I.; Fich, A.; Lamprecht, S. Up-regulation of stromal COX-2 expression by colon carcinoma cells. Unpublished work. 2017. [Google Scholar]
- Rudnick, J.A.; Kuperwasser, C. Stromal biomarkers in breast cancer development and progression. Clin. Exp. Metastasis. 2012, 29, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.; Ringel, A.; Sigal-Batikoff, I.; Abu-Freha, N.; Vaknine, H.; Friah, W.; Reshef, A.; Pinsk, I.; Fich, A.; Lamprecht, S. Human colorectal cancer stage-dependent global DNA hypomethylation of cancer-associated fibroblasts. Anticancer Res. 2016, 36, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, M.; Puig, M.; Carmona, F.J.; Maqueda, M.; Velásquez, A.; Gómez, A.; Labernadie, A.; Lugo, R.; Gabasa, M.; Rigat-Brugarolas, L.G.; et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 2015, 36, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.W.; Hsu, C.H.; Peng, K.L.; Chen, C.C.; Kuo, W.H.; Lee, E.Y.; Shew, J.Y.; Chang, K.J.; Juan, L.J.; Lee, W.H. Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS ONE 2012, 7, e35128. [Google Scholar] [CrossRef] [PubMed]
- Tan Ide, A.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 2013, 133, 2263–2276. [Google Scholar] [PubMed]
- Zong, Y.; Huang, J.; Sankarasharma, D.; Morikawa, T.; Fukayama, M.; Epstein, J.L.; Chada, K.K.; Witte, O.N. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E3395–E3404. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Fusco, A. HMGA and cancer. Biochim. Biophys. Acta 2010, 1799, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, C.; Cataldi, P.; Iop, A.; Isola, M.; Sgarra, R.; Manfioletti, G.; Giancotti, V. The expression of the high-mobility group A2 protein in colorectal cancer and surrounding fibroblasts is linked to tumor invasiveness. Hum. Pathol. 2013, 44, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Norberg, K.J.; Mezheyeuski, A.; Schnittert, J.; Kuninty, P.R.; Moro, C.F.; Paulsson, J.; Schultz, N.A.; Calatayud, D.; Löhr, J.M.; et al. Stroma-regulated HMGA2 is an independent prognostic marker in PDAC and AAC. Br. J. Cancer 2017, 117, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Epigenetic control of the tumor microenvironment. Epigenomics 2016, 8, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.W.; Fujiwara, K.; Che, X.; Zheng, S.; Zheng, L. DNA methylation in the tumor microenvironment. J. Zhejiang Univ. Sci. B. 2017, 18, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Che, G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett. 2017, 13, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harb.Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fässler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-β -crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Scaffidi, A.K.; Hackett, T.L.; Ballard, J.; Shaheen, F.; Thompson, P.J.; Thannickal, V.J.; Knight, D.A. Transforming Growth Factor beta1 induces alphavbeta3 integrin expression in human lung fibroblasts via a beta3 integrin-, c-Src-, and p38 MAPK-dependent pathway. J. Biol. Chem. 2008, 283, 12898–12908. [Google Scholar] [CrossRef] [PubMed]
- Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal modulators of TGF-β in cancer. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Chen, Y.G. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 9, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGF beta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Izzi, L.; Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 2004, 23, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.S.; Massagué, J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat. Cell. Biol. 1999, 1, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Shany, S.; Sigal-Batikoff, I.; Lamprecht, S. Vitamin D and myofibroblasts in fibrosis and cancer: At c ross-purposes with TGF-β/SMAD signaling. Anticancer Res. 2016, 36, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. Impaired Smad7-Smurf-mediated negative regulation of TGF-β signaling in scleroderma fibroblasts. J. Clin. Invest. 2004, 113, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Owens, P.; Moses, H.L. TGF-β, Bone Morphogenetic Protein, and Activin signaling and the tumor microenvironment. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Cukierman, E. Stromal dynamic reciprocity in cancer: Intricacies of fibroblastic-ECM interactions. Curr. Opin. Cell Biol. 2016, 42, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Gharaee-Kermani, M.; Wu, Z.; Phan, S.H. Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am. J. Pathol. 2010, 177, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wu, Z.; Phan, S.H. Smad3 mediates Transforming Growth Factor-beta-induced alpha-smooth muscle actin expression. Am. J. Respir. Cell. Mol. Biol. 2003, 29, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFβ1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, A. Abrogation of TGF-β-1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens--structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force matters: Biomechanical regulation of cell invasion and migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.; de Rond, S.; Rots, M.G.; Bank, R.A. Procollagen lysyl hydroxylase 2 expression is regulated by an alternative downstream Transforming Growth Factor β-1 activation mechanism. J. Biol. Chem. 2015, 290, 28465–28476. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.; Czubryt, M.P. Transcriptional control of collagen I gene expression. Cardiovasc. Hematol. Disord. Drug. Targets 2014, 14, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, Z.; Huang, R.; Yao, Y.; Ma, G. Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS ONE 2013, 8, e60335. [Google Scholar] [CrossRef] [PubMed]
- Hagood, J.S.; Prabhakaran, P.; Kumbla, P.; Salazar, L.; MacEwen, M.W.; Barker, T.H.; Ortiz, L.A.; Schoeb, T.; Siegal, G.P.; Alexander, C.B.; et al. Loss of fibroblast Thy-1 expression correlates with lung fibrogenesis. Am. J. Pathol. 2005, 167, 365–379. [Google Scholar] [CrossRef]
- Neveu, W.A.; Mills, S.T.; Staitieh, B.S.; Sueblinvong, V. TGF-β1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am. J. Physiol.Cell Physiol. 2015, 309, C616–C626. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggiol, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget. 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhao, M.; Liu, J.; He, Z.; Zhang, Y.; You, H.; Huang, J.; Lin, X.; Feng, X.H. Tumor suppressor bromodomain-containing protein 7 cooperates with Smads to promote Transforming Growth Factor-β responses. Oncogene 2017, 36, 362–372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, J.P.X.; Nichols, B.J. Caveolae: One Function or Many? Trends Cell. Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Parat, M.O. The biology of caveolae: Achievements and perspectives. Int. Rev. Cell Mol. Biol. 2009, 273, 117–162. [Google Scholar] [PubMed]
- Sanders, Y.Y.; Liu, H.; Scruggs, A.M.; Duncan, S.R.; Huang, S.K.; Thannickal, V.J. Epigenetic regulation of caveolin-1 gene expression in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2017, 56, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Cui, Z.; Le Saux, C.J.; Horowitz, J.C.; Rangarajan, S.; Kurundkar, A.; Antony, V.B.; Thannickal, V.J. SMAD-independent down-regulation of caveolin-1 by TGF-β: Effects on proliferation and survival of myofibroblasts. PLoS ONE 2015, 10, e0116995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Rajput, A.; Myeroff, L.; Liu, D.F.; Kwon, K.; Willis, J.; Markowitz, S. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998, 58, 3101–3104. [Google Scholar] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Bellam, N.; Pasche, B. Tgf-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010, 155, 85–103. [Google Scholar] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, H.; Kawata, S.; Tamura, S.; Ito, N.; Shirai, Y.; Kiso, S.; Imai, Y.; Shimomukai, H.; Nomura, Y.; Matsuda, Y.; et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: Association with disease progression. Gastroenterology 1996, 110, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Verspaget, H.W.; van der Reijden, J.J.; Jvan der Zon, J.M.; Verheijen, J.H.; Hommes, D.W.; Lamers, C.B.; Sier, C.F. Active TGF-beta1 correlates with myofibroblasts and malignancy in the colorectal adenoma-carcinoma sequence. Cancer Sci. 2009, 100, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Yakicier, M.C.; Irmak, M.B.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 26, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997, 57, 2578–2580. [Google Scholar] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Colorectal carcinogenesis is associated with stromal expression of COL11A1 and COL5A2. Carcinogenesis 2001, 22, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Villa, F.; García-Ocaña, M.; Galván, J.A.; García-Martínez, J.; García-Pravia, P.; Menéndez-Rodríguez, C.; González-del Rey, L.; Barneo-Serra, J.R.; de Los Toyos, J.R. COL11A1/(pro)collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumour Biol. 2015, 36, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; García-Martínez, J.; Vázquez-Villa, F.; García-Ocaña, M.; García-Parvia, C.; Menéndez-Rodríguez, P.; González-del Rey, C.; Barneo-Serra, L.; de los Toyos, J.R. Validation of COL11A1/procollagen 11A1 expression in TGF-β1-activated immortalised human mesenchymal cells and in stromal cells of human colon adenocarcinoma. BMC Cancer 2014, 14, 867. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Islam, A.B.; Herrera, A.; Martín, P.; García, V.; Silva, J.; Garcia, J.M.; Salas, C.; Casal, I.; de Herreros, A.G.; et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin. Cancer Res. 2013, 19, 5914–5926. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, L.; Palmans, S.; Andel, D.; Govaere, O.; Boeckx, B.; Smeets, D.; Galle, E.; Wouters, J.; Barras, D.; Suffiotti, M.; et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br. J. Cancer 2017, 116, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J.; Leyh, B. The impact of tumor stroma on drug response in breast cancer. Semin. Cancer Biol. 2015, 31, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, T. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour-stromal interactions in modifying drugs response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Sahai, E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers 2013, 5, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- De Vlieghere, E.; Verset, L.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-associated fibroblasts as target and tool in cancer therapeutics and diagnostics. Virchows Arch. 2015, 467, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Liu, Q.; Lin, C.M.; Luo, C.; Wang, Y.; Liu, L.; Yin, W.; Hu, S.; Kim, W.Y.; Huang, L. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Res. 2016, 77, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.A.; Varro, A.; Wang, T.C.; Tycko, B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Semin. Cell Dev. Biol. 2010, 21, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Issa, J.-P.J.; Kropf, P. DNA hypomethylating drugs in cancer therapy. Cold Spring Harb. Persp. Med. 2017, 7, 026948. [Google Scholar]
- Shakya, R.; Gonda, T.; Quante, M.; Salas, M.; Kim, S.; Brooks, J.; Hirsch, S.; Davies, J.; Cullo, A.; Olive, K.; et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013, 73, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Yachide, T. Bet bromodomain as a target of epigenetic therapy. Chem. Pharm. Bul. (Tokyo) 2016, 64, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Vakoc, C.R. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. Targeting TGF-β signaling for therapeutic gain. Cold Spring. Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Yan, B.; Vergani, N.; Beyer, B.A.; Chin, E.N.; Zambaldo, C.; Wang, D.; Chatterjee, A.K.; Lairson, L.L.; Schultz, P.G. Small molecule-mediated inhibition of myofibroblast transdifferentiation for the treatment of fibrosis. Proc. Natl. Acad. Sci. USA 2017, 114, 4679–4684. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Han, H.; Von Hoff, D.D. Orchestrating the tumor microenvironment to improve survival for patients with pancreatic cancer: Normalization, not destruction. Cancer J. 2015, 21, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling components of the tumor microenvironment to enhance cancer therapy. Front. Oncol. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.; Caprioli, R.; Chen, X.I.; Walsh, A.J.; Skala, M.C.; Moses, H.L.; et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology 2015, 149, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.S.; Kozlov, P. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Butz, H.; Rácz, K.; Hunyady, L.; Patócs, A. Crosstalk between TGF-β signaling and the microRNA machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamprecht, S.; Sigal-Batikoff, I.; Shany, S.; Abu-Freha, N.; Ling, E.; Delinasios, G.J.; Moyal-Atias, K.; Delinasios, J.G.; Fich, A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts. Cancers 2018, 10, 61. https://doi.org/10.3390/cancers10030061
Lamprecht S, Sigal-Batikoff I, Shany S, Abu-Freha N, Ling E, Delinasios GJ, Moyal-Atias K, Delinasios JG, Fich A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts. Cancers. 2018; 10(3):61. https://doi.org/10.3390/cancers10030061
Chicago/Turabian StyleLamprecht, Sergio, Ina Sigal-Batikoff, Shraga Shany, Naim Abu-Freha, Eduard Ling, George J. Delinasios, Keren Moyal-Atias, John G. Delinasios, and Alexander Fich. 2018. "Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts" Cancers 10, no. 3: 61. https://doi.org/10.3390/cancers10030061
APA StyleLamprecht, S., Sigal-Batikoff, I., Shany, S., Abu-Freha, N., Ling, E., Delinasios, G. J., Moyal-Atias, K., Delinasios, J. G., & Fich, A. (2018). Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts. Cancers, 10(3), 61. https://doi.org/10.3390/cancers10030061