Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer


Abstract
1. Introduction
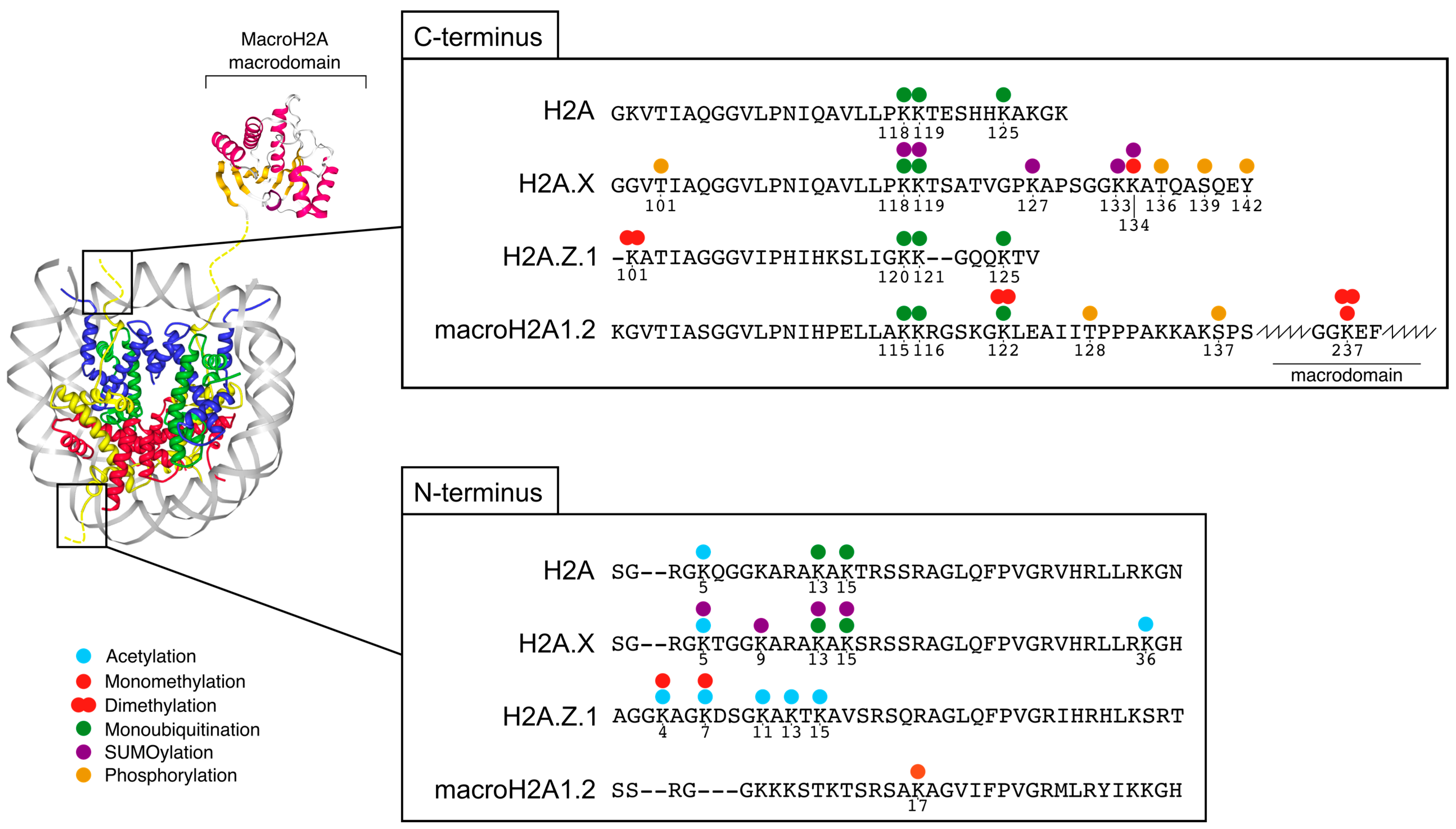
2. Features and Post-Translational Modifications of H2A.Z, H2A.X and macroH2A
2.1. H2A.Z—A Dynamic Protein Regulated by PTMs
2.2. MacroH2A—Stabilizing Histones with a Unique Tripartite Structure
2.3. H2A.X—The Histone Variant at the Core of the DNA Damage Response
3. Role of H2A Variants in Cancer
3.1. H2A.Z Frequently Promotes Pro-Oncogenic Transcription
3.2. MacroH2A Is a Context-Dependent Tumour Suppressor
3.3. H2A.X Is a Tumour Suppressor and a Biomarker
4. Outlook and Conclusions
Acknowledgments
Conflicts of Interest
References
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Schneider, R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta 2016, 1859, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Noh, K.M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Gurard-Levin, Z.A.; Quivy, J.-P.; Almouzni, G. Histone Chaperones: Assisting Histone Traffic and Nucleosome Dynamics. Annu. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Biterge, B.; Schneider, R. Histone variants: Key players of chromatin. Cell Tissue Res. 2014, 356, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- El Kennani, S.; Adrait, A.; Shaytan, A.K.; Khochbin, S.; Bruley, C.; Panchenko, A.R.; Landsman, D.; Pflieger, D.; Govin, J. MS-HistoneDB, a manually curated resource for proteomic analysis of human and mouse histones. Epigenetics Chromatin 2017, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, T.; Takagi, T.; Tsukamoto, D.; Tomaru, C.; Huynh, L.M.; Sivaraman, P.; Kumarevel, T.; Inoue, K.; Nakato, R.; Katou, Y.; et al. Histone Variants Enriched in Oocytes Enhance Reprogramming to Induced Pluripotent Stem Cells. Cell Stem Cell 2014, 14, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.M.; Shinagawa, T.; Ishii, S. Two Histone Variants TH2A and TH2B Enhance Human Induced Pluripotent Stem Cell Generation. Stem Cells Dev. 2016, 25, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Hoghoughi, N.; Barral, S.; Vargas, A.; Rousseaux, S.; Khochbin, S. Histone variants: essential actors in male genome programming. J. Biochem. 2017, 163, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Higashi, M.; Inoue, S.; Ito, T. Core histone H2A ubiquitylation and transcriptional regulation. Exp. Cell Res. 2010, 316, 2707–2712. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Heo, K.; An, W. Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 2009, 37, 5993–6007. [Google Scholar] [CrossRef] [PubMed]
- Altaf, M.; Auger, A.; Monnet-Saksouk, J.; Brodeur, J.; Piquet, S.; Cramet, M.; Bouchard, N.; Lacoste, N.; Utley, R.T.; Gaudreau, L.; et al. NuA4-dependent acetylation of nucleosomal histones H4 and H2A directly stimulates incorporation of H2A.Z by the SWR1 complex. J. Biol. Chem. 2010, 285, 15966–15977. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Davey, G.E.; Nazarov, A.A.; Dyson, P.J.; Davey, C.A. Specific DNA structural attributes modulate platinum anticancer drug site selection and cross-link generation. Nucleic Acids Res. 2011, 39, 8200–8212. [Google Scholar] [CrossRef] [PubMed]
- Moreland, J.L.; Gramada, A.; Buzko, O.V.; Zhang, Q.; Bourne, P.E. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinformatics 2005, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Hothorn, M.; Scheffzek, K.; Ladurner, A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005, 12, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A web application for molecular visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. Web-based molecular graphics for large complexes. In Proceedings of the 21st International Conference on Web3D Technology—Web3D ’16, Anaheim, CA, USA, 22–24 July 2016; ACM Press: New York, NY, USA, 2016; pp. 185–186. [Google Scholar]
- Bonenfant, D.; Coulot, M.; Towbin, H.; Schindler, P.; van Oostrum, J. Characterization of Histone H2A and H2B Variants and Their Post-translational Modifications by Mass Spectrometry. Mol. Cell. Proteomics 2006, 5, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Dryhurst, D.; Rose, K.L.; Shabanowitz, J.; Hunt, D.F.; Ausió, J. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry 2009, 48, 5007–5017. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.; Jaffe, J.D.; Koche, R.P.; Rheinbay, E.; Endoh, M.; Koseki, H.; Carr, S.A.; Bernstein, B.E. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Draker, R.; Ng, M.K.; Sarcinella, E.; Ignatchenko, V.; Kislinger, T. A Combination of H2A.Z and H4 Acetylation Recruits Brd2 to Chromatin during Transcriptional Activation. PLoS Genet. 2012, 8, e1003047. [Google Scholar] [CrossRef] [PubMed]
- Perell, G.T.; Mishra, N.K.; Sudhamalla, B.; Ycas, P.D.; Islam, K.; Pomerantz, W.C.K. Specific Acetylation Patterns of H2A.Z Form Transient Interactions with the BPTF Bromodomain. Biochemistry 2017, 56, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, H.F.; Lyon, N.; Leung, J.W.; Agarwal, P.; Swaim, C.D.; Miller, K.M.; Huibregtse, J.M. Ubiquitin-Activated Interaction Traps (UBAITs) identify E3 ligase binding partners. EMBO Rep. 2015, 16, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Sarcinella, E.; Zuzarte, P.C.; Lau, P.N.I.; Draker, R.; Cheung, P. Monoubiquitylation of H2A.Z Distinguishes Its Association with Euchromatin or Facultative Heterochromatin. Mol. Cell. Biol. 2007, 27, 6457–6468. [Google Scholar] [CrossRef] [PubMed]
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Binda, O.; Sevilla, A.; LeRoy, G.; Lemischka, I.R.; Garcia, B.A.; Richard, S. SETD6 monomethylates H2AZ on lysine 7 and is required for the maintenance of embryonic stem cell self-renewal. Epigenetics 2013, 8, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Chen, Y.J.; Yu, C.J.; Tzeng, S.R.; Wu, I.C.; Kuo, W.H.; Lin, M.C.; Chan, N.L.; Wu, K.J.; Teng, S.C. SMYD3-mediated H2A.Z.1 methylation promotes cell cycle and cancer proliferation. Cancer Res. 2016, 76, 6043–6053. [Google Scholar] [CrossRef] [PubMed]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 Spreading and SUMO-H2A.Z-Dependent Chromosome Fixation in Response to a Persistent DNA Double-Strand Break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, A.; Ikura, M.; Ikura, T.; Sun, J.; Horikoshi, Y.; Shima, H.; Igarashi, K.; Kusakabe, M.; Harata, M.; Horikoshi, N.; et al. SUMO modification system facilitates the exchange of histone variant H2A.Z-2 at DNA damage sites. Nucleus 2017. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Nusinow, D.A.; Chalkley, R.J.; Plath, K.; Panning, B.; Burlingame, A.L. Mapping Post-translational Modifications of the Histone Variant MacroH2A1 Using Tandem Mass Spectrometry. Mol. Cell. Proteomics 2006, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Ono, T.; Wakata, Y.; Okawa, K.; Tagami, H.; Shibahara, K.I. Histone variant macroH2A1.2 is mono-ubiquitinated at its histone domain. Biochem. Biophys. Res. Commun. 2005, 336, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Muñoz, I.; Lund, A.H.; van der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D.A.; Panning, B.; Marahrens, Y.; van Lohuizen, M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7635–7640. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Chan, D.W.; Jung, S.Y.; Chen, Y.; Qin, J.; Wang, Y. The Histone Variant MacroH2A1 Is a BRCA1 Ubiquitin Ligase Substrate. Cell Rep. 2017, 19, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Muratore-Schroeder, T.L.; Diaz, R.L.; Chow, J.C.; Changolkar, L.N.; Shabanowitz, J.; Heard, E.; Pehrson, J.R.; Hunt, D.F.; Allis, C.D. A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis. Proc. Natl. Acad. Sci. USA 2008, 105, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Maiolica, A.; de Medina-Redondo, M.; Schoof, E.M.; Chaikuad, A.; Villa, F.; Gatti, M.; Jeganathan, S.; Lou, H.J.; Novy, K.; Hauri, S.; et al. Modulation of the Chromatin Phosphoproteome by the Haspin Protein Kinase. Mol. Cell. Proteomics 2014, 13, 1724–1740. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Odate, S.; Chandramouly, G.; Scully, R.A. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle 2010, 9, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yu, Y.; Lee, S.C.; Ishibashi, T.; Lees-Miller, S.P.; Ausió, J. Phosphorylation of histone H2A.X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. J. Biol. Chem. 2010, 285, 17778–17788. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX Is Phosphorylated in an ATR-dependent Manner in Response to Replicational Stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Keogh, M.-C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. γ-H2AX Dephosphorylation by Protein Phosphatase 2A Facilitates DNA Double-Strand Break Repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Xu, X.; Zhong, X.; Ahmed, F.; Zhong, J.; Liao, J.; Dykxhoorn, D.M.; Weinstock, D.M.; Pfeifer, G.P.; Lieberman, J. A PP4-Phosphatase Complex Dephosphorylates γ-H2AX Generated during DNA Replication. Mol. Cell 2008, 31, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Chen, G.I.; Gingras, A.C.; Durocher, D. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008, 9, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Zhong, J.; Ye, R.; Moorhead, G.B.G.; Xu, X.; Lees-Miller, S.P. Protein Phosphatase 6 Interacts with the DNA-Dependent Protein Kinase Catalytic Subunit and Dephosphorylates γ-H2AX. Mol. Cell. Biol. 2010, 30, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates γH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Lin, L.; Zhang, X.; Nguyen, T.A.; Darlington, Y.; Waldman, A.S.; Lu, X.; Donehower, L.A. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant γ-H2AX and suppresses DNA double strand break repair. J. Biol. Chem. 2010, 285, 12935–12947. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Jeong, D.G.; Jung, S.K.; Ryu, S.E.; Xiao, A.; Allis, C.D.; Kim, S.J.; Tonks, N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J. Biol. Chem. 2009, 284, 16066–16070. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Gatti, M.; Pinato, S.; Maspero, E.; Soffientini, P.; Polo, S.; Penengo, L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012, 11, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.-C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Nicassio, F.; Corrado, N.; Vissers, J.H.A.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Lilli, D.R.; Patterson-Fortin, J.; Coleman, K.A.; Morrissey, D.E.; Greenberg, R.A. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 3166–3171. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.R.; Densham, R.M.; Jia, J.; Garvin, A.J.; Stone, H.R.; Shah, V.; Weekes, D.; Festy, F.; Beesley, J.; Morris, J.R. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012, 31, 3918–3934. [Google Scholar] [CrossRef] [PubMed]
- Mosbech, A.; Lukas, C.; Bekker-Jensen, S.; Mailand, N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J. Biol. Chem. 2013, 288, 16579–16587. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Diaz, M.R.; Martin, Y.; Berg, A.; Freire, R.; Smits, V.A.J. Dub3 controls DNA damage signalling by direct deubiquitination of H2AX. Mol. Oncol. 2014, 8, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, K.; Mao, Z.; Luo, J.; Gu, W.; Zhao, W. USP11 Is a Negative Regulator to γH2AX Ubiquitylation by RNF8/RNF168. J. Biol. Chem. 2016, 291, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Benlekbir, S.; Fradet-Turcotte, A.; Sherker, A.; Julien, J.-P.; McEwan, A.; Noordermeer, S.M.; Sicheri, F.; Rubinstein, J.L.; Durocher, D. The structural basis of modified nucleosome recognition by 53BP1. Nature 2016, 536, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Salomons, F.A.; Hoogstraten, D.; Groothuis, T.A.M.; De Waard, H.; Wu, J.; Yuan, L.; Citterio, E.; Houtsmuller, A.B.; Neefjes, J.; et al. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev. 2006, 20, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 Is Recruited to DNA Breaks and Contributes to DNA Damage-Induced H2A Ubiquitination and Repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Peng, G.; Hungs, W.C.; Lin, S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 2011, 286, 28599–28607. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Kang, H.Y.; Yang, W.L.; Wu, J.; Jeong, Y.S.; Wang, J.; Chan, C.H.; Lee, S.W.; Zhang, X.; Lamothe, B.; et al. Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J. Biol. Chem. 2011, 286, 30806–30815. [Google Scholar] [CrossRef] [PubMed]
- Kusch, T.; Florens, L.; MacDonald, H.W.; Swanson, S.K.; Glaser, R.L.; Yates, J.R., III; Abamyr, S.M.; Washburn, M.P.; Workman, J.L. Acetylation by Tip60 Is Required for Selective Histone Variant Exchange at DNA Lesions. Science 2004, 306, 2084–2087. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K.; et al. DNA Damage-Dependent Acetylation and Ubiquitination of H2AX Enhances Chromatin Dynamics. Mol. Cell. Biol. 2007, 27, 7028–7040. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xu, Y.; Price, B.D. Acetylation of H2AX on lysine 36 plays a key role in the DNA double-strand break repair pathway. FEBS Lett. 2010, 584, 2926–2930. [Google Scholar] [CrossRef] [PubMed]
- Sone, K.; Piao, L.; Nakakido, M.; Ueda, K.; Jenuwein, T.; Nakamura, Y.; Hamamoto, R. Critical role of lysine 134 methylation on histone H2AX for γ-H2AX production and DNA repair. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, M.K.; Kudithipudi, S.; Jeltsch, A. Investigation of H2AX methylation by the SUV39H2 protein lysine methyltransferase. FEBS Lett. 2016, 590, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Alpert, A.; Leiter, C.; Gong, F.; Jackson, S.P.; Miller, K.M. Systematic Identification of Functional Residues in Mammalian Histone H2AX. Mol. Cell. Biol. 2013, 33, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Faast, R.; Thonglairoam, V.; Schulz, T.C.; Beall, J.; Wells, J.R.E.; Taylor, H.; Matthaei, K.; Rathjen, P.D.; Tremethick, D.J.; Lyons, I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001, 11, 1183–1187. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; González-Romero, R.; Dryhurst, D.; Ishibashi, T.; Ausió, J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, C.; Schneider, K.; Pünzeler, S.; Wiedemann, S.M.; Bielmeier, C.; Bocola, M.; Eberl, H.C.; Kuegel, W.; Neumann, J.; Kremmer, E.; et al. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res. 2012, 40, 5951–5964. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, A.; Binda, O. Post-translational modifications of the histone variant H2AZ. Stem Cell Res. 2014, 12, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Xu, F.; Zhang, K.; Grunstein, M. Acetylation of H2AZ Lys 14 is associated with genome-wide gene activity in yeast. Genes Dev. 2006, 20, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Côté, J. The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 2004, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Auger, A.; Galarneau, L.; Altaf, M.; Nourani, A.; Doyon, Y.; Utley, R.T.; Cronier, D.; Allard, S.; Côté, J. Eaf1 Is the Platform for NuA4 Molecular Assembly That Evolutionarily Links Chromatin Acetylation to ATP-Dependent Exchange of Histone H2A Variants. Mol. Cell. Biol. 2008, 28, 2257–2270. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.; Myers, F.A.; Mantouvalou, E.; Lefevre, P.; Greaves, I.; Bonifer, C.; Tremethick, D.J.; Thorne, A.W.; Crane-Robinson, C. The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005, 33, 5633–5639. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Markoulaki, S.; Levine, S.S.; Hanna, J.; Lodato, M.A.; Sha, K.; Young, R.A.; Jaenisch, R.; Boyer, L.A. H2AZ Is Enriched at Polycomb Complex Target Genes in ES Cells and Is Necessary for Lineage Commitment. Cell 2008, 135, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cui, K.; Northrup, D.; Liu, C.; Wang, C.; Tang, Q.; Ge, K.; Levens, D.; Crane-Robinson, C.; Zhao, K. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2013, 12, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, M.; Nordell Markovits, A.; Rodrigue, S.; Lupien, M.; Jacques, P.É.; Gévry, N. The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res. 2015, 43, 9742–9756. [Google Scholar] [CrossRef] [PubMed]
- Law, C.; Cheung, P. Expression of non-acetylatable H2A.Z in myoblast cells blocks myoblast differentiation through disruption of MyoD expression. J. Biol. Chem. 2015, 290, 13234–13249. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, A.; Meshorer, E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015, 16, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Ouararhni, K.; Papin, C.; Diebold, M.; Padmanabhan, K.; Marek, M.; Stoll, I.; Roy, L.; Reilly, P.T.; Mak, T.W.; et al. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 2014, 505, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z Controls a Critical Chromatin Remodeling Step Required for DNA Double-Strand Break Repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Yuzugullu, O.; Ayrapetov, M.K.; Price, B.D. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Gundimella, S.K.Y.; Caron, C.; Perche, P.; Pehrson, J.R.; Khochbin, S. Structural Characterization of the Histone Variant macroH2A. Mol. Cell. Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef] [PubMed]
- Changolkar, L.N.; Pehrson, J.R. Reconstitution of nucleosomes with histone macroH2A1.2. Biochemistry 2002, 41, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Kustatscher, G.; Buhecha, H.R.; Allen, M.D.; Pugieux, C.; Sait, F.; Bycroft, M.; Ladurner, A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Changolkar, L.N.; Costanzi, C.; Leu, N.A. Mice Without MacroH2A Histone Variants. Mol. Cell. Biol. 2014, 34, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Uribesalgo, I.; Wibowo, I.; Rué, P.; Martin, D.; Gutierrez, A.; Morey, L.; Guigó, R.; López-Schier, H.; Di Croce, L. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 2009, 16, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lv, P.; Yan, G.; Fan, H.; Cheng, L.; Zhang, F.; Dang, Y.; Wu, H.; Wen, B. MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 2015, 5, 17186. [Google Scholar] [CrossRef] [PubMed]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Marjanović, M.P.; Cantari’o, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Posavec, M.; Timinszky, G.; Buschbeck, M. Macro domains as metabolite sensors on chromatin. Cell. Mol. Life Sci. 2013, 70, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Fried, V. MacroH2A, a core histone containing a large nonhistone region. Science 1992, 257, 1398–1400. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, B.P.; Willard, H.F. Histone H2A variants and the inactive X chromosome: identification of a second macroH2A variant. Hum. Mol. Genet. 2001, 10, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, C.; Pehrson, J.R. MACROH2A2, a New Member of the MACROH2A Core Histone Family. J. Biol. Chem. 2001, 276, 21776–21784. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.K.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Ouararhni, K.; Hadj-Slimane, R.; Ait-Si-Ali, S.; Robin, P.; Mietton, F.; Harel-Bellan, A.; Dimitrov, S.; Hamiche, A. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006, 20, 3324–3336. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Jung, B. Differential Regulation and Predictive Potential of MacroH2A1 Isoforms in Colon Cancer. AJPA 2012, 180, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martin-Caballero, J.; Sumoy, L.; et al. MacroH2A1 Regulates the Balance between Self-Renewal and Differentiation Commitment in Embryonic and Adult Stem Cells. Mol. Cell. Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Posavec Marjanović, M.; Hurtado-Bagès, S.; Lassi, M.; Valero, V.; Malinverni, R.; Delage, H.; Navarro, M.; Corujo, D.; Guberovic, I.; Douet, J.; et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD+ consumption. Nat. Struct. Mol. Biol. 2017, 24, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-Substrate Complexes: Insights into Molecular Architectures of BTB-Cul3 Ubiquitin Ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Ausió, J.; Abbott, D.W. The many tales of a tail: Carboxyl-terminal tail heterogeneity specializes histone H2A variants for defined chromatin function. Biochemistry 2002, 41, 5945–5949. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. Double-stranded Brekas Induce Histone H2AX phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Meffre, E.; Difilippantonio, M.J.; Redon, C.; et al. Genomic Instability in Mice Lacking Histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y. Chromatin response to DNA double-strand break damage. Epigenomics 2011, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Pinder, J.B.; Attwood, K.M.; Dellaire, G. Reading, writing, and repair: The role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front. Genet. 2013, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 2009, 186, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Wang, B. RNF8- and Ube2S-Dependent Ubiquitin Lysine 11-Linkage Modification in Response to DNA Damage. Mol. Cell 2017, 66, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ikura, M.; Furuya, K.; Matsuda, S.; Matsuda, R.; Shima, H.; Adachi, J.; Matsuda, T.; Shiraki, T.; Ikura, T. Acetylation of Histone H2AX at Lys 5 by the TIP60 Histone Acetyltransferase Complex Is Essential for the Dynamic Binding of NBS1 to Damaged Chromatin. Mol. Cell. Biol. 2015, 35, 4147–4157. [Google Scholar] [CrossRef] [PubMed]
- Ikura, M.; Furuya, K.; Fukuto, A.; Matsuda, R.; Adachi, J.; Matsuda, T.; Kakizuka, A. Coordinated Regulation of TIP 60 and Poly(ADP-Ribose) Polymerase 1 in Damaged-Chromatin Dynamics. Mol. Cell. Biol. 2016, 36, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Liu, J.; Khramtsov, A.; Tretiakova, M.S.; et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Svotelis, A.; Gévry, N.; Grondin, G.; Gaudreau, L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle 2010, 9, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mora, F.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Nair, S.S.; Patterson, K.I.; Tremethick, D.J.; Stirzaker, C.; Clark, S.J. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 2012, 22, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Dryhurst, D.; McMullen, B.; Fazli, L.; Rennie, P.S.; Ausió, J. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012, 315, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Dryhurst, D.; Ausió, J. Histone H2A.Z deregulation in prostate cancer. Cause or effect? Cancer Metastastis Rev. 2014, 33, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Punj, V.; Choi, J.; Heo, K.; Kim, J.M.; Laird, P.W.; An, W. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenetics Chromatin 2013, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.D.; Kim, P.-J.; Eun, J.W.; Shen, Q.; Kim, H.S.; Shin, W.C.; Ahn, Y.M.; Park, W.S.; Lee, J.Y.; Nam, S.W. Oncogenic potential of histone-variant H2A.Z.1 and its regulatory role in cell cycle and epithelial-mesenchymal transition in liver cancer. Oncotarget 2016, 5, 11412–11423. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Hake, S.B.; Bernstein, E. Histone variant H2A.Z.2: A novel driver of melanoma progression. Mol. Cell. Oncol. 2016, 3, e1073417. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Long, J.; Li, J. MacroH2A suppresses the proliferation of the B16 melanoma cell line. Mol. Med. Rep. 2014, 10, 1845–1850. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Konstantinov, N.K.; Ulff-Møller, C.J.; Dimitrov, S. Histone variants and melanoma: facts and hypotheses. Pigment Cell Melanoma Res. 2016, 29, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Heo, K.; Choi, J.; Kim, K.; An, W. The histone variant MacroH2A regulates Ca2+ influx through TRPC3 and TRPC6 channels. Oncogenesis 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Shim, J.W.; Park, H.S.; Eum, D.-Y.; Park, M.-T.; Mi Yi, J.; Choi, S.H.; Kim, S.D.; Son, T.G.; Lu, W.; et al. MacroH2A1 downregulation enhances the stem-like properties of bladder cancer cells by transactivation of Lin28B. Oncogene 2016, 35, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for Histone MacroH2A1 Isoforms Marks Steatosis-Associated Hepatocellular Carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-Mediated Alternative Splicing of the Histone Variant MacroH2A1 Regulates Cancer Cell Proliferation. Mol. Cell. Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-H.; Miyai, K.; Sporn, J.C.; Luo, L.; Wang, J.Y.J.; Cosman, B.; Ramamoorthy, S. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 2016, 69, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Difilippantonio, S.; Difilippantonio, M.J.; Fernandez-Capetillo, O.; Pilch, D.R.; Sedelnikova, O.A.; Eckhaus, M.; Ried, T.; Bonner, W.M.; Nussenzweig, A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114, 371–383. [Google Scholar] [CrossRef]
- Srivastava, N.; Gochhait, S.; Gupta, P.; Bamezai, R.N.K. Copy number alterations of the H2AFX gene in sporadic breast cancer patients. Cancer Genet. Cytogenet. 2008, 180, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Jurkovic Mlakar, S.; Lopez, G.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. 11q deletion in neuroblastoma: A review of biological and clinical implications. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Kryh, H.; Nethander, M.; Sjöberg, R.-M.; Träger, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.A.; White, J.S.; Huang, X.; Schoppy, D.W.; Baysal, B.E.; Baskaran, R.; Bakkenist, C.J.; Saunders, W.S.; Hsu, L.-C.; Romkes, M.; et al. Loss of Distal 11q Is Associated with DNA Repair Deficiency and Reduced Sensitivity to Ionizing Radiation in Head and Neck Squamous Cell Carcinoma. Genes. Chromosomes Cancer 2007, 46, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Goutham, H.V.; Mumbrekar, K.D.; Vadhiraja, B.M.; Fernandes, D.J.; Sharan, K.; Kanive Parashiva, G.; Kapaettu, S.; Bola Sadashiva, S.R. DNA Double-Strand Break Analysis by γ-H2AX Foci: A Useful Method for Determining the Overreactors to Radiation-Induced Acute Reactions Among Head-and-Neck Cancer Patients. Int. J. Radiat. Oncol. 2012, 84, e607–e612. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, I.P.; Caramelo, F.; Esteves, L.; Menoita, J.; Marques, F.; Barroso, L.; Miguéis, J.; Melo, J.B.; Carreira, I.M. Genomic predictive model for recurrence and metastasis development in head and neck squamous cell carcinoma patients. Sci. Rep. 2017, 7, 13897. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, T.; Skowronska, A. The role of ATM mutations and 11q deletions in disease progression in chronic lymphocytic leukemia. Leuk. Lymphoma 2014, 55, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Knittel, G.; Liedgens, P.; Reinhardt, H.C. Targeting ATM-deficient CLL through interference with DNA repair pathways. Front. Genet. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Van Kuijk, S.J.A.; Sweep, F.C.G.J.; Nagtegaal, I.D.; Hoogerbrugge, N.; Martens, J.W.M.; Timmermans, M.A.; Van Laarhoven, H.W.M.; Bussink, J.; Span, P.N. Constitutive expression of γ-H2AX has prognostic relevance in triple negative breast cancer. Radiother. Oncol. 2011, 101, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Warters, R.L.; Adamson, P.J.; Pond, C.D.; Leachman, S.A. Melanoma Cells Express Elevated Levels of Phosphorylated Histone H2AX Foci. J. Invest. Dermatol. 2005, 124, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Wasco, M.J.; Pu, R.T.; Yu, L.; Su, L.; Ma, L. Expression of γ-H2AX in melanocytic lesions. Hum. Pathol. 2008, 39, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-salomon, A.; Miller, K.M. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Choudhuri, R.; Aziz, T.; Maeda, D.; Boufraqech, M.; Parekh, P.R.; Sethi, T.K.; Kasoji, M.; Abrams, N.; et al. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Sethi, T.K.; Burrell, A.S.; Jailwala, P.; Kasoji, M.; Abrams, N.; Merchant, A.; Bonner, W.M. Twist1 and Slug mediate H2AX-regulated epithelial-mesenchymal transition in breast cells. Cell Cycle 2016, 15, 2398–2404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yager, J.D.; Davidson, N.E. Mechanisms of Disease : Estrogen Carcinogenesis in Breast Cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Yerrum, S.; Safadi, F.F.; Monroy, M.A. The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. J. Cell. Physiol. 2010, 224, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Chevillard-briet, M.; Quaranta, M.; Grézy, A.; Mattera, L.; Courilleau, C.; Philippe, M.; Mercier, P.; Corpet, D.; Lough, J.; Ueda, T.; et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum. Mol. Genet. 2014, 23, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Taty-Taty, G.-C.; Courilleau, C.; Quaranta, M.; Aymard, F.; Trouche, D.; Courilleau, C.; Quaranta, M.; Carayon, A.; Chailleux, C. H2A.Z depletion impairs proliferation and viability but not DNA double-strand breaks repair in human immortalized and tumoral cell lines. Cell Cycle 2014, 13, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Oki, M.; Szyjka, S.J.; Aparicio, O.M.; Kamakaka, R.T. H2A.Z Functions To Regulate Progression through the Cell Cycle. Mol. Cell. Biol. 2006, 26, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, L.; Dalvai, M.; Kocanova, S.; Moutahir, F.; Bystricky, K. Activation of p21 by HDAC Inhibitors Requires Acetylation of H2A.Z. PLoS ONE 2013, 8, e54102. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Domaschenz, R.; Kurscheid, S.; Nekrasov, M.; Han, S.; Tremethick, D.J. The Histone Variant H2A.Z Is a Master Regulator of the Epithelial-Mesenchymal Transition. Cell Rep. 2017, 21, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Giakountis, A.; Moulos, P.; Sarris, M.E.; Hatzis, P.; Talianidis, I. Smyd3-associated regulatory pathways in cancer. Semin. Cancer Biol. 2017, 42, 70–80. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, D.J.; Williamson, S.C.; Alkharaif, D.; Monteiro, C.M.; Goudreault, M.; Gaughan, L.; Robson, C.N.; Gingras, A.-C.; Binda, O. SETD6 controls the expression of estrogen- responsive genes and proliferation of breast carcinoma cells. Epigenetics 2014, 9, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Cantariño, N.; Douet, J.; Buschbeck, M. MacroH2A - An epigenetic regulator of cancer. Cancer Lett. 2013, 336, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yi, P.; Pi, J.; Li, L.; Hui, J.; Wang, F. QKI5-mediated alternative splicing of the histone variant macroH2A1 regulates gastric carcinogenesis. Oncotarget 2016, 7, 32821–32834. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, J.; Shen, Y.; Majer, M.M.; Nelson, C.C.; Parsawar, K.; Heichman, K.A.; Kuwada, S.K. The atypical histone macroH2A1.2 interacts with HER-2 protein in cancer cells. J. Biol. Chem. 2012, 287, 23171–23183. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, A.-C.; Castells, M.; Mermet, J.; Kocanova, S.; Dalvai, M.; Bystricky, K. Increased macroH2A1.1 Expression Correlates with Poor Survival of Triple-Negative Breast Cancer Patients. PLoS ONE 2014, 9, e98930. [Google Scholar] [CrossRef] [PubMed]
- Danan-Gotthold, M.; Golan-Gerstl, R.; Eisenberg, E.; Meir, K.; Karni, R.; Levanon, E.Y. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015, 43, 5130–5144. [Google Scholar] [CrossRef] [PubMed]
- Gamble, M.J.; Frizzell, K.M.; Yang, C.; Krishnakumar, R.; Kraus, W.L. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010, 24, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Posavec, M.; Douet, J.; Buschbeck, M. MacroH2A in stem cells: a story beyond gene repression. Epigenomics 2012, 4, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Cong, R.; Das, S.; Douet, J.; Wong, J.; Buschbeck, M.; Mongelard, F.; Bouvet, P. MacroH2A1 histone variant represses rDNA transcription. Nucleic Acids Res. 2014, 42, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Barzily-Rokni, M.; Friedman, N.; Ron-Bigger, S.; Isaac, S.; Michlin, D.; Eden, A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res. 2011, 39, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Wanessa Rocha, S.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of Cancer Cell Stemness by Depletion of Macrohistone H2A1 in Hepatocellular Carcinoma. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sturgill, D.; Sebastian, R.; Khurana, S.; Tran, A.D.; Edwards, G.B.; Kruswick, A.; Burkett, S.; Hosogane, E.K.; Hannon, W.W.; et al. Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol. Cell 2018, 69, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, Y.; Gursoy-Yuzugullu, O.; Price, B.D. The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett. 2012, 586, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.C.; Makharashvili, N.; Agarwal, P.; Chiu, L.Y.; Pourpre, R.; Cammarata, M.B.; Cannon, J.R.; Sherker, A.; Durocher, D.; Brodbelt, J.S.; et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017, 31, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Palla, V.V.; Karaolanis, G.; Katafigiotis, I.; Anastasiou, I.; Patapis, P.; Dimitroulis, D.; Perrea, D. gamma-H2AX: Can it be established as a classical cancer prognostic factor? Tumor Biol. 2017, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Osawa, M.; Takamura, M.; Yamagiwa, S.; Aoyagi, Y.; Sanpei, A.; Fujimaki, S. DNA damage sensor γ-H2AX is increased in preneoplastic lesions of hepatocellular carcinoma. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.I.; Gong, Y.; Ye, Y.; Lin, J.; Chang, D.W.; Kamat, A.M.; Wu, X. γ-H2AX level in peripheral blood lymphocytes as a risk predictor for bladder cancer. Carcinogenesis 2013, 34, 2543–2547. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Pardini, B.; Allione, A.; Fiorito, G.; Viberti, C.; Guarrera, S.; Russo, A.; Anglesio, S.; Ruo Redda, M.G.; et al. H2AX phosphorylation level in peripheral blood mononuclear cells as an event-free survival predictor for bladder cancer. Mol. Carcinog. 2016, 55, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chang, D.W.; Gong, Y.; Eng, C.; Wu, X. Measurement of DNA damage in peripheral blood by the gamma-H2AX assay as predictor of colorectal cancer risk. DNA Repair 2017, 53, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Pouliliou, S.; Koukourakis, M.I. Gamma histone 2AX (γH2AX) as a predictive tool in radiation oncology. Biomarkers 2014, 19, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Halaby, M.-J.; Hakem, A.; Cardoso, R.; El Ghamrasni, S.; Harding, S.; Chan, N.; Bristow, R.; Sanchez, O.; Durocher, D.; et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 2010, 207, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Bohgaki, T.; Bohgaki, M.; Cardoso, R.; Panier, S.; Zeegers, D.; Li, L.; Stewart, G.S.; Sanchez, O.; Hande, M.P.; Durocher, D.; et al. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet. 2011, 7, e1001381. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Li, L.; Guo, L.; Su, Y.; Wang, Y.; Xu, Y.; Wang, X.; Meng, S.; Lei, L.; Xu, L.; Shao, G. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, H.; Wang, C.; Sun, H.; Sun, G.; Sun, N.; Zeng, K.; Song, H.; Zou, R.; Zhou, T.; et al. RNF8 identified as a co-activator of estrogen receptor α promotes cell growth in breast cancer. Biochim. Biophys. Acta 2017, 1863, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Csizmok, V.; Forman-Kay, J.D. Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications. Curr. Opin. Struct. Biol. 2018, 48, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Torrano, V.; Martín-Martín, N.; Carracedo, A. Metabolism and Transcription in Cancer: Merging Two Classic Tales. Front. Cell Dev. Biol. Cell Dev. Biol. 2018, 5, 1–8. [Google Scholar] [CrossRef]


{kind=link}
| Histone | Modification | Residues | Writers | Erasers | Readers | References |
|---|---|---|---|---|---|---|
| H2A.Z | Acetylation | K4, K7, K11, K13, K15 | TIP60 | Uncharacterised histone deacetylases (HDACs) | BPTF, Brd2 | [19,25,26,27,28,29] |
| Monoubiquitination | Unknown | RNF168 | Unknown | Unknown | [30] | |
| Monoubiquitination | K120, K121, K125 | RING1B (PRC1) | USP10 | Unknown | [27,31,32] | |
| Methylation | K4, K7 | SETD6, SMYD3 | Unknown | Unknown | [33] | |
| Dimethylation | K101 | SMYD3 | Unknown | Unknown | [34] | |
| SUMOylation | K126, K133 (Yeast) | PIAS4 | Unknown | Unknown | [35,36] | |
| MacroH2A | Monoubiquitination | K115, K116 | PRC1, CULLIN3/SPOP | Unknown | Unknown | [17,37,38,39] |
| Monoubiquitination | K122 | BRCA1 | Unknown | Unknown | [40] | |
| Methylation | K17 (mono), K122 (di), K237 (mono/di) | Unknown | Unknown | Unknown | [37] | |
| Phosphorylation | T128 | Unknown | Unknown | Unknown | [37] | |
| Phosphorylation | S137 | Cdk1/cyclinB, Cdk2/cyclinE, Haspin kinase | Unknown | Unknown | [41,42] | |
| H2A.X | Phosphorylation | T101 | Unknown | Unknown | Unknown | [43] |
| Phosphorylation | T136 | DNA-PK | Unknown | Unknown | [44] | |
| Phosphorylation | S139 | ATM, ATR, DNA-PK | PP2A, PP4, PP6, Wip1 | MDC1 | [45,46,47,48,49,50,51,52,53] | |
| Phosphorylation | Y142 | WSTF | EYA | Impairs MDC1 | [53,54,55,56] | |
| Monoubiquitination | K13, K15 | RNF168 | USP3, USP11, USP16, USP44, BRCC36, Dub3 | 53BP1 | [57,58,59,60,61,62,63,64,65,66,67,68,69,70] | |
| Monoubiquitination | K118, K119 | RNF2-BMI1 (PRC1) | Unknown | Unknown | [71,72,73,74,75] | |
| Acetylation | K5 | TIP60 | Uncharacterised HDACs | Unknown | [76,77] | |
| Acetylation | K36 | CBP/p300 | Unknown | Unknown | [78] | |
| Methylation | K134 | SUV39H2 | Unknown | Unknown | [79,80] | |
| SUMOylation | K5, K9, K13, K15, K118, K119, K127, K133, K134 | PIAS4 | Unknown | Unknown | [81] |
| Histone | Cancer Type | Alteration | Observation/Function | PTM Involvement | References |
|---|---|---|---|---|---|
| H2A.Z | Breast | Upregulation | Correlates with poor survival, promotes proliferation, involved in gene activation by estrogen signalling | Dimethylation on K101 promotes proliferation by activating cyclinA1 H2A.Z acetylation may be responsible for increased p21 expression in ER-negative p53−/− breast cancer cells | [34,92,134,135,136] |
| Prostate | Upregulation | Involved in gene activation by androgen signalling, poises PSA activation | Acetylation and deubiquitination are necessary for oncogenic hormone-mediated activation | [32,137,138,139,140] | |
| Bladder | Upregulation | Promotes cell proliferation and oncogene transcription by recruiting WDR5 (MLL complex) and BPTF (NuRD complex) | Unknown | [141] | |
| H2A.Z.1 | Hepatocellular carcinoma | Upregulation | Correlates with poor survival, promotes tumour growth and EMT | Unknown | [142] |
| H2A.Z.2 | Melanoma | Upregulation | Correlates with poor survival, its depletion sensitizes cells to chemotherapy | Unknown | [143,144] |
| macroH2A1, macroH2A2 | Melanoma | Downregulation | Promotes disease progression and metastasis | Unknown | [145,146,147] |
| macroH2A1, macroH2A2 | Bladder cancer | Downregulation | Correlates with disease progression, promotes cell growth, stemness and invasiveness | Unknown | [148,149] |
| macroH2A1 | Hepatocellular carcinoma | Upregulation | Higher immunopositivity in steatosis-associated hepatocellular carcinoma, prevents chemotherapy-induced senescence | Unknown | [150,151] |
| macroH2A1.1, macroH2A1.2 | Breast cancer | Increased macroH2A1.2/macroH2A1.1 ratio | Observed in highly proliferative tumours, correlates with poor survival, promotes tumour growth and metastasis | Unknown | [152] |
| macroH2A1.1 | Colorectal cancer | Downregulation | Correlates with poor survival, promotes proliferation and metastasis | Unknown | [112] |
| macroH2A1.1 | Lung cancer | Downregulation | Correlates with higher risk of tumour recurrence | Unknown | [153,154] |
| macroH2A2 | Anal neoplasm | Downregulation | Correlates with disease progression | Unknown | [155] |
| H2A.X | Sporadic breast cancer | Deletion | Proposed to increase genomic instability and tumorigenesis as observed in KO mice | Does not apply | [118,119,156,157] |
| Neuroblastoma | Deletion | Correlates with disease progression and poor prognosis | Does not apply | [158,159] | |
| Head and neck squamous cell carcinoma | Deletion | Associated with higher genomic instability and reduced radiosensitivity, included in predictive model for recurrence and metastasis risk | Does not apply | [160,161,162] | |
| Chronic lymphocytic leukaemia | Deletion | Associated with higher genomic instability, correlates with poor prognosis | Does not apply | [163,164] | |
| Triple negative breast cancer | Upregulation | High levels of γ-H2A.X correlate with poor prognosis | S139 Phosphorylation | [165] | |
| Melanoma | Upregulation | High levels of γ-H2A.X observed in melanocytic lesions | S139 Phosphorylation | [166,167] | |
| Breast cancer | Downregulation | Correlates with better prognosis | Chemotherapy induces H2A.X degradation mediated by polyubiquitination at K13 and K15 | [168] | |
| Colon cancer cells | Downregulation | Promotes EMT | Unknown | [169] | |
| Breast cancer cells | Downregulation | Promotes EMT | Unknown | [170] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corujo, D.; Buschbeck, M. Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers 2018, 10, 59. https://doi.org/10.3390/cancers10030059
Corujo D, Buschbeck M. Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers. 2018; 10(3):59. https://doi.org/10.3390/cancers10030059
Chicago/Turabian StyleCorujo, David, and Marcus Buschbeck. 2018. "Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer" Cancers 10, no. 3: 59. https://doi.org/10.3390/cancers10030059
APA StyleCorujo, D., & Buschbeck, M. (2018). Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers, 10(3), 59. https://doi.org/10.3390/cancers10030059