Mitochondrial Hyperactivation and Enhanced ROS Production are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling

 , , ,
, , ,
Abstract
1. Introduction
2. Results
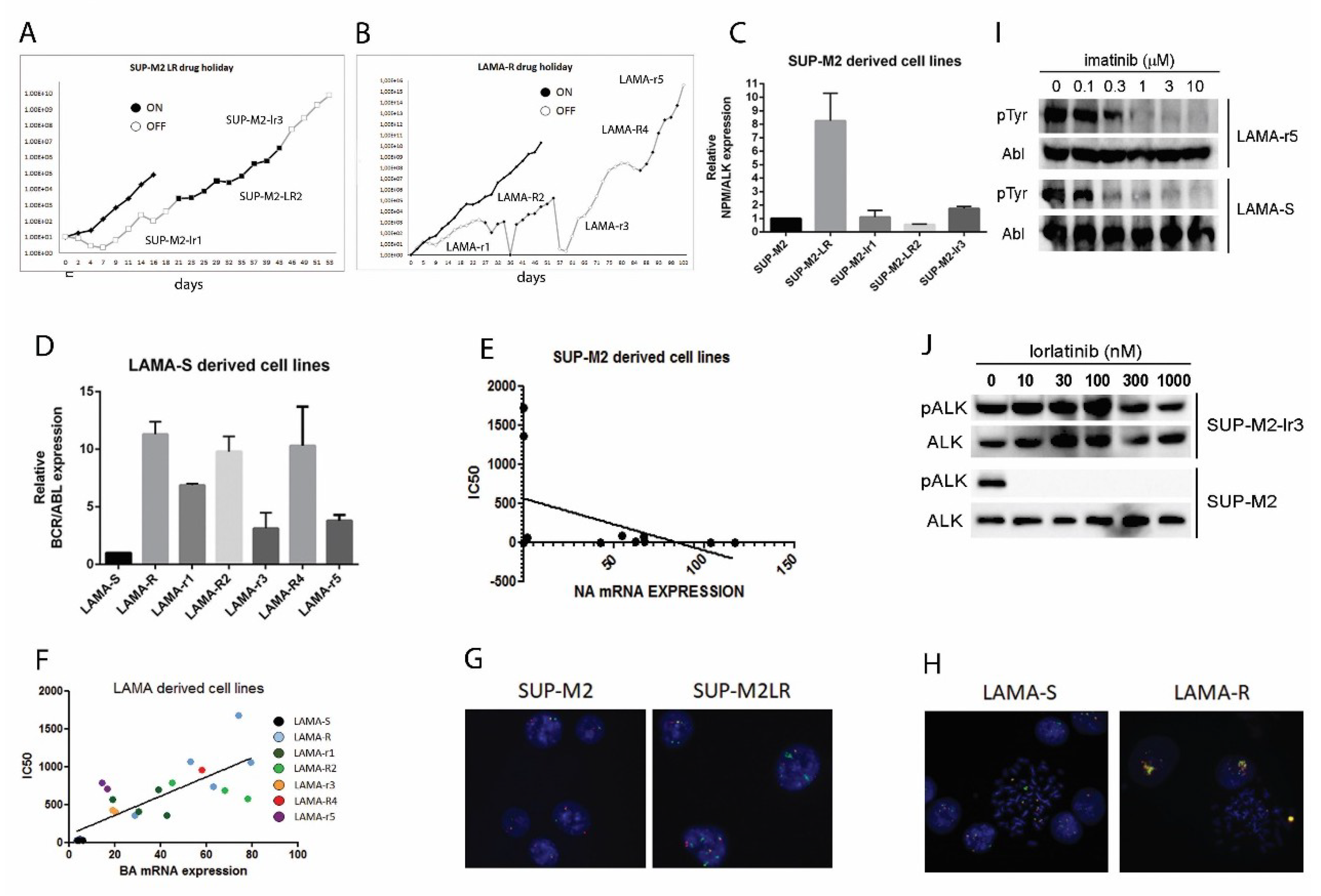
2.1. Drug Holiday Schedule Delays the Growth of Drug Addicted Cell Lines
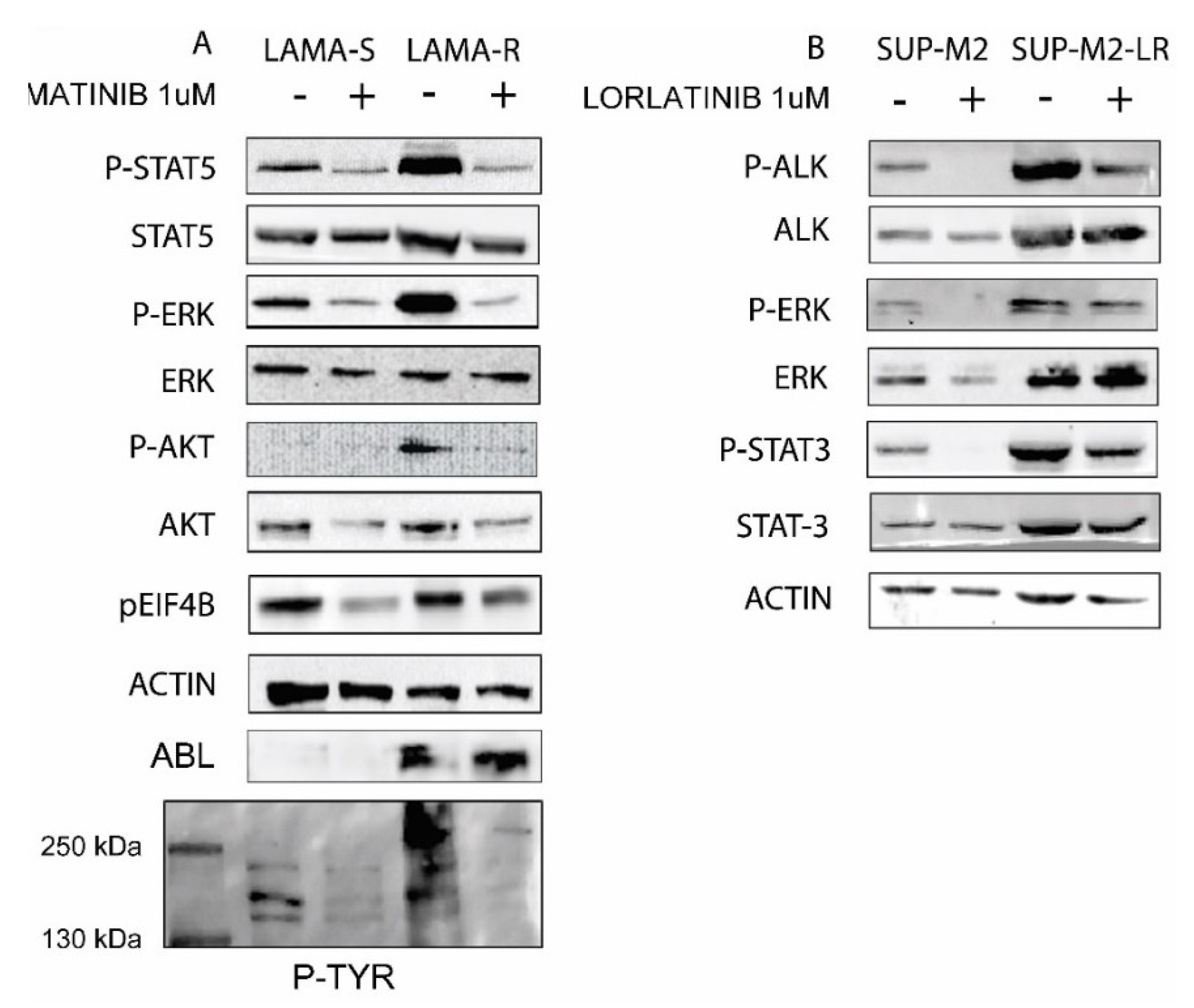
2.2. Drug Withdrawal in Drug Addicted Cell Lines Enhances Oncogene Driven Signaling
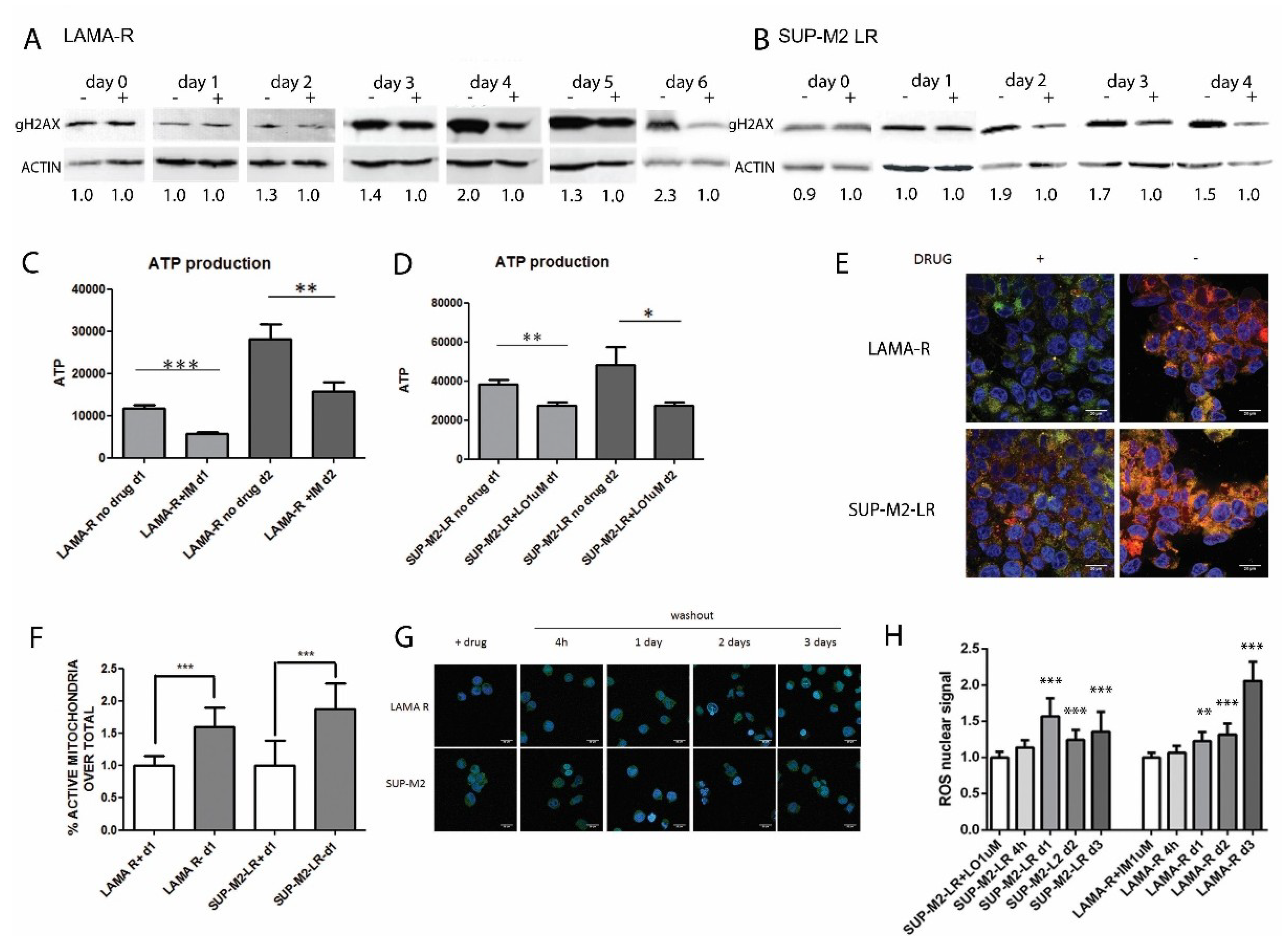
2.3. Enhanced Oncogenic Signaling Increases Mitochondria Activity and ROS Production, Inducing Genotoxic Stress
2.4. Enhanced Oncogenic Signaling Causes Cell Death
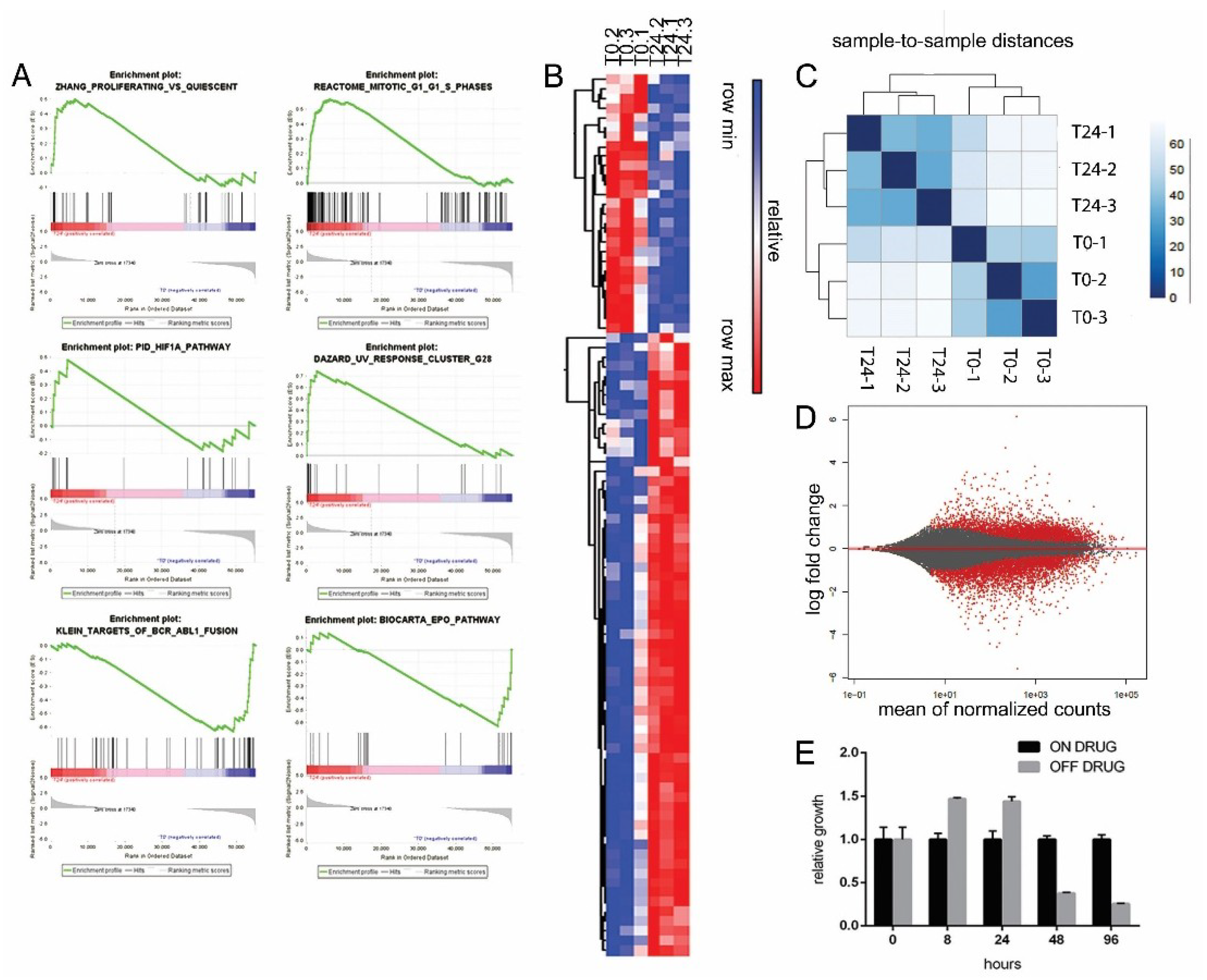
2.5. Excess of ProliferAtive Stimuli and ROS Production Induces Stress Response Activation and Oncogene Effectors Downregulation
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Drugs and RNA
4.2. Q-RT PCR and Sequencing
4.3. Western Blot
4.4. ATP Detection, Apoptosis, MTS Assay and Cell Proliferation Assay
4.5. Fluorescence Imaging and FISH
4.6. Software, Statistical Analysis and Bioinformatic
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Lavallade, H.; Apperley, J.F.; Khorashad, J.S.; Milojkovic, D.; Reid, A.G.; Bua, M.; Szydlo, R.; Olavarria, E.; Kaeda, J.; Goldman, J.M.; et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: Incidence of sustained responses in an intention-to-treat analysis. J. Clin. Oncol. 2008, 26, 3358–3363. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Tamura, T.; Kiura, K.; Seto, T.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yoshioka, H.; Harada, M.; Ohe, Y.; et al. Three-Year Follow-Up of an Alectinib Phase I/II Study in ALK-Positive Non-Small-Cell Lung Cancer: AF-001JP. J. Clin. Oncol. 2017, 35, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Tipping, A.J.; Mahon, F.X.; Lagarde, V.; Goldman, J.M.; Melo, J.V. Restoration of sensitivity to STI571 in STI571-resistant chronic myeloid leukemia cells. Blood 2001, 98, 3864–3867. [Google Scholar] [CrossRef]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–3865. [Google Scholar] [CrossRef]
- Amin, A.D.; Rajan, S.S.; Liang, W.S.; Pongtornpipat, P.; Groysman, M.J.; Tapia, E.O.; Peters, T.L.; Cuyugan, L.; Adkins, J.; Rimsza, L.M.; et al. Evidence suggesting that discontinuous dosing of ALK kinase inhibitors may prolong control of ALK+ tumors. Cancer Res. 2015, 75, 2916–2927. [Google Scholar] [CrossRef]
- Funakoshi, Y.; Mukohara, T.; Tomioka, H.; Ekyalongo, R.C.; Kataoka, Y.; Inui, Y.; Kawamori, Y.; Toyoda, M.; Kiyota, N.; Fujiwara, Y.; et al. Excessive MET signaling causes acquired resistance and addiction to MET inhibitors in the MKN45 gastric cancer cell line. Investig. New Drugs 2013, 31, 1158–1168. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Tomizawa, K.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Conversion from the “oncogene addiction” to “drug addiction” by intensive inhibition of the EGFR and MET in lung cancer with activating EGFR mutation. Lung Cancer 2012, 76, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Koop, A.; Satzger, I.; Alter, M.; Kapp, A.; Hauschild, A.; Gutzmer, R. Intermittent BRAF-inhibitor therapy is a feasible option: Report of a patient with metastatic melanoma. Br. J. Dermatol. 2014, 170, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Van Obberghen-Schilling, E.; Chambard, J.C. Prolonged activation of ERK1,2 induces FADD-independent caspase 8 activation and cell death. Apoptosis 2006, 11, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Geukes Foppen, M.H.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sic. USA 2009, 106, 22229–22232. [Google Scholar]
- Yang, R.; Rincon, M. Mitochondrial Stat3, the need for design thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment Efficacy and Resistance Mechanisms Using the Second-Generation ALK Inhibitor AP26113 in Human NPM-ALK-Positive Anaplastic Large Cell Lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef]
- Mologni, L.; Ceccon, M.; Pirola, A.; Chiriano, G.; Piazza, R.; Scapozza, L.; Gambacorti-Passerini, C. NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative ALK inhibitors but succumb to the novel compound PF-06463922. Oncotarget 2015, 6, 5720–5734. [Google Scholar] [CrossRef] [PubMed]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Mologni, L.; Rostagno, R.; Piazza, R.; Magistroni, V.; Ceccon, M.; Viltadi, M.; Flynn, D.; Gambacorti-Passerini, C. Three novel patient-derived BCR/ABL mutants show different sensitivity to second and third generation tyrosine kinase inhibitors. Am. J. Hematol. 2012, 87, E125–E128. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Treiber, N.; Huber, M.A.; Schneider, L.A.; Scharffetter-Kochanek, K.; Schultz, E.; Debus, D. Intermittent vemurafenib therapy in malignant melanoma. J. Dtsch Dermatol. Ges. 2017, 15, 451–454. [Google Scholar] [CrossRef]
- Hehlmann, R. How I treat CML blast crisis. Blood 2012, 120, 737–747. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef]
- Zimmermann, J.; Buchdunger, E.; Mett, H.; Meyer, T.; Lydon, N.B. Potent and selective inhibitors of the Abl-kinase: Phenylamino-pyrimidine (PAP) derivatives. Bioorg. Med. Chem. Lett. 1997, 7, 187–192. [Google Scholar] [CrossRef]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 132. [Google Scholar] [CrossRef]
- Mologni, L.; Costanza, M.; Sharma, G.G.; Viltadi, M.; Massimino, L.; Citterio, S.; Purgante, S.; Raman, H.; Pirola, A.; Zucchetti, M.; et al. Concomitant BCORL1 and BRAF Mutations in Vemurafenib-Resistant Melanoma Cells. Neoplasia 2018, 20, 467–477. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Drug | IC50 [nM] | RR Index | Relative Transcript Level |
|---|---|---|---|---|
| LAMA-S | IMATINIB | 22 | 1 | 1 |
| LAMA-R | IMATINIB | 773 | 35 | 11 |
| LAMA-r1 | IMATINIB | 490 | 22 | 7 |
| LAMA-R2 | IMATINIB | 714 | 32 | 10 |
| LAMA-r3 | IMATINIB | 413 | 19 | 4 |
| LAMA-R4 | IMATINIB | 953 | 43 | 10 |
| LAMA-r5 | IMATINIB | 744 | 34 | 4 |
| SUP-M2 | LORLATINIB | 2.13 | 1 | 1 |
| SUP-M2LR | LORLATINIB | >3000 | >1400 | 8 |
| SUP-M2lr1 | LORLATINIB | 67 | 3 | 1 |
| SUP-M2LR2 | LORLATINIB | 3114 | 141 | 1 |
| SUP-M2lr3 | LORLATINIB | 1530 | 69 | 2 |
| Cell Line | Mutations | ||
|---|---|---|---|
| SUP-M2 | - | - | - |
| SUP-M2 LR | C1156F | L1198F | - |
| SUP-M2 lr1 | - | - | - |
| SUP-M2 LR2 | C1156F | L1198F | D1203N |
| SUP-M2 lr3 | C1156F | L1198F | D1203N |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccon, M.; Mauri, M.; Massimino, L.; Giudici, G.; Piazza, R.; Gambacorti-Passerini, C.; Mologni, L. Mitochondrial Hyperactivation and Enhanced ROS Production are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling. Cancers 2018, 10, 509. https://doi.org/10.3390/cancers10120509
Ceccon M, Mauri M, Massimino L, Giudici G, Piazza R, Gambacorti-Passerini C, Mologni L. Mitochondrial Hyperactivation and Enhanced ROS Production are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling. Cancers. 2018; 10(12):509. https://doi.org/10.3390/cancers10120509
Chicago/Turabian StyleCeccon, Monica, Mario Mauri, Luca Massimino, Giovanni Giudici, Rocco Piazza, Carlo Gambacorti-Passerini, and Luca Mologni. 2018. "Mitochondrial Hyperactivation and Enhanced ROS Production are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling" Cancers 10, no. 12: 509. https://doi.org/10.3390/cancers10120509
APA StyleCeccon, M., Mauri, M., Massimino, L., Giudici, G., Piazza, R., Gambacorti-Passerini, C., & Mologni, L. (2018). Mitochondrial Hyperactivation and Enhanced ROS Production are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling. Cancers, 10(12), 509. https://doi.org/10.3390/cancers10120509