Tumor-Targeted Immunotherapy by Using Primary Adipose-Derived Stem Cells and an Antigen-Specific Protein Vaccine

 and
and 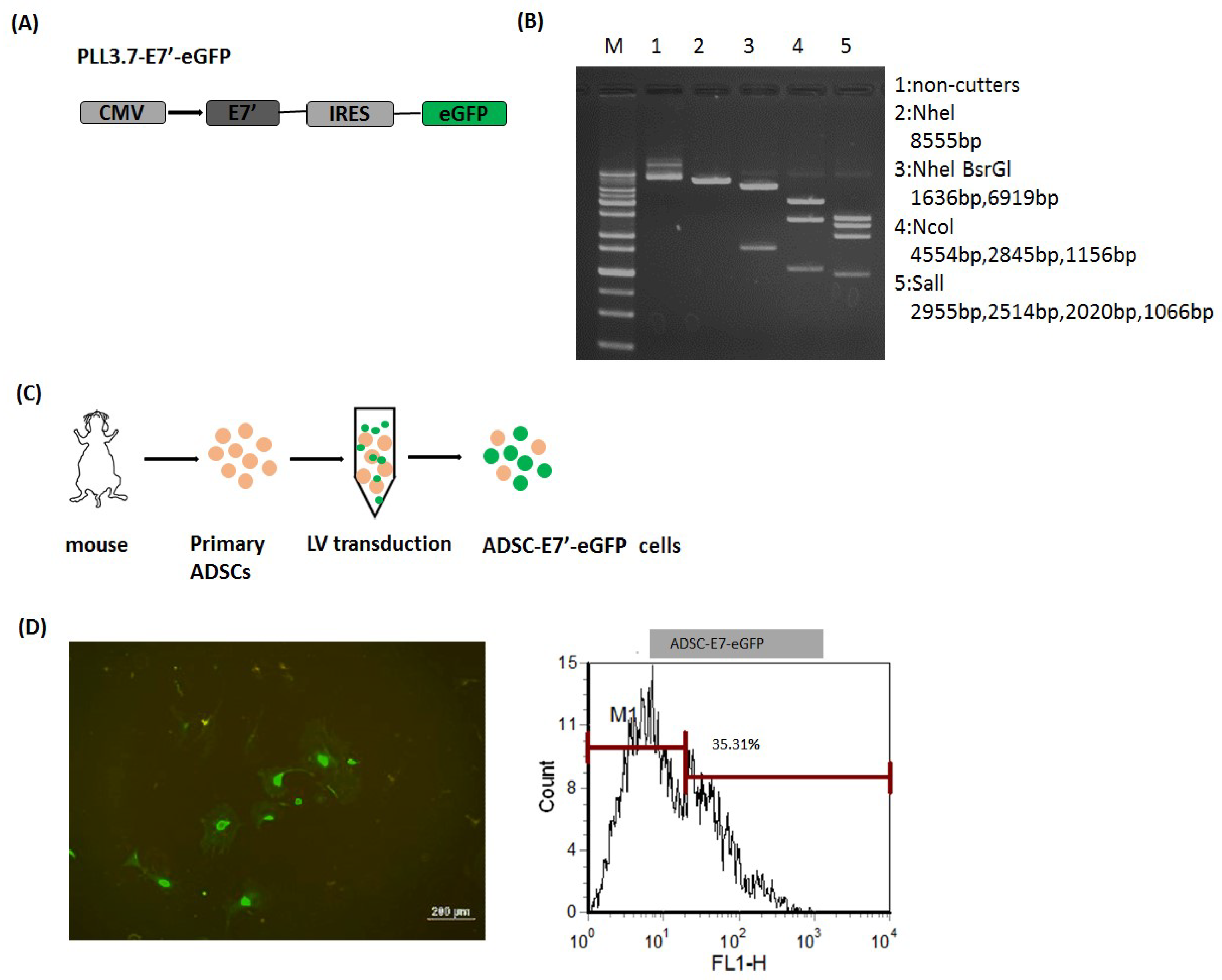{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Modified-E7 (E7’) Transduction of Primary ADSCs with a Lentiviral Vector
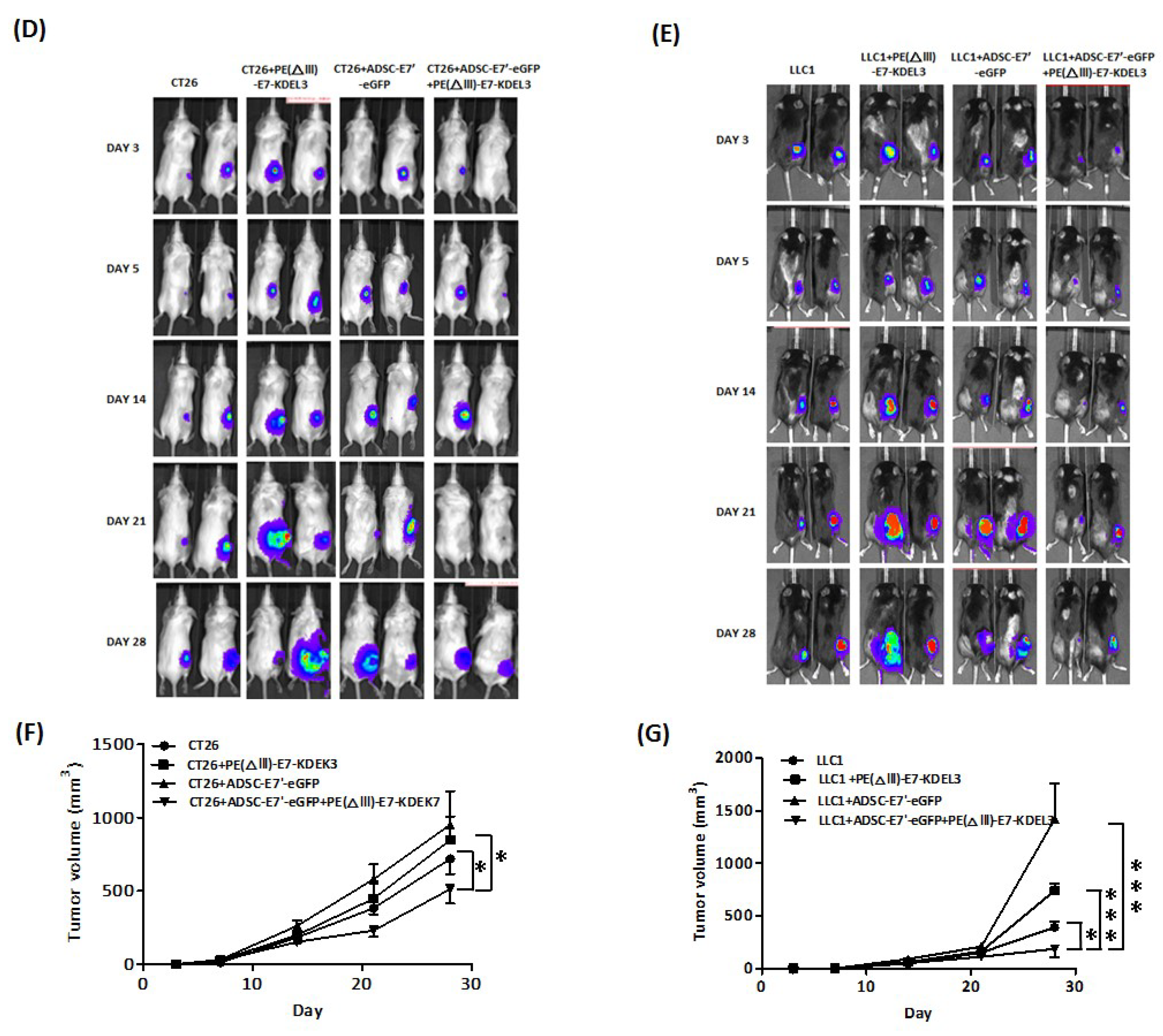
2.2. ADSC-E7’-eGFP–PE(ΔIII)-E7-KDEL3 Combined Treatment Inhibits the Tumor Growth of Colon and Lung Cancer Cells in Mice
2.3. Systemic Administration of ADSC-E7’-eGFP in Combination with PE(ΔIII)-E7-KDEL3 Reduces the Growth of Colon and Lung Cancer Cell Induced Tumors
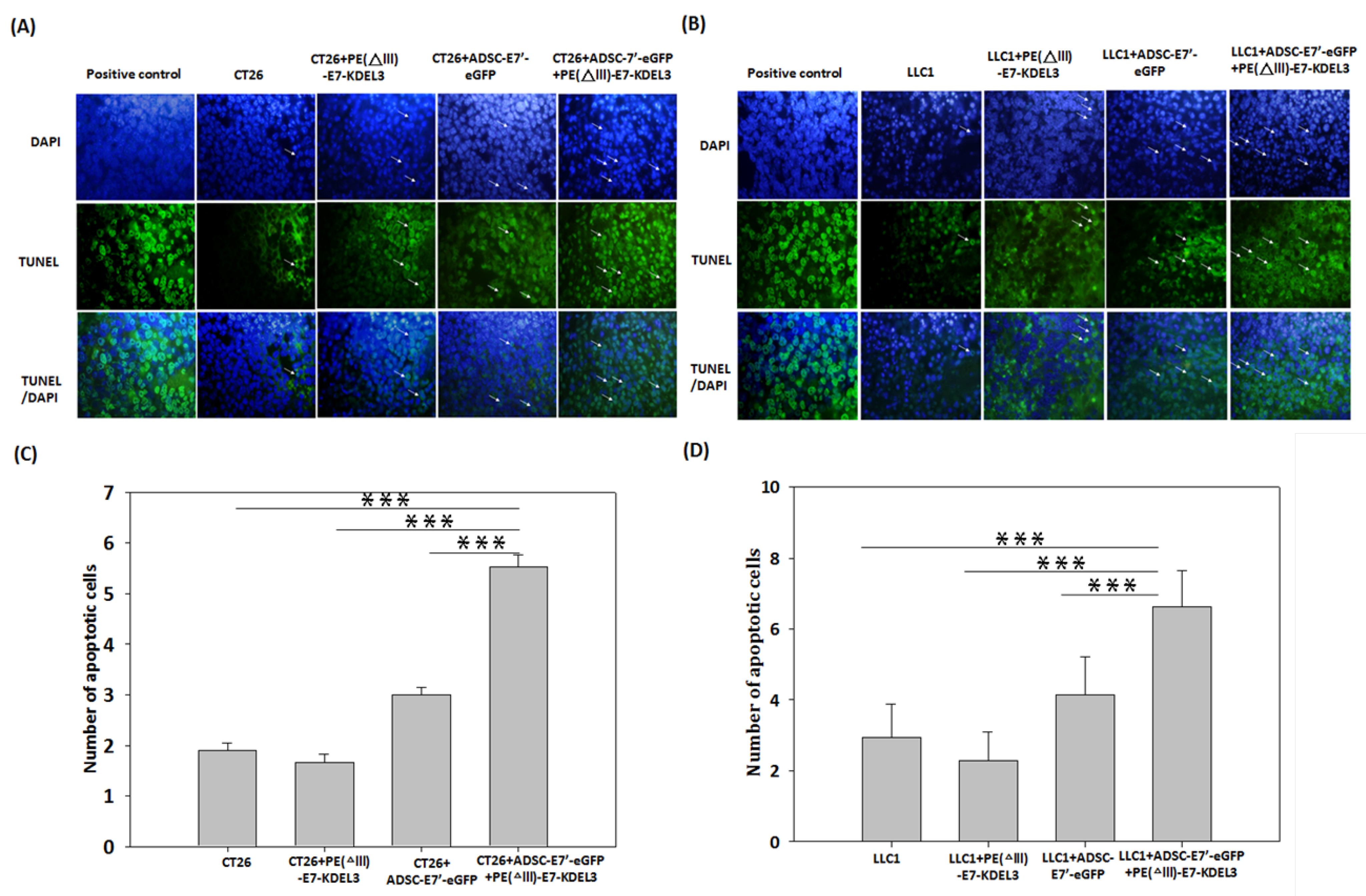
2.4. Systemic Administration of ADSC-E7’-eGFP in Combination with PE(ΔIII)-E7-KDEL3 Enhances Apoptosis in Tumors
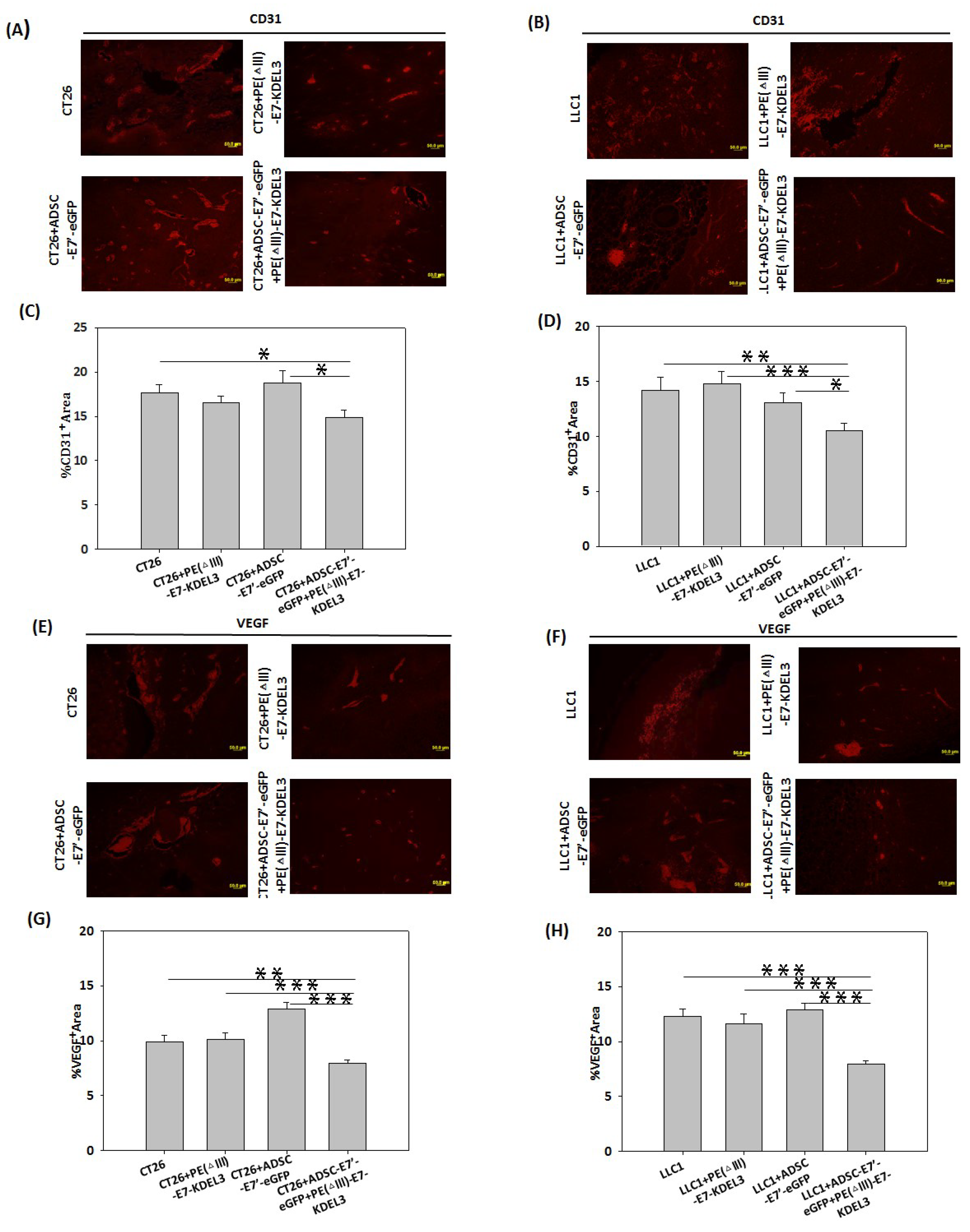
2.5. Inhibition of Tumor Angiogenesis by the ADSC-E7’-eGFP–PE (ΔIII)-E7-KDEL3 Combined Treatment
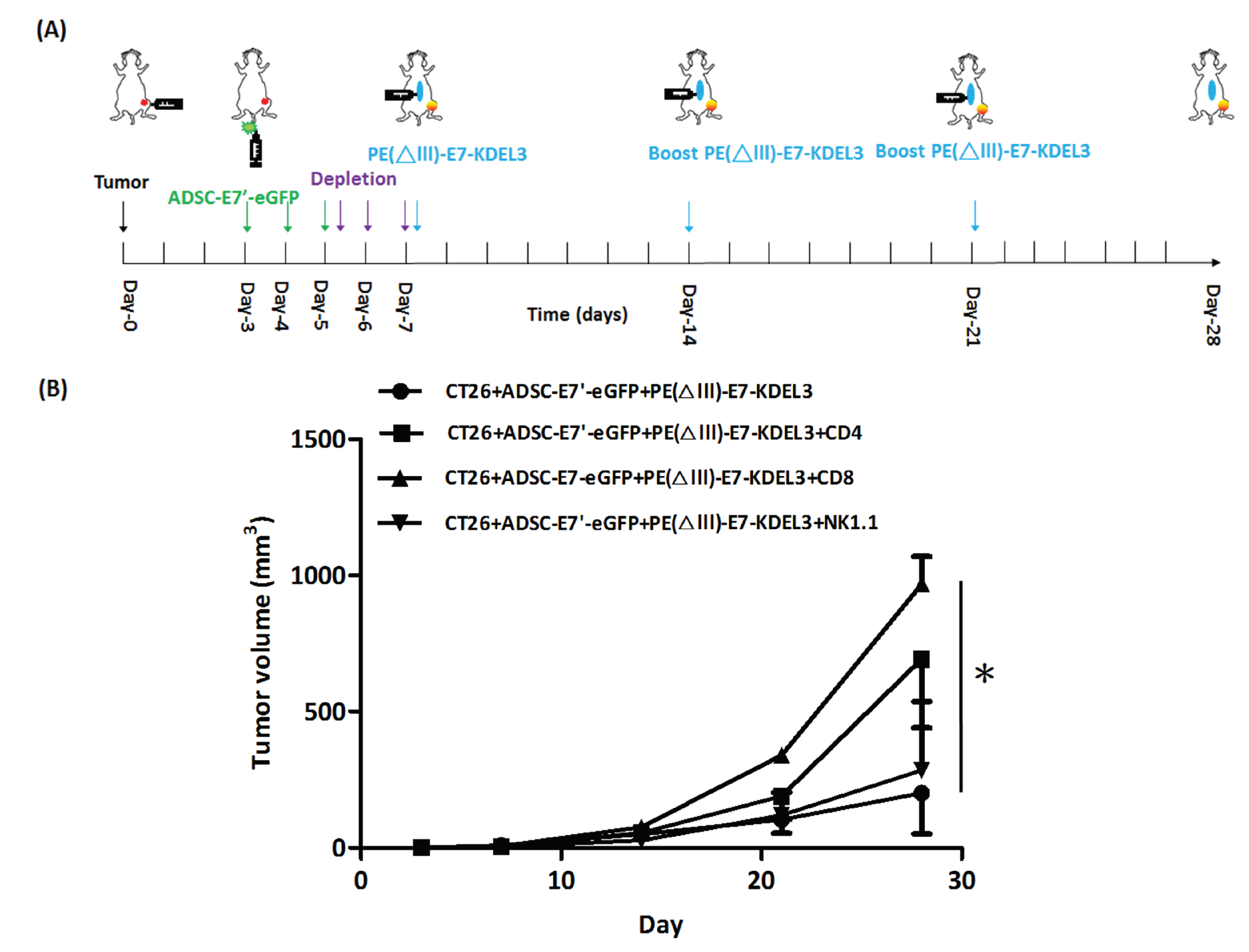
2.6. ADSC-E7’-eGFP–PE(ΔIII)-E7-KDEL3 Combined Treatment Contributes to Tumor Regression by Immune System Activation
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Isolation, Culture, and Lentiviral Transduction of ADSCs
4.3. Preparation and Vaccination of Protein Vaccines
4.4. Animal Studies
4.5. Bioluminescence Imaging (BLI)
4.6. Histology
4.7. TUNEL Assay
4.8. In Vivo Antibody Depletion
4.9. Statistical Analysis and Replicates
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ward, U. Biological therapy in the treatment of cancer. Br. J. Nurs. 1995, 4, 869–872, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Deng, W.P.; Yang, W.K.; Liu, R.S.; Lee, C.C.; Su, T.C.; Lin, R.J.; Yang, D.M.; Chang, C.W.; Chen, W.H.; et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin. Cancer Res. 2005, 11, 7749–7756. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Komarova, S.; Kawakami, Y.; Stoff-Khalili, M.A.; Curiel, D.T.; Pereboeva, L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol. Cancer Ther. 2006, 5, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Studeny, M.; Marini, F.C.; Champlin, R.E.; Zompetta, C.; Fidler, I.J.; Andreeff, M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002, 62, 3603–3608. [Google Scholar] [PubMed]
- Kucerova, L.; Matuskova, M.; Pastorakova, A.; Tyciakova, S.; Jakubikova, J.; Bohovic, R.; Altanerova, V.; Altaner, C. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J. Gene Med. 2008, 10, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Kobune, M.; Nakamura, K.; Kawano, Y.; Kato, K.; Honmou, O.; Houkin, K.; Matsunaga, T.; Niitsu, Y. Mesenchymal stem cells (MSC) as therapeutic cytoreagents for gene therapy. Cancer Sci. 2005, 96, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Nakamura, K.; Tamiya, T.; Kawano, Y.; Ishii, K.; Kobune, M.; Hirai, S.; Uchida, H.; Sasaki, K.; Ito, Y.; et al. Mesenchymal stem cells that produce neurotrophic factors reduce ischemic damage in the rat middle cerebral artery occlusion model. Mol. Ther. 2005, 11, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Wu, A.T.; Hsu, C.H.; Lin, Y.P.; Cheng, W.F.; Su, C.H.; Chiu, W.T.; Whang-Peng, J.; Douglas, F.L.; Deng, W.P. The development of a novel cancer immunotherapeutic platform using tumor-targeting mesenchymal stem cells and a protein vaccine. Mol. Ther. 2011, 19, 2249–2257. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Yang, D.M.; Chang, C.F.; Lin, R.J.; Wang, J.S.; Low-Tone Ho, L.; Yang, W.K. Immortalization without neoplastic transformation of human mesenchymal stem cells by transduction with HPV16 E6/E7 genes. Int. J. Cancer 2004, 110, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Frese, L.; Dijkman, P.E.; Hoerstrup, S.P. Adipose tissue-derived stem cells in regenerative medicine. Transfus. Med. Hemother. 2016, 43, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.W.; Chen, C.A.; Lee, C.N.; Su, Y.N.; Chang, M.C.; Syu, M.H.; Hsieh, C.Y.; Cheng, W.F. Fusion protein vaccine by domains of bacterial exotoxin linked with a tumor antigen generates potent immunologic responses and antitumor effects. Cancer Res. 2005, 65, 9089–9098. [Google Scholar] [CrossRef] [PubMed]
- Mundra, V.; Gerling, I.C.; Mahato, R.I. Mesenchymal stem cell-based therapy. Mol. Pharm 2013, 10, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Shah, K. Mesenchymal stem cells engineered for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell. Tissue Kinet. 1970, 3, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.R.; West, C.C.; Hardy, W.R.; James, A.W.; Park, T.S.; Nguyen, A.; Tawonsawatruk, T.; Lazzari, L.; Soo, C.; Peault, B. Natural history of mesenchymal stem cells, from vessel walls to culture vessels. Cell. Mol. Life Sci. 2014, 71, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Hamabe, A.; Hasegawa, S.; Ogawa, H.; Fukusumi, T.; Nishikawa, S.; Ohta, K.; Kano, Y.; Ozaki, M.; Noguchi, Y.; et al. Adipose-derived mesenchymal stem cells and regenerative medicine. Dev. Growth Differ. 2013, 55, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lee, H.; Seo, K.; Kang, S.; Ra, J.; Youn, H. Anti-tumor effect of adipose tissue derived-mesenchymal stem cells expressing interferon-beta and treatment with cisplatin in a xenograft mouse model for canine melanoma. PLoS ONE 2013, 8, e74897. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Fitch, S.; Wang, C.; Wilson, C.; Li, J.; Grant, G.A.; Yang, F. Nanoparticle engineered trail-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proc. Natl. Acad. Sci. USA 2016, 113, 13857–13862. [Google Scholar] [CrossRef] [PubMed]
- Guiho, R.; Biteau, K.; Grisendi, G.; Taurelle, J.; Chatelais, M.; Gantier, M.; Heymann, D.; Dominici, M.; Redini, F. Trail delivered by mesenchymal stromal/stem cells counteracts tumor development in orthotopic ewing sarcoma models. Int. J. Cancer 2016, 139, 2802–2811. [Google Scholar] [CrossRef] [PubMed]
- Grisendi, G.; Bussolari, R.; Cafarelli, L.; Petak, I.; Rasini, V.; Veronesi, E.; De Santis, G.; Spano, C.; Tagliazzucchi, M.; Barti-Juhasz, H.; et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010, 70, 3718–3729. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.X.; Duan, D.J.; Zhou, H.; Hu, Q.M.; Lei, T.C. Adipose-derived mesenchymal stem cell-facilitated trail expression in melanoma treatment in vitro. Mol. Med. Rep. 2016, 14, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Salehi, H.; Oskuee, R.K.; Mohammadpour, A.; Mirzaei, H.R.; Sharifi, M.R.; Salarinia, R.; Darani, H.Y.; Mokhtari, M.; Masoudifar, A.; et al. The therapeutic potential of human adipose-derived mesenchymal stem cells producing CXCL10 in a mouse melanoma lung metastasis model. Cancer Lett. 2018, 419, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, P.; Liu, X.; Lv, P. Expression of interleukin-12 by adipose-derived mesenchymal stem cells for treatment of lung adenocarcinoma. Thorac. Cancer 2015, 6, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zolochevska, O.; Shearer, J.; Ellis, J.; Fokina, V.; Shah, F.; Gimble, J.M.; Figueiredo, M.L. Human adipose-derived mesenchymal stromal cell pigment epithelium-derived factor cytotherapy modifies genetic and epigenetic profiles of prostate cancer cells. Cytotherapy 2014, 16, 346–356. [Google Scholar] [CrossRef] [PubMed]
- de Melo, S.M.; Bittencourt, S.; Ferrazoli, E.G.; da Silva, C.S.; da Cunha, F.F.; da Silva, F.H.; Stilhano, R.S.; Denapoli, P.M.; Zanetti, B.F.; Martin, P.K.; et al. The anti-tumor effects of adipose tissue mesenchymal stem cell transduced with HSV-Tk gene on U-87-driven brain tumor. PLoS ONE 2015, 10, e0128922. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar] [CrossRef] [PubMed]
- Cavarretta, I.T.; Altanerova, V.; Matuskova, M.; Kucerova, L.; Culig, Z.; Altaner, C. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol. Ther. 2010, 18, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Lee, J.Y.; Wang, K.C.; Phi, J.H.; Song, S.H.; Song, G.; Kim, S.K. Human adipose tissue-derived mesenchymal stem cells: Characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur. J. Cancer 2012, 48, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Gonzales, M.I.; Parkhurst, M.; Li, Y.F.; Southwood, S.; Sette, A.; Rosenberg, S.A.; Robbins, P.F. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR-restricted tyrosinase epitopes. J. Exp. Med. 1996, 183, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Manici, S.; Sturniolo, T.; Imro, M.A.; Hammer, J.; Sinigaglia, F.; Noppen, C.; Spagnoli, G.; Mazzi, B.; Bellone, M.; Dellabona, P.; et al. Melanoma cells present a mage-3 epitope to CD4(+) cytotoxic T cells in association with histocompatibility leukocyte antigen DR11. J. Exp. Med. 1999, 189, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Pieper, R.; Christian, R.E.; Gonzales, M.I.; Nishimura, M.I.; Gupta, G.; Settlage, R.E.; Shabanowitz, J.; Rosenberg, S.A.; Hunt, D.F.; Topalian, S.L. Biochemical identification of a mutated human melanoma antigen recognized by CD4(+) T cells. J. Exp. Med. 1999, 189, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F.; Wang, X.; Rosenberg, S.A. Identification of a novel major histocompatibility complex class ii-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4(+) T cells. J. Exp. Med. 1999, 189, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Boel, P.; Wildmann, C.; Sensi, M.L.; Brasseur, R.; Renauld, J.C.; Coulie, P.; Boon, T.; van der Bruggen, P. Bage: A new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995, 2, 167–175. [Google Scholar] [CrossRef]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Tang, K.C.; Patel, A.P.; Bonilla, L.M.; Pierobon, N.; Ponzio, N.M.; Rameshwar, P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006, 107, 4817–4824. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Pommey, S.; Eliopoulos, N.; Galipeau, J. Interferon-gamma-stimulated marrow stromal cells: A new type of nonhematopoietic antigen-presenting cell. Blood 2006, 107, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Romieu-Mourez, R.; Stock-Martineau, S.; Boivin, M.N.; Bramson, J.L.; Galipeau, J. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood 2009, 114, 2632–2638. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Zeng, R.; Lu, J.H.; Lai, W.F.; Chen, W.H.; Liu, H.Y.; Chang, Y.T.; Deng, W.P. Adipose-derived stem cells promote tumor initiation and accelerate tumor growth by interleukin-6 production. Oncotarget 2015, 6, 7713–7726. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.-H.; Peng, B.-Y.; Chang, C.-C.; Dubey, N.K.; Lo, W.-C.; Cheng, H.-C.; Wang, J.R.; Wei, H.-J.; Deng, W.-P. Tumor-Targeted Immunotherapy by Using Primary Adipose-Derived Stem Cells and an Antigen-Specific Protein Vaccine. Cancers 2018, 10, 446. https://doi.org/10.3390/cancers10110446
Lu J-H, Peng B-Y, Chang C-C, Dubey NK, Lo W-C, Cheng H-C, Wang JR, Wei H-J, Deng W-P. Tumor-Targeted Immunotherapy by Using Primary Adipose-Derived Stem Cells and an Antigen-Specific Protein Vaccine. Cancers. 2018; 10(11):446. https://doi.org/10.3390/cancers10110446
Chicago/Turabian StyleLu, Jui-Hua, Bou-Yue Peng, Chun-Chao Chang, Navneet Kumar Dubey, Wen-Cheng Lo, Hsin-Chung Cheng, Joseph R. Wang, Hong-Jian Wei, and Win-Ping Deng. 2018. "Tumor-Targeted Immunotherapy by Using Primary Adipose-Derived Stem Cells and an Antigen-Specific Protein Vaccine" Cancers 10, no. 11: 446. https://doi.org/10.3390/cancers10110446
APA StyleLu, J.-H., Peng, B.-Y., Chang, C.-C., Dubey, N. K., Lo, W.-C., Cheng, H.-C., Wang, J. R., Wei, H.-J., & Deng, W.-P. (2018). Tumor-Targeted Immunotherapy by Using Primary Adipose-Derived Stem Cells and an Antigen-Specific Protein Vaccine. Cancers, 10(11), 446. https://doi.org/10.3390/cancers10110446