Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review

 ,
, 
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy and Study Selection
2.2. Data Extraction
2.3. Data Analysis
3. Results
3.1. Localized Disease
3.2. Mixed Population (Locally Advanced and Metastatic Disease)
3.3. Metastatic Disease
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CYVADIC | cyclophosphamide, vincristine, doxorubicin, and dacarbazine |
| Dox + Ifo | doxorubicin + ifosfamide |
| FNCLCC | Federation Nationale des Centres de Lutte Contre le Cancer |
| ICD | International statistical Classification of Disease and related health problems |
| SLR | systematic literature review |
| SS | synovial sarcoma |
| STS | soft tissue sarcoma |
References
- National Comprehensive Cancer Network (NCCN). Soft Tissue Sarcoma (Version 1.2018). Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 15 February 2018).
- The ESMO/European Sarcoma Network Working Group. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii102–iii112. [Google Scholar] [CrossRef] [PubMed]
- De Vita, A.; Mercatali, L.; Recine, F.; Pieri, F.; Riva, N.; Bongiovanni, A.; Liverani, C.; Spadazzi, C.; Miserocchi, G.; Amadori, D.; et al. Current classification, treatment options, and new perspectives in the management of adipocytic sarcomas. OncoTargets Ther. 2016, 9, 6233–6246. [Google Scholar] [CrossRef] [PubMed]
- Eilber, F.C.; Brennan, M.F.; Eilber, F.R.; Eckardt, J.J.; Grobmyer, S.R.; Riedel, E.; Forscher, C.; Maki, R.G.; Singer, S. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann. Surg. 2007, 246, 105–113. [Google Scholar] [CrossRef] [PubMed]
- De Necochea-Campion, R.; Zuckerman, L.M.; Mirshahidi, H.R.; Khosrowpour, S.; Chen, C.-S.; Mirshahidi, S. Metastatic biomarkers in synovial sarcoma. Biomarker Res. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Poulin, N.M.; Ladanyi, M. Synovial sarcoma: Recent discoveries as a roadmap to new avenues for therapy. Cancer Discov. 2015, 5, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Sun, T.; Hou, B.; Hong, G.; Mallampati, S.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; et al. Survival changes in Patients with Synovial Sarcoma, 1983–2012. J. Cancer 2017, 8, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Kerouanton, A.; Jimenez, I.; Cellier, C.; Laurence, V.; Helfre, S.; Pannier, S.; Mary, P.; Freneaux, P.; Orbach, D. Synovial sarcoma in children and adolescents. J. Pediatr. Hematol. Oncol. 2014, 36, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Fisher, C. Synovial sarcoma: Defining features and diagnostic evolution. Ann. Diagn. Pathol. 2014, 18, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Van Tine, B.A. Synovial Sarcoma: Current Concepts and Future Perspectives. J. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.; Stiller, C.; Grimer, R.; Dennis, N.; Broggio, J.; Francis, M. Outcome and the effect of age and socioeconomic status in 1318 patients with synovial sarcoma in the English National Cancer Registry: 1985–2009. Clin. Sarcoma Res. 2016, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; de Salvo, G.L.; Brennan, B.; van Noesel, M.M.; De Paoli, A.; Casanova, M.; Francotte, N.; Kelsey, A.; Alaggio, R.; Oberlin, O.; et al. Synovial sarcoma in children and adolescents: The European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann. Oncol. 2015, 26, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene 2001, 20, 5755–5762. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Sambri, A.; Righi, A.; Dei Tos, A.P.; Picci, P.; Donati, D. Histology and grading are important prognostic factors in synovial sarcoma. Eur. J. Surg. Oncol. 2017, 43, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Shilatifard, A. Epigenetic ConFUSION: SS18-SSX Fusion Rewires BAF Complex to Activate Bivalent Genes in Synovial Sarcoma. Cancer Cell 2018, 33, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Nicholaou, T.; Ebert, L.; Davis, I.D.; Robson, N.; Klein, O.; Maraskovsky, E.; Chen, W.; Cebon, J. Directions in the immune targeting of cancer: Lessons learned from the cancer-testis Ag NY-ESO-1. Immunol. Cell Biol. 2006, 84, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, A.A.; Antonescu, C.R.; Busam, K.J.; Iversen, K.; Kolb, D.; Coplan, K.; Chen, Y.T.; Stockert, E.; Ladanyi, M.; Old, L.J. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int. J. Cancer 2001, 94, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Gnjatic, S.; Nishikawa, H.; Jungbluth, A.A.; Gure, A.O.; Ritter, G.; Jager, E.; Knuth, A.; Chen, Y.T.; Old, L.J. NY-ESO-1: Review of an immunogenic tumor antigen. Adv. Cancer Res. 2006, 95, 1–30. [Google Scholar] [PubMed]
- Komarov, Y.; Barchuk, A.; Semenova, A.I.; Semiglazova, T.; Imianitov, E.; Iyevleva, A.; Baldueva, I.; Novik, A.V.; Nehaeva, T.L.; Pipia, N.; et al. NY-ESO-1 antigen expression as a prognostic factor for soft tissue sarcomas. J. Clin. Oncol. 2017, 35, 11075. [Google Scholar] [CrossRef]
- Van Tine, B.A. Multiple Molecular Subtypes of Sarcoma Allow for Orphan Drug Development. J. Target. Ther. Cancer 2016. Available online: https://www.targetedonc.com/publications/targeted-therapies-cancer/2016/2016-october/multiple-molecular-subtypes-of-sarcoma-allow-for-orphan-drug-development?p=2 (accessed on 1 November 2018).
- Pasquali, S.; Gronchi, A. Neoadjuvant chemotherapy in soft tissue sarcomas: Latest evidence and clinical implications. Ther. Adv. Med. Oncol. 2017, 9, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.; Baldini, E.H.; Demetri, G.D.; Fletcher, J.A.; Corson, J.M. Synovial sarcoma: Prognostic significance of tumor size, margin of resection, and mitotic activity for survival. J. Clin. Oncol. 1996, 14, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Beaino, M.E.; Araujo, D.M.; Gopalakrishnan, V.; Lazar, A.J.; Lin, P.P. Prognosis of T1 synovial sarcoma depends upon surgery by oncologic surgeons. J.Surg. Oncol. 2016, 114, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Martin Broto, J.; Le Cesne, A.; Reichardt, P. The importance of treating by histological subtype in advanced soft tissue sarcoma. Future Oncol. 2017, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Spurrell, E.L.; Fisher, C.; Thomas, J.M.; Judson, I.R. Prognostic factors in advanced synovial sarcoma: An analysis of 104 patients treated at the Royal Marsden Hospital. Ann. Oncol. 2005, 16, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Systematic Reviews. CRD’s Guidance for Undertaking Reviews in Health Care; Centre for Reviews and Dissemination, University of York: York, UK, 2009. [Google Scholar]
- Hutton, B.; Salanti, G.; Caldwell, D.M.; Chaimani, A.; Schmid, C.H.; Cameron, C.; Ioannidis, J.P.A.; Straus, S.; Thorlund, K.; Jansen, J.P.; et al. The PRISMA Extension Statement for Reporting of Systematic Reviews Incorporating Network Meta-analyses of Health Care Interventions: Checklist and Explanations. Ann. Intern. Med. 2015, 162, 777. [Google Scholar] [CrossRef] [PubMed]
- Vlenterie, M.; Litiere, S.; Rizzo, E.; Marreaud, S.; Judson, I.; Gelderblom, H.; Le Cesne, A.; Wardelmann, E.; Messiou, C.; Gronchi, A.; et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; Setting a new landmark for studies in this entity. Eur. J. Cancer 2016, 58, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Benhattar, J.; Bonichon, F.; Gallagher, G.; Terrier, P.; Stauffer, E.; De Saint Aubain Somerhausen, N.; Michels, J.J.; Jundt, G.; Vince, D.R.; et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: A multicenter, retrospective analysis. J. Clin. Oncol. 2004, 22, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Ueda, T.; Naka, N.; Araki, N.; Hashimoto, N.; Myoui, A.; Ozaki, T.; Nakayama, T.; Toguchida, J.; Tanaka, K.; et al. Prognostic implication of SYT-SSX fusion type in synovial sarcoma: A multi-institutional retrospective analysis in Japan. Oncol. Rep. 2008, 19, 467–476. [Google Scholar] [CrossRef] [PubMed]
- De Silva, M.V.C.; McMahon, A.D.; Reid, R. Prognostic Factors Associated with Local Recurrence, Metastases, and Tumor-Related Death in Patients with Synovial Sarcoma. Am. J. Clin. Oncol. 2004, 27, 113–121. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/classifications/icd/icdonlineversions/en/ (accessed on 19 April 2018).
- Vining, C.C.; Sinnamon, A.J.; Ecker, B.L.; Kelz, R.R.; Fraker, D.L.; Roses, R.E.; Karakousis, G.C. Adjuvant chemotherapy in resectable synovial sarcoma. J. Surg. Oncol. 2017, 116, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, H.; Hogg, D.; Blackstein, M.E.; O’Sullivan, B.; Catton, C.N.; Chung, P.W.; Griffin, A.M.; Hodgson, D.; Hopyan, S.; Kandel, R.; et al. Clinical features, treatment, and outcome in 102 adult and pediatric patients with localized high-grade synovial sarcoma. Sarcoma 2011, 2011, 231789. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Penel, N.; Robin, Y.M.; Bui, B.; Le Cesne, A.; Piperno-Neumann, S.; Tubiana-Hulin, M.; Bompas, E.; Chevreau, C.; Isambert, N.; et al. Neo/adjuvant chemotherapy does not improve outcome in resected primary synovial sarcoma: A study of the French Sarcoma Group. Ann. Oncol. 2009, 20, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Orbach, D.; McDowell, H.; Rey, A.; Bouvet, N.; Kelsey, A.; Stevens, M.C. Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: Experience of the International Society of pediatric oncology, malignant mesenchymal tumors (SIOP-MMT) Working Group. Pediatr. Blood Cancer 2011, 57, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Dantonello, T.; Hallmen, E.; Blank, B.; Sparber-Sauer, M.; Vokuhl, C.; Leuschner, I.; Munter, M.W.; von Kalle, T.; Bielack, S.S.; et al. Synovial Sarcoma Recurrence in Children and Young Adults. Ann. Surg. Oncol. 2016, 23, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.H.; Hefti, F.; Speth, B.M.; Jundt, G.; Guillou, L.; Exner, U.G.; von Hochstetter, A.R.; Cserhati, M.D.; Fuchs, B.; Mouhsine, E.; et al. Synovial sarcomas usually metastasize after >5 years: A multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Ann. Oncol. 2011, 22, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Indelicato, D.J.; Morris, C.G.; Scarborough, M.T.; Gibbs, C.P.; Zlotecki, R.A. Long-term treatment outcomes for patients with synovial sarcoma: A 40-year experience at the university of Florida. Am. J. Clin. Oncol. 2013, 36, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Trassard, M.; Le Doussal, V.; Hacene, K.; Terrier, P.; Ranchere, D.; Guillou, L.; Fiche, M.; Collin, F.; Vilain, M.O.; Bertrand, G.; et al. Prognostic factors in localized primary synovial sarcoma: A multicenter study of 128 adult patients. J. Clin. Oncol. 2001, 19, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Brecht, I.B.; Ferrari, A.; Int-Veen, C.; Schuck, A.; Mattke, A.C.; Casanova, M.; Bisogno, G.; Carli, M.; Koscielniak, E.; Treuner, J. Grossly-resected synovial sarcoma treated by the German and Italian pediatric soft tissue sarcoma cooperative groups: Discussion on the role of adjuvant therapies. Pediatr. Blood Cancer 2006, 46, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.J.; Qin, L.X.; Maki, R.G.; Brennan, M.F.; Ladanyi, M.; Singer, S. A synovial sarcoma-specific preoperative nomogram supports a survival benefit to ifosfamide-based chemotherapy and improves risk stratification for patients. Clin. Cancer Res. 2008, 14, 8191–8197. [Google Scholar] [CrossRef] [PubMed]
- Vlenterie, M.; Ho, V.K.Y.; Kaal, S.E.J.; Vlenterie, R.; Haas, R.; Van Der Graaf, W.T.A. Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br. J. Cancer 2015, 113, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Setsu, N.; Kohashi, K.; Fushimi, F.; Endo, M.; Yamamoto, H.; Takahashi, Y.; Yamada, Y.; Ishii, T.; Yokoyama, K.; Iwamoto, Y.; et al. Prognostic impact of the activation status of the Akt/mTOR pathway in synovial sarcoma. Cancer 2013, 119, 3504–3513. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.; Mankin, H.J.; Singer, S. Synovial Sarcoma: The Importance of Size and Location for Survival. Clin. Orthop. Relat. Res. 2004, 419, 155–161. [Google Scholar] [CrossRef]
- Palmerini, E.; Staals, E.L.; Alberghini, M.; Zanella, L.; Ferrari, C.; Benassi, M.S.; Picci, P.; Mercuri, M.; Bacci, G.; Ferrari, S. Synovial sarcoma: Retrospective analysis of 250 patients treated at a single institution. Cancer 2009, 115, 2988–2998. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Gronchi, A.; Casanova, M.; Meazza, C.; Gandola, L.; Collini, P.; Lozza, L.; Bertulli, R.; Olmi, P.; Casali, P.G. Synovial sarcoma: A retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 2004, 101, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Corey, R.M.; Swett, K.; Ward, W.G. Epidemiology and survivorship of soft tissue sarcomas in adults: A national cancer database report. Cancer Med. 2014, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, R.; Dileo, P.; Blay, J.Y.; Constantinidou, A.; Le Cesne, A.; Benson, C.; Vizzini, L.; Contu, M.; Baldi, G.G.; Dei Tos, A.P.; et al. Trabectedin in advanced synovial sarcomas: A multicenter retrospective study from four European institutions and the Italian Rare Cancer Network. Anticancer Drugs 2015, 26, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Savina, M.; Le Cesne, A.; Blay, J.Y.; Ray-Coquard, I.; Mir, O.; Toulmonde, M.; Cousin, S.; Terrier, P.; Ranchere-Vince, D.; Meeus, P.; et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: The METASARC observational study. BMC Med. 2017, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Gronchi, A.; Ferrari, S.; Quagliuolo, V.; Broto, J.M.; Pousa, A.L.; Grignani, G.; Basso, U.; Blay, J.Y.; Tendero, O.; Beveridge, R.D.; et al. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): An international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol. 2017, 18, 812–822. [Google Scholar] [CrossRef]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; PharmD, G.D.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer 2014, 120, 2448–2456. [Google Scholar] [PubMed]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schoffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; De Brauwer, A.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar] [PubMed]
- Pappo, A.S.; Devidas, M.; Jenkins, J.; Rao, B.; Marcus, R.; Thomas, P.; Gebhardt, M.; Pratt, C.; Grier, H.E. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J. Clin. Oncol. 2005, 23, 4031–4038. [Google Scholar] [CrossRef] [PubMed]
- Chugh, R.; Wathen, J.K.; Maki, R.G.; Benjamin, R.S.; Patel, S.R.; Meyers, P.A.; Priebat, D.A.; Reinke, D.K.; Thomas, D.G.; Keohan, M.L.; et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. 2009, 27, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Araki, N.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: A randomised, open-label, phase 2 study. Lancet Oncol. 2015, 16, 406–416. [Google Scholar] [CrossRef]
- Mir, O.; Brodowicz, T.; Italiano, A.; Wallet, J.; Blay, J.Y.; Bertucci, F.; Chevreau, C.; Piperno-Neumann, S.; Bompas, E.; Salas, S.; et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1732–1742. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Le Cesne, A.; Whelan, J.S.; Schoffski, P.; Bui, B.N.; Verweij, J.; Marreaud, S.; Van Glabbeke, M.; Hogendoorn, P.; Blay, J.Y. A phase II study of gefitinib for patients with advanced HER-1 expressing synovial sarcoma refractory to doxorubicin-containing regimens. Oncologist 2008, 13, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Ray-Coquard, I.L.; Cioffi, A.; Bui, N.B.; Bauer, S.; Hartmann, J.T.; Krarup-Hansen, A.; Grünwald, V.; Sciot, R.; Dumez, H.; et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol. 2011, 12, 1045–1052. [Google Scholar] [CrossRef]
- Schoffski, P.; Adkins, D.; Blay, J.Y.; Gil, T.; Elias, A.D.; Rutkowski, P.; Pennock, G.K.; Youssoufian, H.; Gelderblom, H.; Willey, R.; et al. An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur. J. Cancer 2013, 49, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Grosso, F.; Jones, R.L.; Demetri, G.D.; Judson, I.R.; Blay, J.Y.; Le Cesne, A.; Sanfilippo, R.; Casieri, P.; Collini, P.; Dileo, P.; et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar] [CrossRef]
- Gronchi, A.; Bui, B.N.; Bonvalot, S.; Pilotti, S.; Ferrari, S.; Hohenberger, P.; Hohl, R.J.; Demetri, G.D.; Le Cesne, A.; Lardelli, P.; et al. Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann. Oncol. 2012, 23, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Widemann, B.C.; Reinke, D.K.; Helman, L.J.; Ludwig, J.A.; Schuetze, S.; Staddon, A.P.; Milhem, M.M.; Rushing, D.A.; Moertel, C.L.; Goldman, S.; et al. SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)-associated high-grade malignant peripheral nerve sheath tumors (MPNSTs). J. Clin. Oncol. 2013, 31, 10522. [Google Scholar]
- Demetri, G.D.; Schoffski, P.; Grignani, G.; Blay, J.Y.; Maki, R.G.; Van Tine, B.A.; Alcindor, T.; Jones, R.L.; D’Adamo, D.R.; Guo, M.; et al. Activity of Eribulin in Patients With Advanced Liposarcoma Demonstrated in a Subgroup Analysis From a Randomized Phase III Study of Eribulin Versus Dacarbazine. J. Clin. Oncol. 2017, 35, 3433–3439. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Del-Muro, X.; Lopez-Pousa, A.; Maurel, J.; Martin, J.; Martinez-Trufero, J.; Casado, A.; Gomez-Espana, A.; Fra, J.; Cruz, J.; Poveda, A.; et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: A Spanish Group for Research on Sarcomas study. J. Clin. Oncol. 2011, 29, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: Results of a randomized phase II study of two different schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef] [PubMed]
- Recine, F.; Bongiovanni, A.; Riva, N.; Fausti, V.; De Vita, A.; Mercatali, L.; Liverani, C.; Miserocchi, G.; Amadori, D.; Ibrahim, T. Update on the role of trabectedin in the treatment of intractable soft tissue sarcomas. OncoTargets Ther. 2017, 10, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Ducoulombier, A.; Cousin, S.; Kotecki, N.; Penel, N. Gemcitabine-based chemotherapy in sarcomas: A systematic review of published trials. Crit. Rev. Oncol. Hematol. 2016, 98, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.; Forscher, C.; Lowenbraun, S.; Eilber, F.; Eckardt, J.; Holmes, C.; Fu, Y.S. Synovial sarcoma. Uniform response of metastases to high dose ifosfamide. Cancer 1994, 73, 2506–2511. [Google Scholar] [CrossRef]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Tap, W.D.; Jones, R.L.; Van Tine, B.A.; Chmielowski, B.; Elias, A.D.; Adkins, D.; Agulnik, M.; Cooney, M.M.; Livingston, M.B.; Pennock, G.; et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 2016, 388, 488–497. [Google Scholar] [CrossRef]
- Vincenzi, B.; Badalamenti, G.; Napolitano, A.; Spalato Ceruso, M.; Pantano, F.; Grignani, G.; Russo, A.; Santini, D.; Aglietta, M.; Tonini, G. Olaratumab: PDGFR-alpha inhibition as a novel tool in the treatment of advanced soft tissue sarcomas. Crit. Rev. Oncol. Hematol. 2017, 118, 1–6. [Google Scholar] [CrossRef] [PubMed]


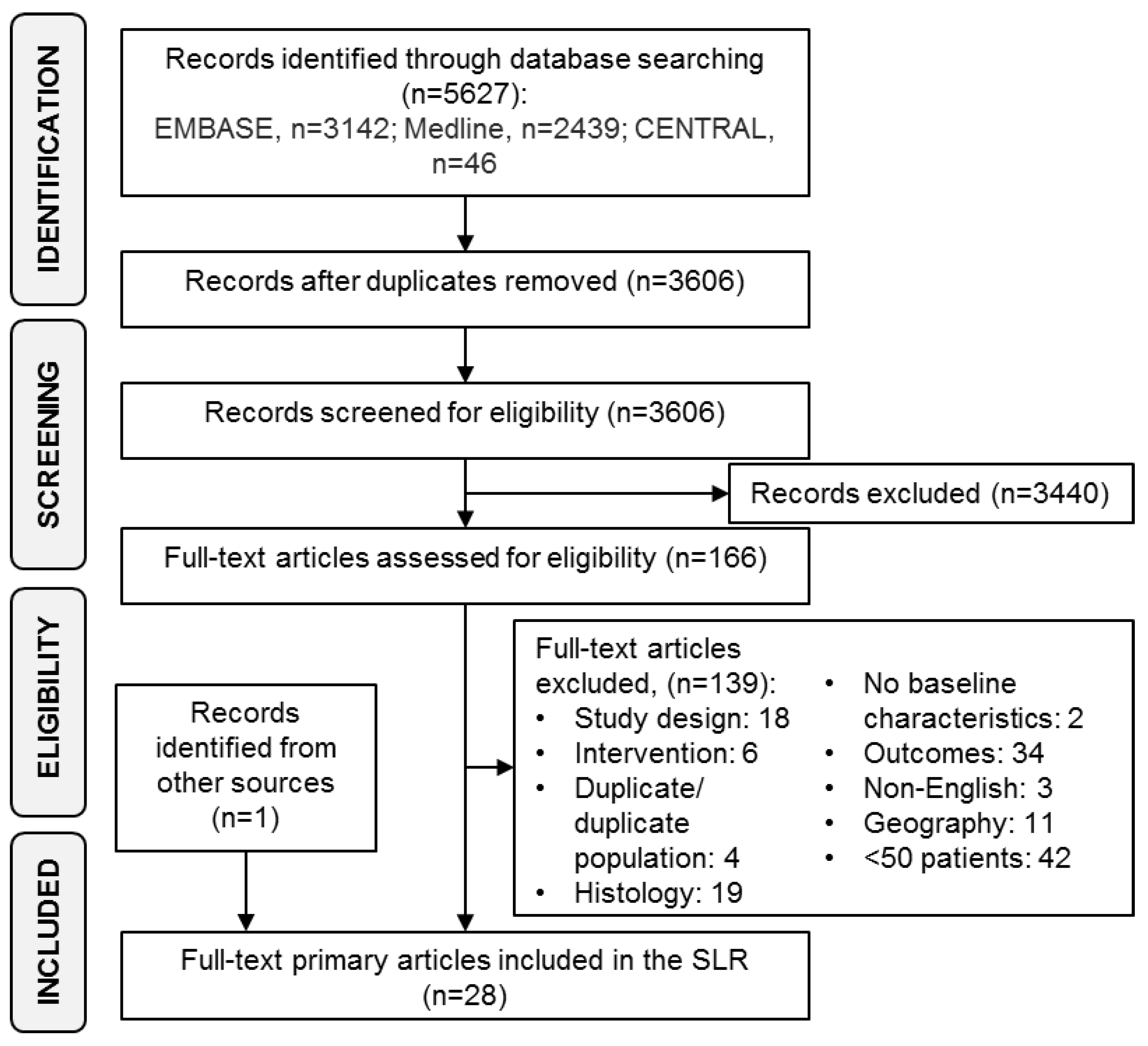{kind=link}
| Patients | Patients with reported SS (adult or pediatric) |
| Interventions/Comparators | Systemic anti-cancer therapy |
| Outcomes | OS, PFS, response (including overall, complete and partial response and stable and progressive disease), duration of response, time to next treatment |
| Study Design | Clinical trials (randomized or non-randomized) or observational study, excluding case reports and case series; minimum number of SS patients: 50 |
| Geography/Language | Europe, North America, Japan, Australia or New Zealand; published in English |
| Publication Year | 2000–2018 |
| Author, Publication Year | Country | Number of Participants | Study Design | Follow-Up in Months Median (Range) |
|---|---|---|---|---|
| Localized | ||||
| de Silva, 2004 [31] | Scotland | 51 | Retrospective, Cohort | 99 |
| Scheer, 2016 [37] | Germany | 52 | Retrospective, Cohort | -- |
| Krieg, 2011 [38] | Switzerland | 62 | Retrospective, Cohort | 136.8 (3.6–331.2) |
| Beaino, 2016 [23] | USA | 63 | Retrospective, Cohort | 85 * (13–210) |
| Orbach, 2011 [36] | Europe | 88 | Retrospective, Cohort | 102 (3–168) |
| Shi, 2013 [39] | USA | 92 | Retrospective, Cohort | 62.4 |
| Eilber, 2007 [4] | USA | 101 | Retrospective, Cohort | 58 (12–185) |
| Al-Hussaini, 2011 [34] | Canada | 102 | Retrospective, Cohort | 67.2 (3.1–216) |
| Trassard, 2001 [40] | France | 128 | Retrospective, Cohort | 128 |
| Ferrari, 2015 [12] | Multi-national | 138 | Prospective trial | 52.1 (13.8–104.4) |
| Brecht, 2006 [41] | Germany, Italy | 150 | Retrospective, Cohort | 80 (6–250) |
| Italiano, 2009 [35] | France, Switzerland | 237 | Retrospective, Cohort | 58 (1–321) |
| Canter, 2008 [42] | USA | 255 | Retrospective, Cohort | 72 (0–287) |
| Vlenterie, 2015 [43] | Netherlands | 461 | Retrospective, Cohort | -- |
| Vining, 2017 [33] | USA | 544 | Retrospective, Cohort | 49.2 |
| Gronchi, 2017 [51] | France, Italy, Poland, Spain | 70 † | Prospective trial | 12.3 |
| Locally advanced or metastatic | ||||
| Takenaka, 2008 [30] | Japan | 108 | Retrospective, Cohort | 54 |
| Setsu, 2013 [44] | Japan | 112 | Retrospective, Cohort | 51 |
| Deshmukh, 2004 [45] | USA | 135 | Retrospective, Cohort | 78 (20–420) |
| Guillou, 2004 [29] | France, Switzerland, Belgium | 165 | Retrospective, Cohort | 37 (2–302) |
| Palmerini, 2009 [46] | Italy | 250 | Retrospective, Cohort | 66 |
| Ferrari, 2004 [47] | Italy | 271 | Retrospective, Cohort | 65 (12–250) |
| Vlenterie, 2016 [28] | Europe | 313 | Retrospective, Cohort | -- |
| Brennan, 2016 [11] | England | 1318 | Retrospective, Cohort | -- |
| Corey, 2014 [48] | USA | 3756 | Retrospective, Database | -- |
| Spurrell, 2005 [25] | UK | 104 | Retrospective, Cohort | -- |
| Metastatic disease | ||||
| Sanfilippo, 2015 [49] | Italy, France, UK | 61 | Retrospective, Cohort | -- |
| Savina, 2017 [50] | France | 188 † | Prospective, Cohort | 61 (1–300) |
| Author, Publication Year | Subgroup | N Baseline | Age (Years) | % Female | Treatments (%) | SS Histology (%) | Primary Tumor Site (%) | Resectable (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median (Range) | RT | Chemo | Surgery | Monophasic | Biphasic | Extremities | Upper Extremities | Lower Extremities | |||||
| Localised | |||||||||||||
| de Silva, 2004 [31] | All SS patients | 51 | 33 (9–77) | 49 | 25 | 27 | 100 | 18 | 77 | 92 | -- | 75 | 100 |
| Scheer, 2016 [37] | All SS patients, pediatric and adolescent | 52 | 13.9 (0.9–20.9) | 46 | 73 | 94 | 100 | 65 | 33 | -- | -- | -- | -- |
| Krieg, 2011 [38] | All SS patients | 62 | -- (6–82) | 58 | -- | -- | -- | 56 | 44 | 76 | -- | -- | -- |
| Beaino, 2016 [23] | All SS patients | 63 | 33 (4–74) | 59 | 56 | 14 | -- | 59 | 38 | 17 | 57 | 100 | |
| Orbach, 2011 [36] | All SS patients, pediatric and adolescent | 88 | 12 (2–17) | 47 | -- | 93 | 74 | -- | -- | -- | 20 | 46 | -- |
| IRS I + Tumor size ≤ 5 | 17 | -- | -- | 12 | 65 | 88 | -- | -- | -- | -- | -- | -- | |
| IRS I + Tumor size > 5 | 4 | -- | -- | -- | 100 | 100 | -- | -- | -- | -- | -- | -- | |
| IRS II + Tumor size ≤ 5 | 16 | -- | -- | 13 | 88 | 94 | -- | -- | -- | -- | -- | -- | |
| IRS II + Tumor size > 5 | 12 | -- | -- | 33 | 92 | 92 | -- | -- | -- | -- | -- | -- | |
| IRS III | 39 | -- | -- | 56 | 97 | 79 | -- | -- | -- | -- | -- | -- | |
| Shi, 2013 [39] | All SS patients | 92 | 35.3 | 50 | -- | 23 | -- | -- | -- | -- | 24 | 58 | 100 |
| Eilber, 2007 [4] | All SS patients | 101 | 34 (16–75) | 51 | 95 | 67 | -- | 68 | 32 | -- | 30 | 70 | -- |
| IFO-based | 68 | 33 (16–64) | 54 | 97 | 100 | -- | 68 | 32 | -- | 37 | 63 | -- | |
| No chemo | 33 | 38 (17–75) | 46 | 91 | 0 | -- | 70 | 30 | -- | 15 | 85 | -- | |
| Al-Hussaini, 2011 [34] | All SS patients | - | - | 45 | 78 | 25 | 100 | -- | -- | -- | 27 | 57 | 100 |
| Adult patients | 87 | 37.6 (15–76) | 48.3 | 83 | 14 | 100 | -- | -- | -- | 30 | 60 | 100 | |
| Pediatric patients | 15 | 14 (0.4–18) | 26.7 | 53 | 87 | 100 | -- | -- | -- | 13 | 33 | 100 | |
| Trassard, 2001 [40] | All SS patients | 128 | 33 (15–76) | 58 | 80 | 57 | 99 | 57 | 35 | 76 | -- | -- | 99.2 |
| Ferrari, 2015 [12] | All SS patients, pediatric and adolescent | 138 | 13.7 | 44 | -- | -- | -- | -- | -- | -- | -- | -- | -- |
| IRS I, tumor < 5 cm, no chemo | 24 | -- | -- | -- | -- | 100 | -- | -- | -- | -- | -- | -- | |
| IRS 1 tumor > 5 cm or IRSII, three to six courses of adjuvant chemo and RT | 37 | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | |
| IRS III or N1 tumor, six courses of chemo, delayed surgery, RT | 77 | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | |
| Brecht, 2006 [41] | All SS patients, pediatric and adolescent | 150 | 12 (1–21) | 45 | 71 | 97 | 51 | 41 | 35 | 84 | -- | -- | 50.7 |
| Italiano, 2009 [35] | All SS patients | 237 | 35 (15–76) | 51 | -- | -- | 100 | -- | -- | -- | 21 | 65 | 100 |
| Canter, 2008 [42] | All SS patients | 255 | 34 | 50 | -- | -- | -- | 66 | 34 | -- | 23 | 58 | -- |
| SYT-SSX1 | 73 | 41 (16–80) | 36 | 66 | 47 | -- | 56 | 44 | -- | 20 | 59 | -- | |
| SYT-SSX2 | 59 | 35 (18–78) | 63 | 75 | 40 | -- | 85 | 15 | -- | 19 | 56 | -- | |
| Vlenterie, 2015 [43] | All SS patients | 461 | 38 (2–89) | 46 | -- | -- | -- | 22 | 24 | 66 | -- | -- | 90.2 |
| Vining, 2017 [33] | All SS patients | 544 | 42 (29–55) | 49.5 | -- | -- | -- | 41.5 | 22.6 | -- | 18.4 | 47.2 | 100 |
| Gronchi, 2017* [51] | SS patients receiving standard chemotherapy | 36 | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | 100 |
| SS patients receiving histotype-tailored therapy | 34 | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | 100 | |
| Mixed population (localized and metastatic) | |||||||||||||
| Takenaka, 2008 [30] | All SS patients | 108 | 37 (8–74) | 59.3 | 21 | 77 | 93 | 63 | 30 | 67 | -- | -- | 96.3 |
| Setsu, 2013 [44] | All SS patients | 112 | -- | 61 | 15 | 24 | -- | 67 | 29 | -- | -- | -- | -- |
| Deshmukh, 2004 [45] | All SS patients | 135 | 31 (8–81) | 37 | 71 | 38 | 90 | -- | -- | -- | 16 | 65 | -- |
| Palmerini, 2009 [46] | Metastatic | 46 | 40 (13–79) | 39 | -- | -- | 89 | 74 | 22 | -- | -- | -- | -- |
| Localized | 204 | 36 (7–83) | 54 | -- | 48 | 100 | 60 | 36 | -- | -- | -- | -- | |
| Ferrari, 2004 [47] | All SS patients | 271 | 32 (5–87) | 47 | -- | 41 | -- | 38 | 43 | 22 | 63 | -- | |
| Vlenterie, 2016 [28] | All SS patients | 313 | 40 (18–81) | 39 | 41 | 100 | 43 | -- | -- | 56 | 14 | 41 | -- |
| Brennan, 2016 [11] | All SS patients | 1318 | -- | 48 | -- | -- | 71 | -- | -- | 65 | -- | -- | -- |
| Adults | 1136 | -- | 48 | -- | -- | 70 | -- | -- | 66 | -- | -- | -- | |
| Children and adolescents | 182 | -- | 43 | -- | -- | 77 | -- | -- | 63 | -- | -- | -- | |
| Corey, 2014 [48] | SS, biphasic | 732 | 40 | 47 | -- | -- | -- | -- | 100 | -- | -- | -- | -- |
| SS, histology not specified | 1820 | 41 | 48 | -- | -- | -- | -- | -- | -- | -- | -- | -- | |
| SS, spindle cell | 1204 | 41 | 49 | -- | -- | -- | -- | -- | -- | -- | -- | -- | |
| Spurrell, 2005 [25] | All SS patients | 104 | 33 | 50 | 67 | 30 | 87 | 40 | 38 | 66 | 18 | 48 | -- |
| Metastatic disease | |||||||||||||
| Sanfilippo, 2015 [49] | All SS patients | 61 | 37 (18–68) | 58 | -- | 100 | -- | -- | -- | 57 | -- | -- | 0 |
| Savina, 2017 * [50] | All SS patients | 188 | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- |
| Study ID | Population | N | PFS | OS | ||
|---|---|---|---|---|---|---|
| % PFS (95% CI) | Median, Months (95%CI) | % OS (95% CI) | Median, Months (95% CI) | |||
| Localized | ||||||
| Time point 1 year | ||||||
| Italiano, 2009 [35] | All SS patients | 237 | -- | -- | 85 (82, 88) | 136 (70, 204) |
| Time point 3 years | ||||||
| Ferrari, 2015 [12] | Tumor site-axial | 39 | 77.7 (60.2–88.2) | -- | 100 (-) | -- |
| Tumor site-extremities | 99 | 83.8 (74.4–89.9) | -- | 96 (88.2–98.7) | -- | |
| Time point 5 years | ||||||
| de Silva, 2004 [31] | All SS patients | 51 | -- | -- | 56 (-) | 40.1 (-) |
| Scheer, 2016 [37] | All SS patients | 52 | 26 (-) | -- | 40 (-) | -- |
| Tumor site-axial | 11 | 18.2 (-) | -- | 22.7 (-) | -- | |
| Tumor site-non-axial | 41 | 28.4 (-) | -- | 44.7 (-) | -- | |
| Krieg, 2011 [38] | All SS patients | 62 | -- | -- | 74.2 (-) | -- |
| FNCLCC Grade 2 | 32 | -- | -- | 97 (-) | -- | |
| FNCLCC Grade 3 | 11 | -- | -- | 18 (-) | -- | |
| Orbach, 2011 [36] | All SS patients, pediatric | 88 | 68 (-) | -- | 85 (-) | -- |
| Shi, 2013 [39] | All SS patients | 92 | 56 (-) | -- | 61 (-) | -- |
| Chemo | 21 | 67 (-) | -- | 65 (-) | -- | |
| No Chemo | 71 | 49 (-) | -- | 53 (-) | -- | |
| Tumor site-extremities | 75 | 57 (-) | -- | 60 (-) | -- | |
| Tumor site-trunk | 11 | 52 (-) | -- | 63 (-) | -- | |
| Al-Hussaini, 2011 [34] | All SS patients | 102 | 69.3 (-) | -- | 80.3 (-) | -- |
| Adult | 87 | 68.3 (-) | -- | 76.9 (-) | -- | |
| Pediatric | 15 | 74.9 (-) | -- | 100 (-) | -- | |
| Chemo (all SS patients) | 25 | 62.6 (-) | -- | -- | -- | |
| No Chemo | 77 | 71.5 (-) | -- | -- | -- | |
| Ferrari, 2015 [12] | All SS patients | 138 | 80.7 (72.5, 86.7) | -- | 90.7 (82, 95.3) | -- |
| Brecht, 2006 [41] | All SS patients | 150 | 77 (-) | -- | 89 (-) | -- |
| Tumor site-extremities | 129 | 79 (-) | -- | 90 (-) | -- | |
| Tumor site-other (not extremities) | 21 | 68 (-) | -- | 88 (-) | -- | |
| Tumor status-T1 | 94 | 88 (-) | -- | 96 (-) | -- | |
| Italiano, 2009 [35] | All SS patients | 237 | -- | -- | 64 (59, 69) | -- |
| Vlenterie, 2015 [43] | All SS patients | 461 | -- | -- | 63.5 (-) | -- |
| Takenaka, 2008 [30] | Localized | 91 | -- | -- | 81 (-) | -- |
| Deshmukh, 2004 [45] | Localized (primary + locally recurrent) | 108 | -- | -- | 69 (-) | -- |
| Palmerini, 2009 [46] | Localized | 204 | 58 (51, 66) | -- | 76 (69, 82) | -- |
| Time point 9 years | ||||||
| Italiano, 2009 [35] | All SS patients | 237 | -- | -- | 46 (40, 52) | -- |
| Time point 10 years | ||||||
| de Silva, 2004 [31] | All SS patients | 51 | -- | -- | 45 (-) | -- |
| Krieg, 2011 [38] | All SS patients | 62 | -- | -- | 61.2 (-) | -- |
| FNCLCC Grade 2 | 32 | -- | -- | 84 (-) | -- | |
| FNCLCC Grade 3 | 11 | -- | -- | 0 (-) | -- | |
| Shi, 2013 [39] | All SS patients | 92 | 53 (-) | -- | 56 (-) | -- |
| Chemo | 21 | 67 (-) | -- | 65 (-) | -- | |
| No Chemo | 71 | 49 (-) | -- | 48 (-) | -- | |
| Tumor site-extremities | 75 | 55 (-) | -- | 55 (-) | -- | |
| Tumor site-trunk | 11 | 41 (-) | -- | 63 (-) | -- | |
| Brecht, 2006 [41] | All SS patients | 150 | 89 (-) | -- | 78 (-) | -- |
| Vlenterie, 2015 [43] | All SS patients | 461 | -- | -- | 53.8 (-) | -- |
| Deshmukh, 2004 [45] | All SS patients | 108 | -- | -- | 51 (-) | |
| Localized (primary + locally recurrent) | 108 | -- | -- | 51 (-) | -- | |
| Mixed population localized/locally advanced / metastatic | ||||||
| Time point 2 years | ||||||
| Corey, 2014 [48] | SS, biphasic | 732 | -- | -- | 85 (-) | -- |
| SS, NOS | 1820 | -- | -- | 71 (-) | -- | |
| SS, spindle cell | 1204 | -- | -- | 77 (-) | -- | |
| Time point 1 year | ||||||
| Vlenterie, 2016 [28] | All SS patients | 313 | 18.7 (14.6, 23.3) | 6.3 (5.9, 7.0) | 63.7 (57.9, 68.8) | 15 (13.9, 16.4) |
| Anthracycline | 121 | 17.5 (11.2, 24.9) | 5.06 (4.3, 6.1) | 62.6 (53.1, 70.8) | 14.85 (12.2, 16.2) | |
| Dox + IFO | 112 | 21.4 (14.4, 29.4) | 7.47 (6.5, 8.7) | 66.3 (56.7, 74.3) | 14.98 (12.9, 18.9) | |
| CYVADIC | 30 | 23.3 (10.3, 39.4) | 6.08 (3.0, 10.8) | 58.6 (38.7, 74.1) | 15.8 (8.4, 23.1) | |
| IFO | 42 | 15.5 (6.4, 28.5) | 7.2 (5.9, 9.2) | 65.6 (48.9, 78) | 15.34 (11.7, 19.7) | |
| Other chemotherapy | 8 | 0 (-) | 2.27 (1.0, 9.0) | 29.2 (1, 71.9) | 10.45 (0.9, NR) | |
| Tumor site-extremity b | 174 | 16.1 (11–21.9) | 6.21 (5.3–7.1) | 63.1 (55.3–69.69) | 15.5 (13.3–18.1) | |
| Tumor site-other c | 52 | 22.0 (11.9–34.2) | 7.13 (5.1–8.9) | 63.6 (48.6–75.3) | 14.4 (11.6–18.7) | |
| Time point 3 years | ||||||
| Ferrari, 2015 [12] | IRS I, tumor <5 cm, no chemo | 24 | 91.7 (70.6, 97.8) | -- | 100 (-) | -- |
| IRS 1 tumor >5 cm or IRSII, three to six courses of adjuvant chemo and RT | 37 | 91.2 (75.1, 97.1) | -- | 100 (-) | -- | |
| IRS III or N1 tumor, six courses of chemo, delayed surgery, RT | 77 | 77.7 (60.2–88.2) | -- | 100 (-) | -- | |
| Time point 5 years | ||||||
| Takenaka, 2008 [30] | All SS patients | 108 | -- | -- | 69.8 (-) | -- |
| Tumor site-extremities | 72 | -- | -- | 57.4 (-) | -- | |
| Tumor site-trunk | 36 | -- | -- | 78.1 (-) | -- | |
| Setsu, 2013 [44] | All SS patients | 65 | -- | 24 (-) | 62 (-) | -- |
| Tumor site-distal extremities | 39 | 48.3 (-) | -- | 79.8 (-) | -- | |
| Tumor site-proximal extremities | 63 | 36.5 (-) | -- | 50.9 (-) | -- | |
| Deshmukh, 2004 [45] | All SS patients | 135 | -- | -- | 61 (-) | -- |
| Palmerini, 2009 [46] | All SS patients | - | -- | -- | 68 (-) | -- |
| Tumor site-lower extremities | - | 59 (-) | -- | 75 (-) | -- | |
| Tumor site-upper extremities | - | 76 (-) | -- | 78 (-) | -- | |
| Ferrari, 2004 [47] | All SS patients | 271 | 36.8 (-) | -- | 64.3 (-) | -- |
| Adjuvant chemo | 61 | 55.3 (-) | -- | 71.5 (-) | -- | |
| No adjuvant chemo | 154 | 35.2 (-) | -- | 70.3 (-) | -- | |
| IFO + Dox/Epi | - | 52.3 (-) | -- | -- | -- | |
| Other chemo | - | 59.4 (-) | -- | -- | -- | |
| Locally advanced disease a | 40 | 31.9 (-) | -- | 49.6 (-) | -- | |
| Corey, 2014 [48] | SS, biphasic | 732 | -- | -- | 65 (-) | -- |
| SS, histology not specified | 1820 | -- | -- | 52 (-) | -- | |
| SS, spindle cell | 1204 | -- | -- | 56 (-) | -- | |
| Time point 10 years | ||||||
| Deshmukh, 2004 [45] | All SS patients | 135 | -- | -- | 42 (-) | -- |
| Primary tumor | 99 | -- | -- | 55 (-) | -- | |
| Local recurrence | 9 | -- | -- | 11 (-) | -- | |
| Ferrari, 2004 [47] | All SS patients | 271 | 29.8 (-) | -- | -- | -- |
| Time point not reported | ||||||
| Spurrell, 2005 [25] | All SS patients | 104 | -- | -- | -- | 22 (-) |
| Metastatic disease | ||||||
| Time point 0.5 years | ||||||
| Sanfilippo, 2015 [49] | All SS patients | 61 | 23 (-) | 3 (-) | -- | -- |
| Time point 5 years | ||||||
| Takenaka, 2008 [30] | Metastatic | 17 | -- | -- | 0 (-) | |
| Palmerini, 2009 [46] | Metastatic | 46 | -- | -- | 10 (-) | -- |
| Brecht, 2006 [41] | Tumor status-T2 | 53 | 60 (-) | -- | 78 (-) | -- |
| Savina 2017 [50] | Metastatic | 188 | 7.14 (-) | 19.7 | -- | -- |
| Time point 10 years | ||||||
| Deshmukh, 2004 [45] | Metastatic | 27 | -- | -- | 15 (-) | -- |
| Study ID | Total Cohort (N) | Synovial Sarcoma Cohort (N) | Treatment Arm | Progression Free Survival (PFS) | Overall Survival (OS) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median, Months (95% CI) | Comparator vs. Reference for | HR (95% CI) | Time, Months | Rate, % (95% CI) | Median, Months (95% CI) | Rate, % (95% CI) | ||||
| Relapse/refractory | ||||||||||
| Pappo, 2014 [52] | 163 | 23 | R1507 | 1.3 (1.2, 1.3) | -- | -- | -- | -- | -- | 17 |
| Sleijfer, 2011 (EORTC Study 62043) [53] | 142 | 38 | Pazopanib | 5.3 (2.6, 6.3) | -- | -- | -- | -- | 10.2 (7.5, 13.3) | NR |
| Locally advanced or metastatic | ||||||||||
| Pappo, 2005 [54] | 43 | 16 | Neoadjuvant vincristine, ifosfamide, and doxorubicin a | -- | -- | -- | 36 | 56.3 (SD: 12) | -- | 81.3 (SD:10) |
| Metastatic disease | ||||||||||
| Chugh, 2009 [55] | 185 | 22 | Imatinib | 1.92 (1.92, 3.96) | -- | -- | -- | -- | -- | -- |
| Kawai, 2015 [56] | 76 | 7 | Trabectedin | NR | Trabectedin vs. BSC | 0.14 (0.03, 0.68) | -- | -- | -- | -- |
| Mir, 2016 (REGOSARC) [57] | 90 | 13 | Regorafenib | 5.6 (1.4, 11.6) | Regorafenib vs. placebo | 0.10 (0.03, 0.35) | 3 | 77 (42, 92) | 13.4 (5.3, NR) | 92 |
| Regorafenib | -- | -- | -- | 6 | 38 (14, 63) | -- | 77 | |||
| Regorafenib | -- | -- | -- | 9 | 38 (14, 63) | -- | -- | |||
| 92 | 14 | Placebo | 1.0 (0.8, 1.4) | -- | -- | 3 | 0 | 6.7 (2.2, NR) | 85 | |
| Placebo | -- | -- | -- | 6 | 0 | -- | 64 | |||
| Placebo | -- | -- | -- | 9 | 0 | -- | -- | |||
| Ray—Coquard, 2008 [58] | 48 b | 46 | Gefitinib | 1.4 | -- | -- | -- | -- | ||
| Schoffski, 2011 [59] | 128 | 19 | Eribulin mesylate | 2.6 (2.3, 4.3) | -- | -- | 3 | 21.1 | -- | 71.1 (43.7, 86.8) |
| Schoffski, 2013 [60] | 111 | 17 | Cixutumumab | 1.5 (1.3, 2.6) | -- | -- | 3 | 21.4 (5.2, 44.8) | 13.0 (5.1, 16.5) | 94.1 (65, 99.1) |
| Van der Graaf, 2012 (PALETTE) [61] | 369 | 30 | Pazopanib | -- | SS vs. other STS | 0.82 (0.51, 1.32) | -- | -- | -- | -- |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedel, R.F.; Jones, R.L.; Italiano, A.; Bohac, C.; Thompson, J.C.; Mueller, K.; Khan, Z.; Pollack, S.M.; Van Tine, B.A. Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review. Cancers 2018, 10, 417. https://doi.org/10.3390/cancers10110417
Riedel RF, Jones RL, Italiano A, Bohac C, Thompson JC, Mueller K, Khan Z, Pollack SM, Van Tine BA. Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review. Cancers. 2018; 10(11):417. https://doi.org/10.3390/cancers10110417
Chicago/Turabian StyleRiedel, Richard F., Robin L. Jones, Antoine Italiano, Chet Bohac, Juliette C. Thompson, Kerstin Mueller, Zaeem Khan, Seth M. Pollack, and Brian A. Van Tine. 2018. "Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review" Cancers 10, no. 11: 417. https://doi.org/10.3390/cancers10110417
APA StyleRiedel, R. F., Jones, R. L., Italiano, A., Bohac, C., Thompson, J. C., Mueller, K., Khan, Z., Pollack, S. M., & Van Tine, B. A. (2018). Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review. Cancers, 10(11), 417. https://doi.org/10.3390/cancers10110417