The Development of an Effective Bacterial Single-Cell Lysis Method Suitable for Whole Genome Amplification in Microfluidic Platforms



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
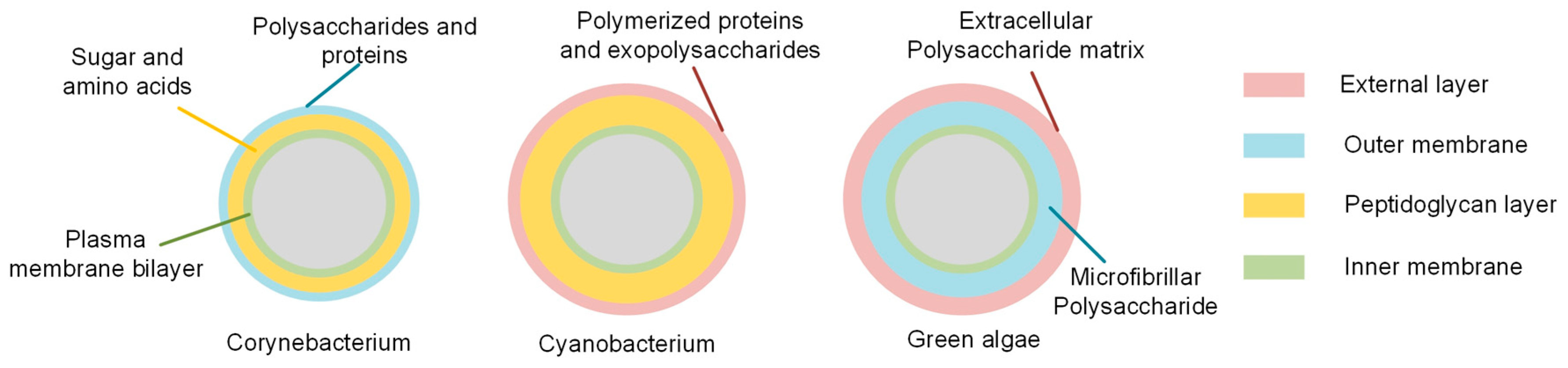
2.1. Cell Wall Components of Chosen Bacterial Cells
2.2. Cell Preparation
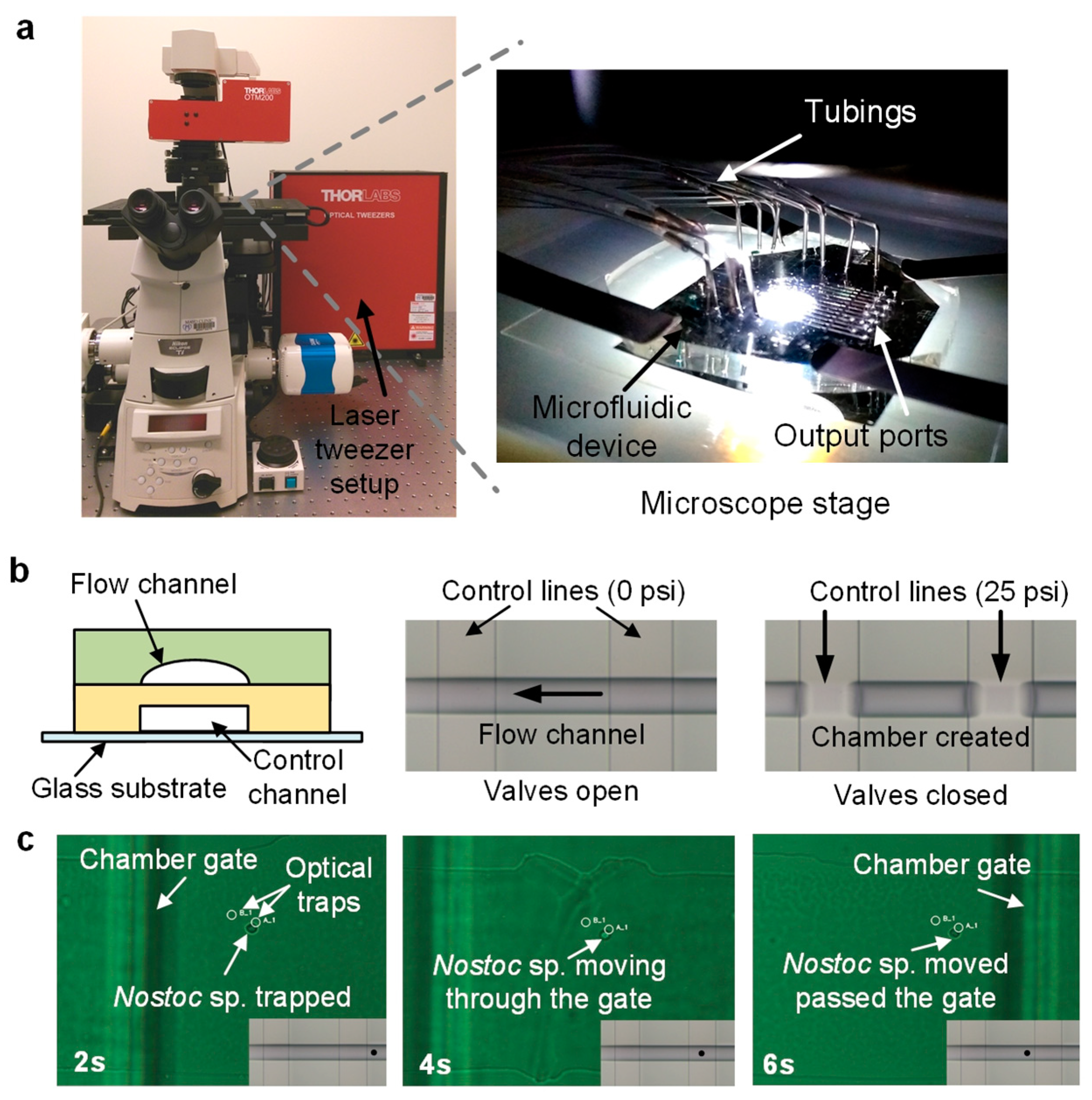
2.3. Microfluidic Experimental Setup
2.4. Choice of Lysis Buffer Components
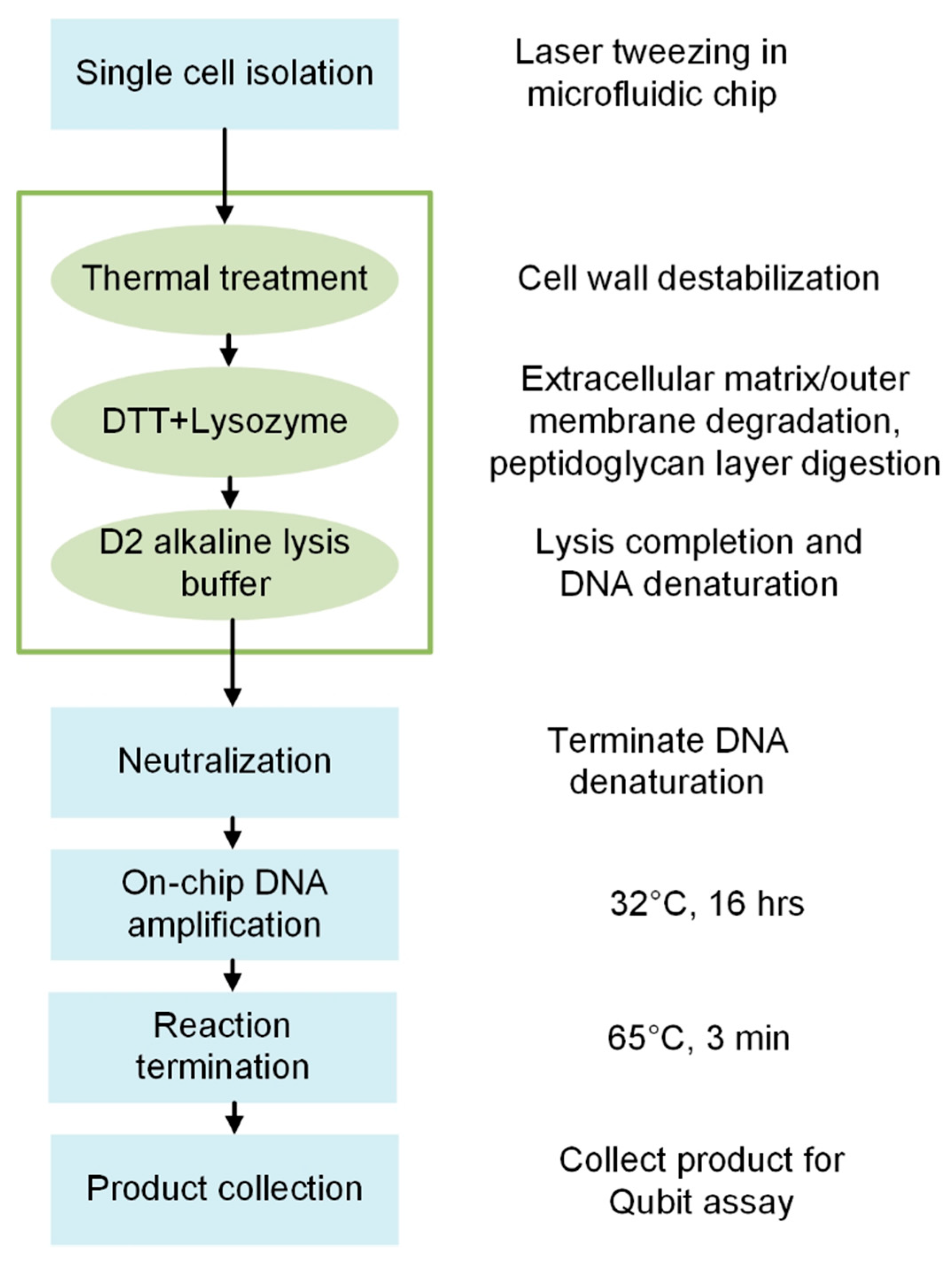
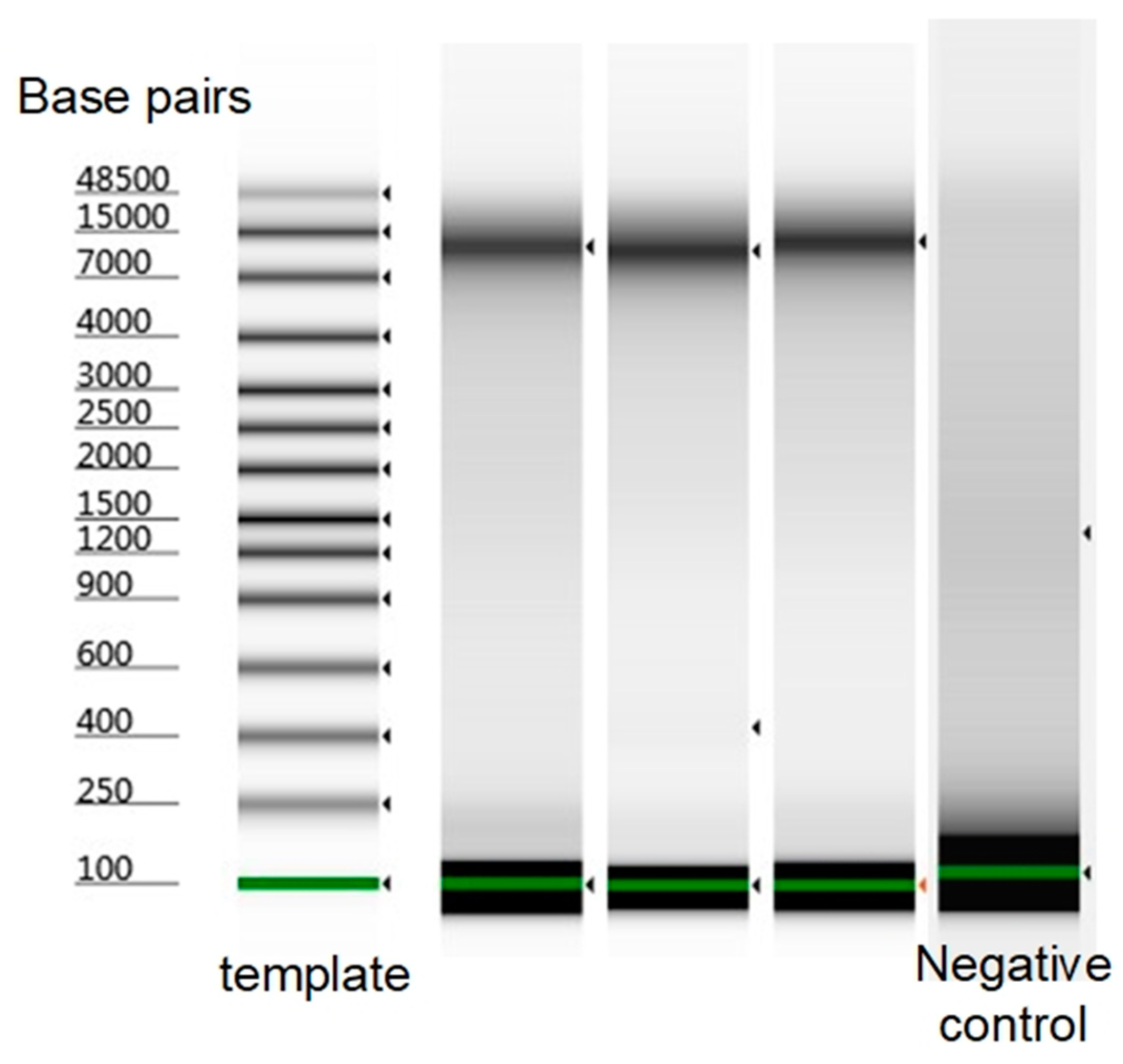
2.5. Microfluidic Bacterial Lysis for SC-WGA Workflow
3. Results and Discussion
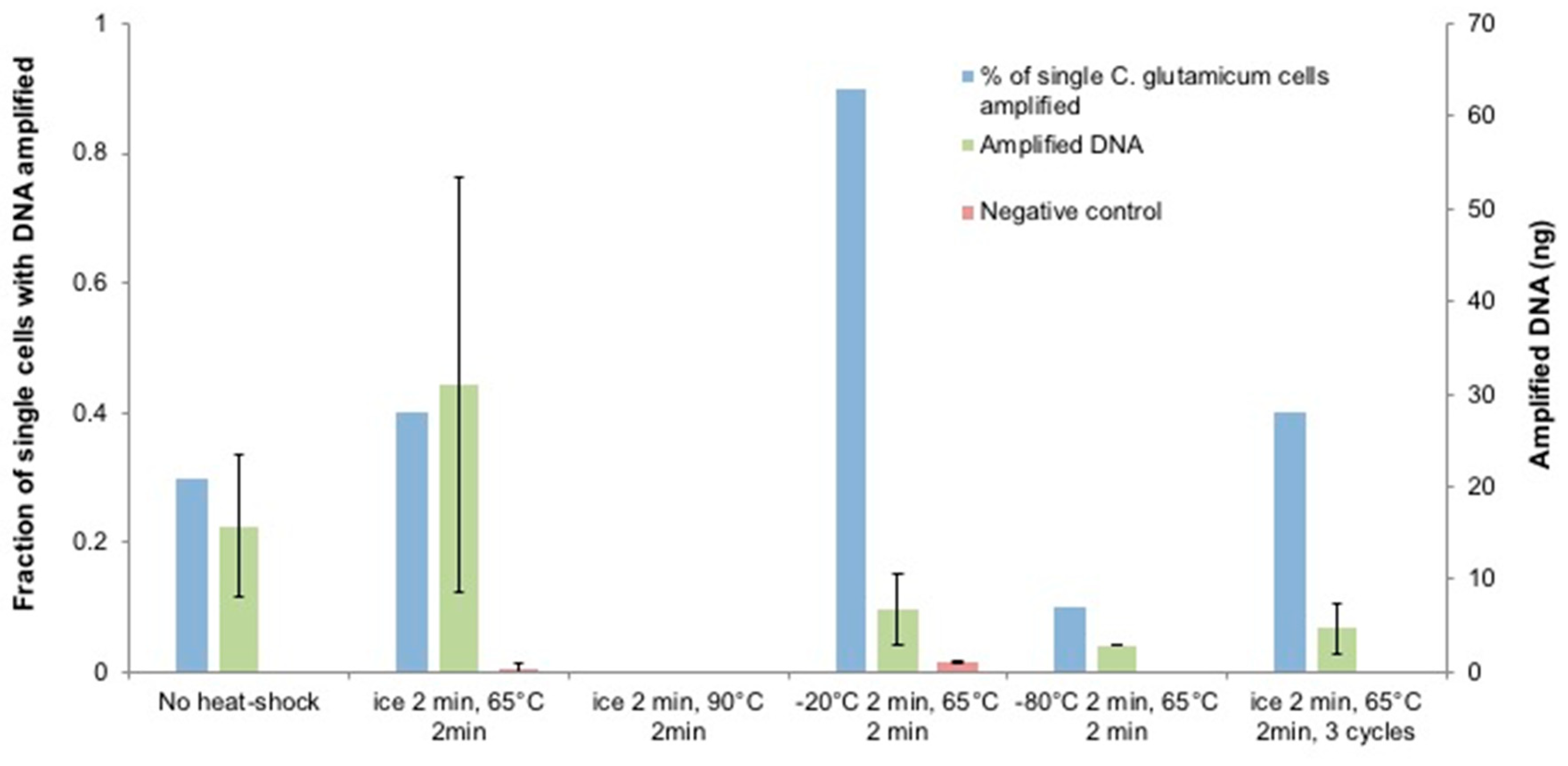
3.1. Optimization of Heat-Shock Treatment on C. glutamicum
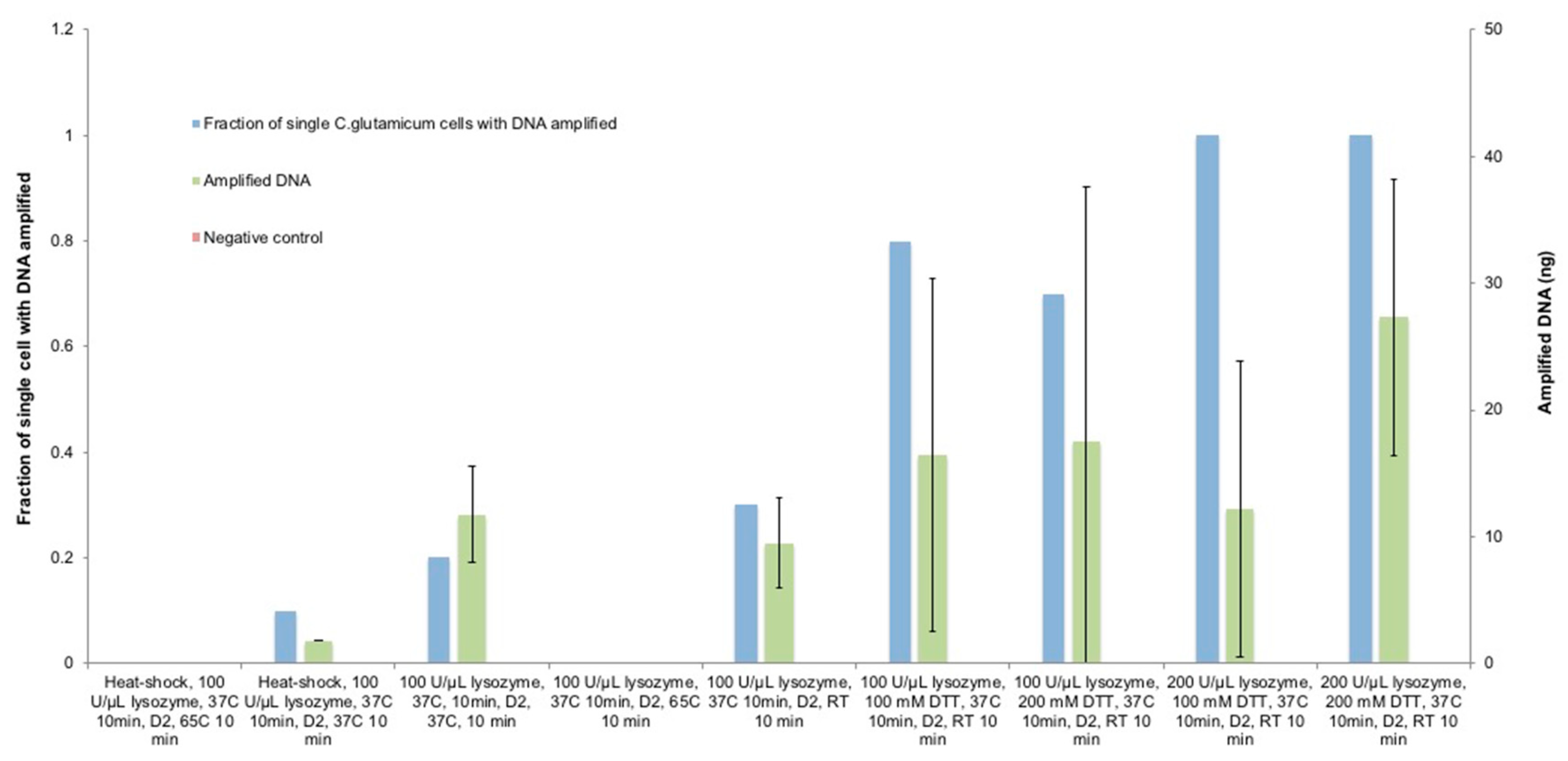
3.2. Optimization of Lysozyme Treatment on C. glutamicum
3.3. Optimization of Lysozyme Combined with DTT Treatment on C. glutamicum
3.4. The Cyanobacteria Species’ Cell Wall Description
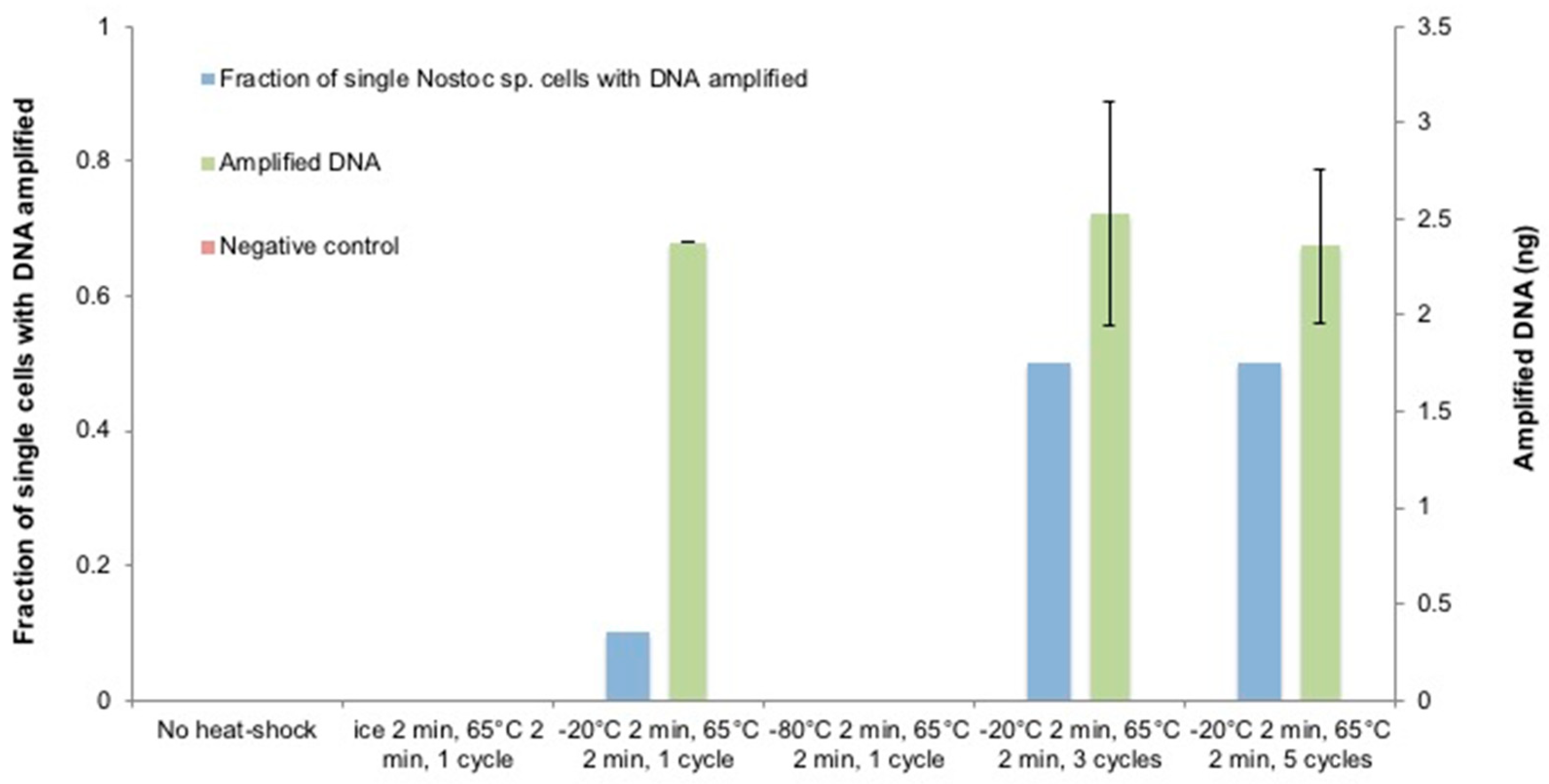
3.5. The Optimization of the Heat-Shock Treatment on Nostoc sp.
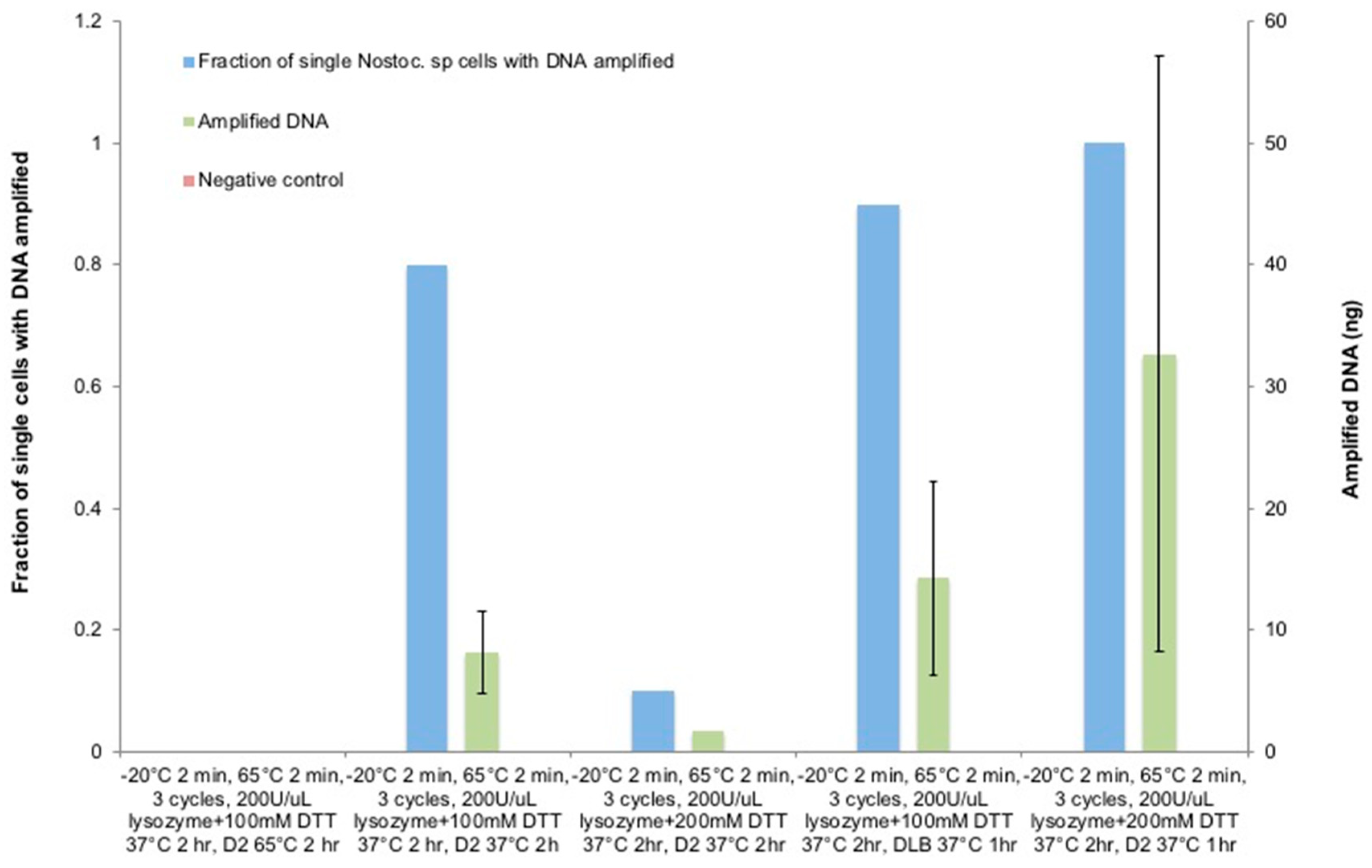
3.6. The Optimization of Lysozyme Combined with the DTT Treatment on Nostoc sp.
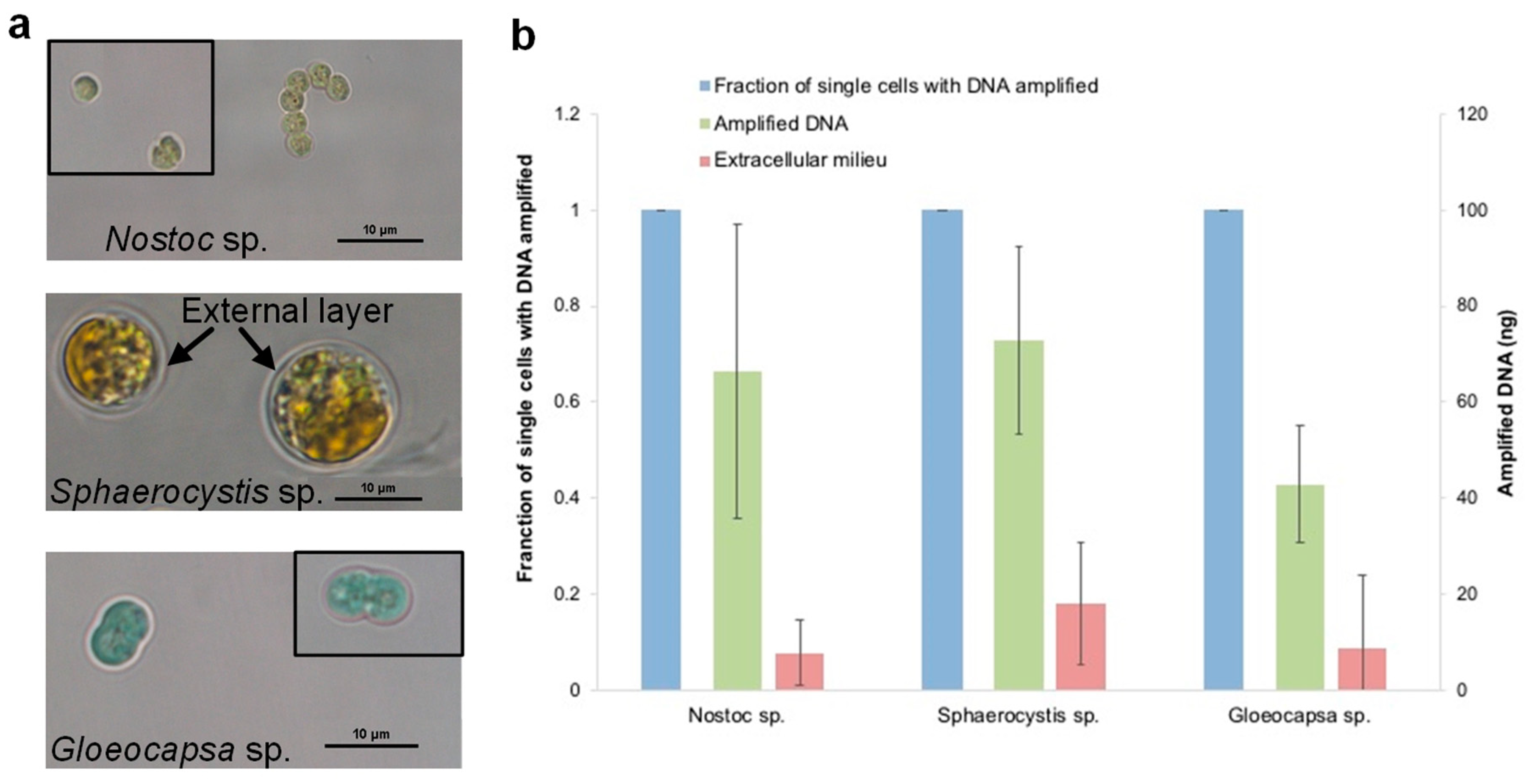
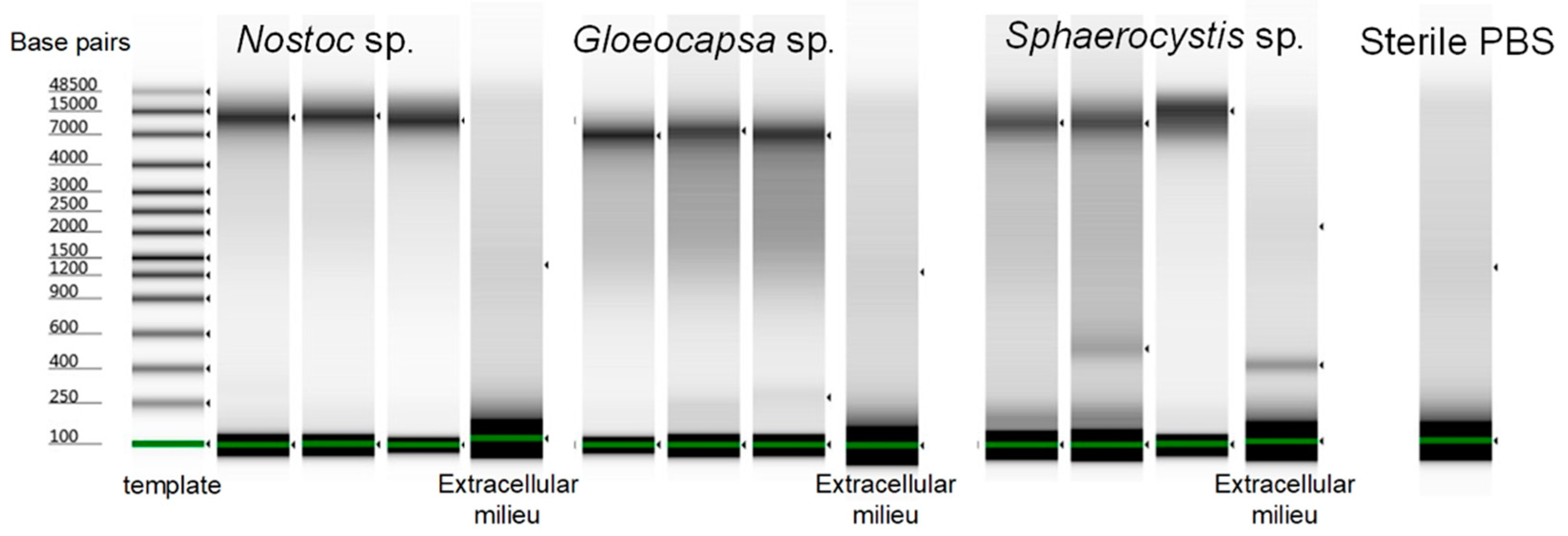
3.7. The Evaluation of the Optimized Lysis Protocol Using Gloeocapsa sp. and Sphaerocystis sp.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Johnston, J.C.; Sui, S.J.H.; Cook, V.J.; Shah, L.; Brodkin, E.; Rempel, S.; Moore, R.; Zhao, Y.; Holt, R. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 2011, 364, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Robert, L.; Pelletier, J.; Dang, W.L.; Taddei, F.; Wright, A.; Jun, S. Robust growth of Escherichia coli. Curr. Biol. 2010, 20, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 2016, 62, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Bamford, R.A.; Smith, A.; Metz, J.; Glover, G.; Titball, R.W.; Pagliara, S. Investigating the physiology of viable but non-culturable bacteria by microfluidics and time-lapse microscopy. BMC Biol. 2017, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Wakamoto, Y.; Dhar, N.; Chait, R.; Schneider, K.; Signorino-Gelo, F.; Leibler, S.; McKinney, J.D. Dynamic persistence of antibiotic-stressed mycobacteria. Science 2013, 339, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R. Single cell genomics: An individual look at microbes. Curr. Opin. Microbiol. 2012, 15, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Invited review: Microbial ecology in the age of genomics and metagenomics: Concepts, tools, and recent advances. Mol. Ecol. 2006, 15, 1713–1731. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: Beyond the genome. Ann. Rev. Mar. Sci. 2010, 3, 347–371. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Kvist, T.; Ahring, B.K.; Lasken, R.S.; Westermann, P. Specific single-cell isolation and genomic amplification of uncultured microorganisms. Appl. Microbiol. Biotechnol. 2007, 74, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R.; Sieracki, M.E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. PNAS 2007, 104, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- Kalisky, T.; Quake, S.R. Single-cell genomics. Nat. Methods 2011, 8, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cui, X.; Schmitt, K.; Hubert, R.; Navidi, W.; Arnheim, N. Whole genome amplification from a single cell: Implications for genetic analysis. PNAS 1992, 89, 5847–5851. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Teh, S.-Y.; Lin, R.; Hung, L.-H.; Lee, A.P. Droplet microfluidics. Lab Chip 2008, 8, 198–220. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Tufenkji, N.; Moraes, C. Microfluidics in microbiology: Putting a magnifying glass on microbes. Integr. Biol. 2016, 8, 914–917. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lagus, T.P.; Edd, J.F. A review of the theory, methods and recent applications of high-throughput single-cell droplet microfluidics. J. Phys. D Appl. Phys. 2013, 46, 114005. [Google Scholar] [CrossRef]
- Autebert, J.; Coudert, B.; Bidard, F.-C.; Pierga, J.-Y.; Descroix, S.; Malaquin, L.; Viovy, J.-L. Microfluidic: An innovative tool for efficient cell sorting. Methods 2012, 57, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Walther-Antonio, M. Microfluidics: A new tool for microbial single cell analyses in human microbiome studies. Biomicrofluidics 2017, 11, 061501. [Google Scholar] [CrossRef]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Landry, Z.C.; Giovanonni, S.J.; Quake, S.R.; Blainey, P.C. Optofluidic cell selection from complex microbial communities for single-genome analysis. Methods Enzymol. 2013, 531, 61–90. [Google Scholar] [PubMed]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Marcy, Y.; Ishoey, T.; Lasken, R.S.; Stockwell, T.B.; Walenz, B.P.; Halpern, A.L.; Beeson, K.Y.; Goldberg, S.M.; Quake, S.R. Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet. 2007, 3, e155. [Google Scholar] [CrossRef] [PubMed]
- Zare, R.N.; Kim, S. Microfluidic platforms for single-cell analysis. Annu. Rev. Biomed. Eng. 2010, 12, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Binga, E.K.; Lasken, R.S.; Neufeld, J.D. Something from (almost) nothing: The impact of multiple displacement amplification on microbial ecology. ISME J. 2008, 2, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Motley, S.T.; Picuri, J.M.; Crowder, C.D.; Minich, J.J.; Hofstadler, S.A.; Eshoo, M.W. Improved multiple displacement amplification (iMDA) and ultraclean reagents. BMC Genom. 2014, 15, 443. [Google Scholar] [CrossRef] [PubMed]
- De Bourcy, C.F.; De Vlaminck, I.; Kanbar, J.N.; Wang, J.; Gawad, C.; Quake, S.R. A quantitative comparison of single-cell whole genome amplification methods. PLoS ONE 2014, 9, e105585. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Song, P.; Zou, D.; Hu, X.; Zhao, S.; Gao, S.; Ling, F. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in single-cell sequencing. PLoS ONE 2014, 9, e114520. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.K.; Evitt, N.H.; Swartz, J.R. Chemical lysis of cyanobacteria. J. Biol. Eng. 2015, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Geciova, J.; Bury, D.; Jelen, P. Methods for disruption of microbial cells for potential use in the dairy industry—A review. Int. Dairy J. 2002, 12, 541–553. [Google Scholar] [CrossRef]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001, 56, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Moré, M.I.; Herrick, J.B.; Silva, M.C.; Ghiorse, W.C.; Madsen, E.L. Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl. Environ. Microbiol. 1994, 60, 1572–1580. [Google Scholar] [PubMed]
- Kim, I.S.; Nguyen, G.H.; Kim, S.; Lee, J.; Yu, H.-W. Evaluation of methods for cyanobacterial cell lysis and toxin (microcystin-LR) extraction using chromatographic and mass spectrometric analyses. Environ. Eng. Res. 2009, 14, 250–254. [Google Scholar] [CrossRef]
- Liu, X.; Curtiss, R. Nickel-inducible lysis system in Synechocystis sp. PCC 6803. PNAS 2009, 106, 21550–21554. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.W.; Kim, S.; Appadoo, V.; Zare, R.N. Lysis of a single cyanobacterium for whole genome amplification. Micromachines 2013, 4, 321–332. [Google Scholar] [CrossRef]
- He, J.; Du, S.; Tan, X.; Arefin, A.; Han, C.S. Method summary. Biotechniques 2016, 60, 129–135. [Google Scholar] [PubMed]
- Rees, P.; Watson, J.; Cumming, R.; Liddell, J.; Turner, P. Effects of heat shock on gram-negative bacteria: Use of lysis by sodium dodecyl sulphate as a probe for the integrity of DNA. Bioseparation 1996, 6, 125–132. [Google Scholar] [PubMed]
- Salazar, O.; Asenjo, J.A. Enzymatic lysis of microbial cells. Biotechnol. Lett. 2007, 29, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Cohen, D.B.; Ravel, J.; Abdo, Z.; Forney, L.J. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE 2012, 7, e33865. [Google Scholar] [CrossRef] [PubMed]
- Cloninger, C.; Felton, M.; Paul, B.; Hirakawa, Y.; Metzenberg, S. Control of pH during plasmid preparation by alkaline lysis of Escherichia coli. Anal. Biochem. 2008, 378, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Listwan, P.; Pédelacq, J.-D.; Lockard, M.; Bell, C.; Terwilliger, T.C.; Waldo, G.S. The optimization of in vitro high-throughput chemical lysis of Escherichia coli. Application to ACP domain of the polyketide synthase ppsC from Mycobacterium tuberculosis. J. Struct. Funct. Geonom. 2010, 11, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Grindberg, R.V.; Ishoey, T.; Brinza, D.; Esquenazi, E.; Coates, R.C.; Liu, W.-T.; Gerwick, L.; Dorrestein, P.C.; Pevzner, P.; Lasken, R. Single cell genome amplification accelerates identification of the apratoxin biosynthetic pathway from a complex microbial assemblage. PLoS ONE 2011, 6, e18565. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, C.; Greenwood-Quaintance, K.E.; Hanssen, A.D.; Patel, R. Corynebacterium prosthetic joint infection. J. Clin. Microbiol. 2012, 50, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Alatoom, A.A.; Cazanave, C.J.; Cunningham, S.A.; Ihde, S.M.; Patel, R. Identification of non-diphtheriae Corynebacterium by use of matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 2012, 50, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zarka, A.; Boussiba, S. A simplified protocol for preparing DNA from filamentous cyanobacteria. Plant Mol. Biol. Rep. 2000, 18, 385–392. [Google Scholar] [CrossRef]
- Puech, V.; Chami, M.; Lemassu, A.; Lanéelle, M.-A.; Schiffler, B.; Gounon, P.; Bayan, N.; Benz, R.; Daffé, M. Structure of the cell envelope of corynebacteria: Importance of the non-covalently bound lipids in the formation of the cell wall permeability barrier and fracture plane. Microbiology 2001, 147, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- Hoiczyk, E.; Hansel, A. Cyanobacterial cell walls: News from an unusual prokaryotic envelope. J. Bacteriol. 2000, 182, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Domozych, D.S. Wiley Online Library: Algal Cell Walls. 2011. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9780470015902.a0000315.pub3 (accessed on 24 July 2018).
- Nicolai, E.; Preston, R. Cell-wall studies in the Chlorophyceae. I. A general survey of submicroscopic structure in filamentous species. Proc. R. Soc. Lond. Ser. B 1952, 140, 244–274. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Demaree, B.; Ahmed, N.; Abate, A.R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 2017, 35, 640. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Nishikawa, Y.; Kogawa, M.; Takeyama, H. Massively parallel whole genome amplification for single-cell sequencing using droplet microfluidics. Sci. Rep. 2017, 7, 5199. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Light, Y.K.; Meagher, R.J.; Singh, A.K. Digital droplet multiple displacement amplification (ddMDA) for whole genome sequencing of limited DNA samples. PLoS ONE 2016, 11, e0153699. [Google Scholar] [CrossRef] [PubMed]
- Neuman, K.C.; Chadd, E.H.; Liou, G.F.; Bergman, K.; Block, S.M. Characterization of photodamage to Escherichia coli in optical traps. Biophys. J. 1999, 77, 2856–2863. [Google Scholar] [CrossRef]
- Ericsson, M.; Hanstorp, D.; Hagberg, P.; Enger, J.; Nyström, T. Sorting out bacterial viability with optical tweezers. J. Bacteriol. 2000, 182, 5551–5555. [Google Scholar] [CrossRef] [PubMed]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. JoVE 2007, 6, 253. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadeh, M.; Sadeghizadeh, M.; Najafi, F.; Arab, S.; Mobasheri, H. Impact of heat shock step on bacterial transformation efficiency. Mol. Bio. Res. Commun. 2016, 5, 257. [Google Scholar]
- Singh, M.; Yadav, A.; Ma, X.; Amoah, E. Plasmid DNA transformation in Escherichia Coli: Effect of heat shock temperature, duration, and cold incubation of CaCl2 treated cells. Int. J. Biotechnol. Biochem. 2010, 6, 561–568. [Google Scholar]
- Membrillo-Hernández, J.; La Mora, A.N.D.; Rio-Albrechtsen, D.; Camacho-Carranza, R.; Gomez-Eichelmann, M.C. Thermally-induced cell lysis in Escherichia coli K12. J. Basic Microbiol. 1995, 35, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Karni, M.; Zidon, D.; Polak, P.; Zalevsky, Z.; Shefi, O. Thermal degradation of DNA. DNA Cell Biol. 2013, 32, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, G.; Bhyravabhotla, J. A phenomenological model for predicting melting temperatures of DNA sequences. PLoS ONE 2010, 5, e12433. [Google Scholar] [CrossRef] [PubMed]
- Röder, B.; Frühwirth, K.; Vogl, C.; Wagner, M.; Rossmanith, P. Impact of long-term storage on stability of standard DNA for nucleic acid-based methods. J. Clin. Microbiol. 2010, 48, 4260–4262. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, S.G.; De Vecchi, E.; Pagani, C.; Zambelli, A.; Di Gregorio, A.; Bosisio, E.; Vanelli, P.; Scrofani, R.; Gismondo, M.R.; Cagnoni, G. Use of Dithiothreitol to Dislodge Bacteria From the Biofilm on an Aortic Valve in the Operating Theatre: A Case of Infective Endocarditis Caused by Staphylococcus aureus and Proteus mirabilis. Ann. Thorac. Surg. 2016, 102, e357–e359. [Google Scholar] [CrossRef] [PubMed]
- Kendoff, D.; Morgan-Jones, R.; Haddad, F.S. Periprosthetic Joint Infections: Changing Paradigms; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Drago, L.; Signori, V.; De Vecchi, E.; Vassena, C.; Palazzi, E.; Cappelletti, L.; Romanò, D.; Romanò, C.L. Use of dithiothreitol to improve the diagnosis of prosthetic joint infections. J. Orthop. Res. 2013, 31, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Hoiczyk, E.; Baumeister, W. Envelope structure of four gliding filamentous cyanobacteria. J. Bacteriol. 1995, 177, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, U.; Drews, G.; Weckesser, J. Primary structure of the peptidoglycan from the unicellular cyanobacterium Synechocystis sp. strain PCC 6714. J. Bacteriol. 1983, 154, 471–478. [Google Scholar] [PubMed]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef] [PubMed]
- Goodman, H.; Pollock, J.J.; Iacono, V.J.; Wong, W.; Shockman, G.D. Peptidoglycan loss during hen egg white lysozyme-inorganic salt lysis of Streptococcus mutans. J. Bacteriol. 1981, 146, 755–763. [Google Scholar] [PubMed]
- Stano, N.M.; Patel, S.S. T7 lysozyme represses T7 RNA polymerase transcription by destabilizing the open complex during initiation. J. Biol. Chem. 2004, 279, 16136–16143. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Schulze-Makuch, D.; De Vera, J.-P.; Cockell, C.; Leya, T.; Baqué, M.; Walther-Antonio, M. The Development of an Effective Bacterial Single-Cell Lysis Method Suitable for Whole Genome Amplification in Microfluidic Platforms. Micromachines 2018, 9, 367. https://doi.org/10.3390/mi9080367
Liu Y, Schulze-Makuch D, De Vera J-P, Cockell C, Leya T, Baqué M, Walther-Antonio M. The Development of an Effective Bacterial Single-Cell Lysis Method Suitable for Whole Genome Amplification in Microfluidic Platforms. Micromachines. 2018; 9(8):367. https://doi.org/10.3390/mi9080367
Chicago/Turabian StyleLiu, Yuguang, Dirk Schulze-Makuch, Jean-Pierre De Vera, Charles Cockell, Thomas Leya, Mickael Baqué, and Marina Walther-Antonio. 2018. "The Development of an Effective Bacterial Single-Cell Lysis Method Suitable for Whole Genome Amplification in Microfluidic Platforms" Micromachines 9, no. 8: 367. https://doi.org/10.3390/mi9080367
APA StyleLiu, Y., Schulze-Makuch, D., De Vera, J.-P., Cockell, C., Leya, T., Baqué, M., & Walther-Antonio, M. (2018). The Development of an Effective Bacterial Single-Cell Lysis Method Suitable for Whole Genome Amplification in Microfluidic Platforms. Micromachines, 9(8), 367. https://doi.org/10.3390/mi9080367