High-Precision Solvent Vapor Annealing for Block Copolymer Thin Films


Abstract
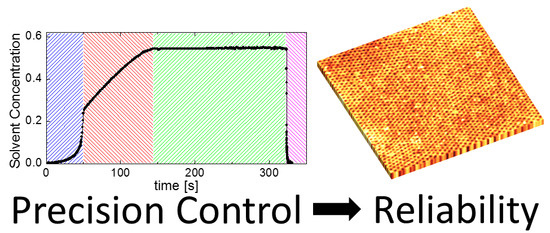
:
1. Introduction
2. Materials and Methods
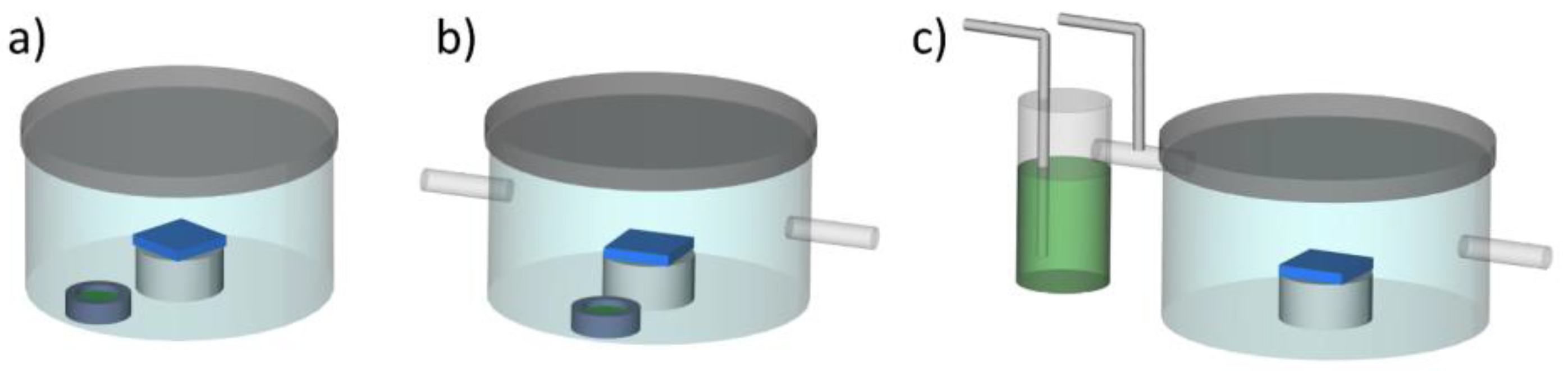
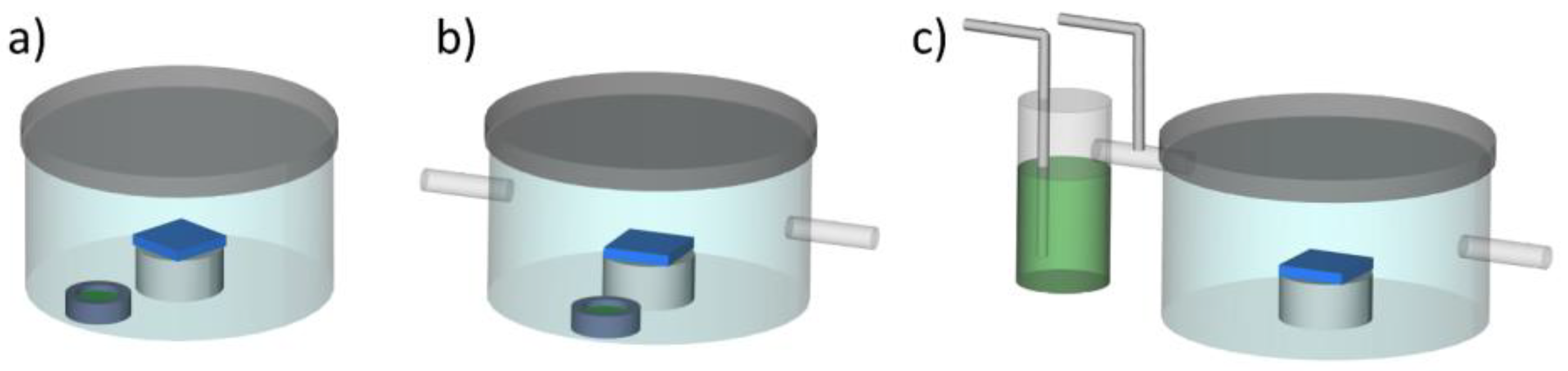
2.1. Solvent Vapor Annealing
2.2. Synthesis of Poly(Styrene)-Block-Poly(Lactide)
2.3. Thin Film Preparation
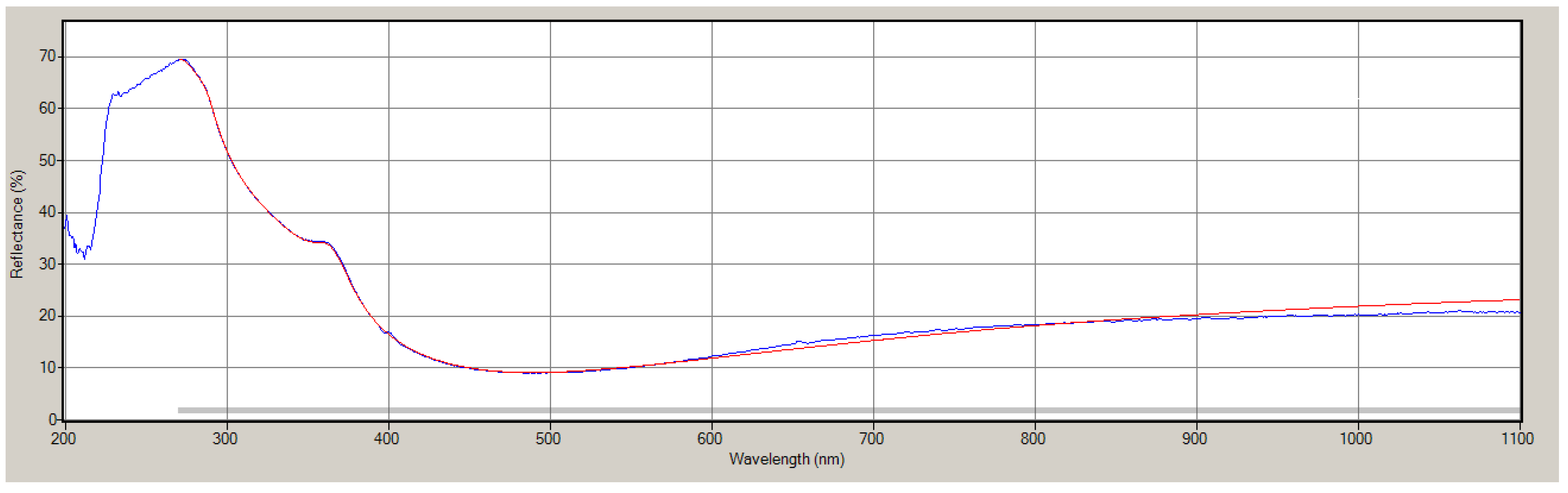
2.4. Measuring Film Thickness
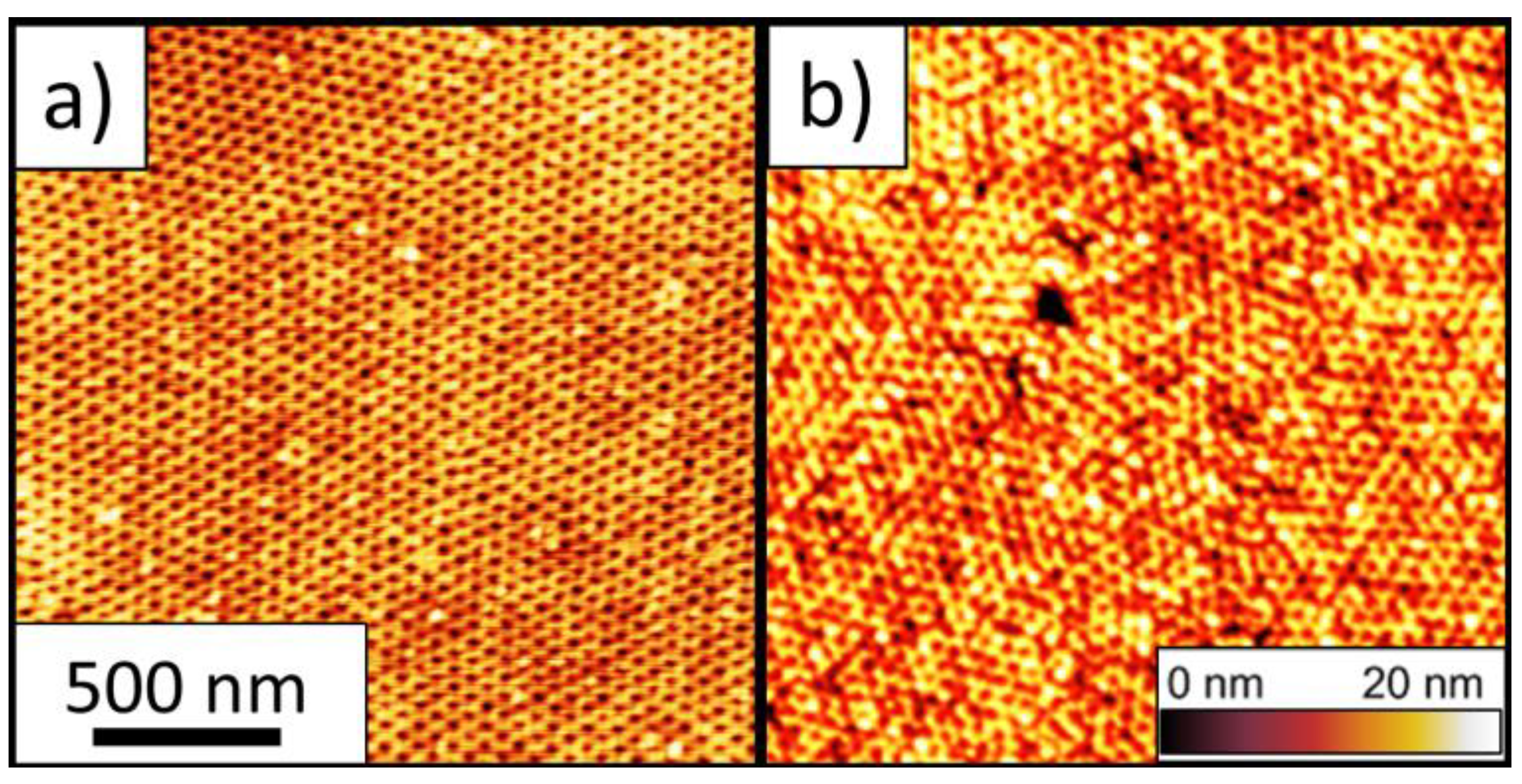
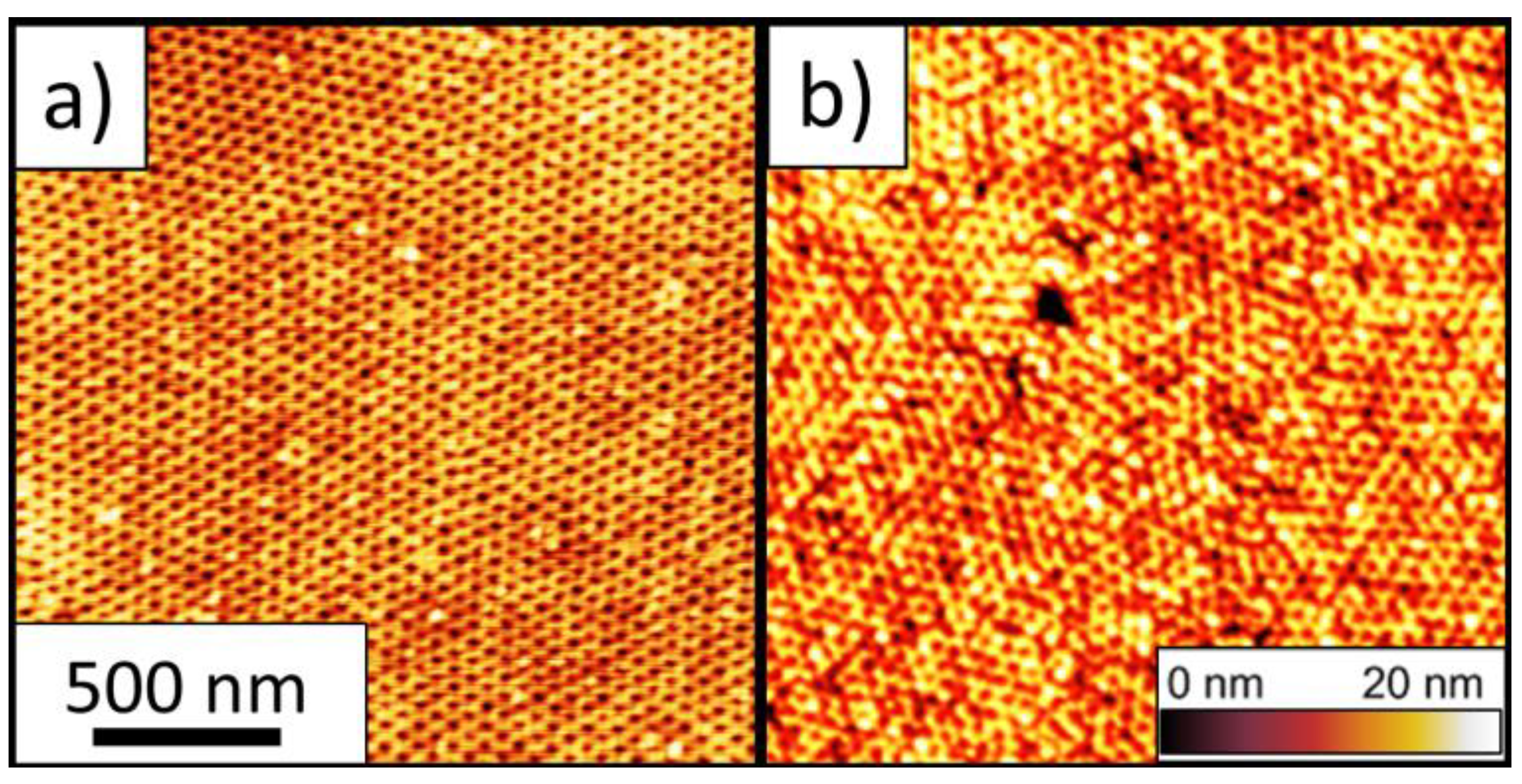
2.5. Atomic Force Microscopy (AFM)
3. Results
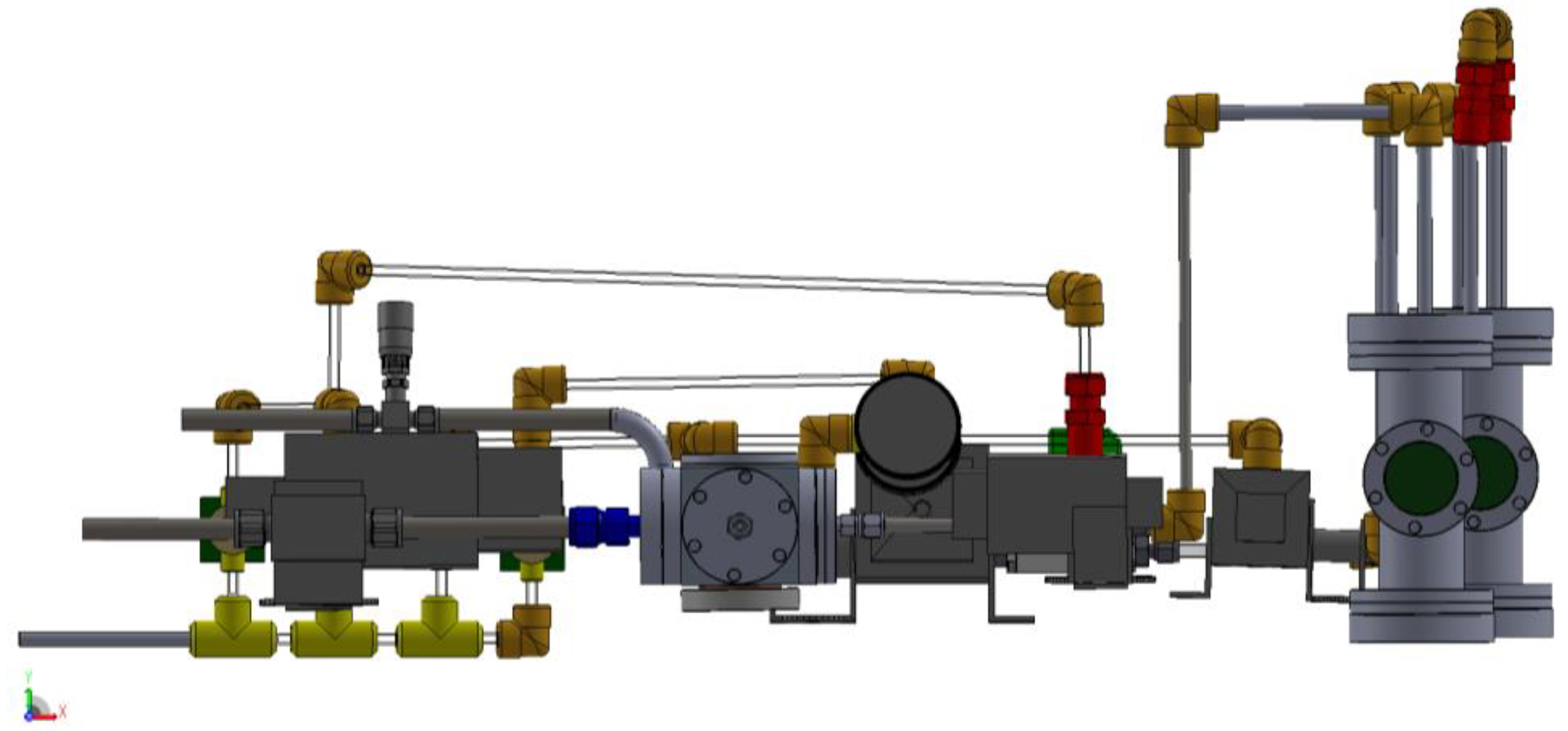
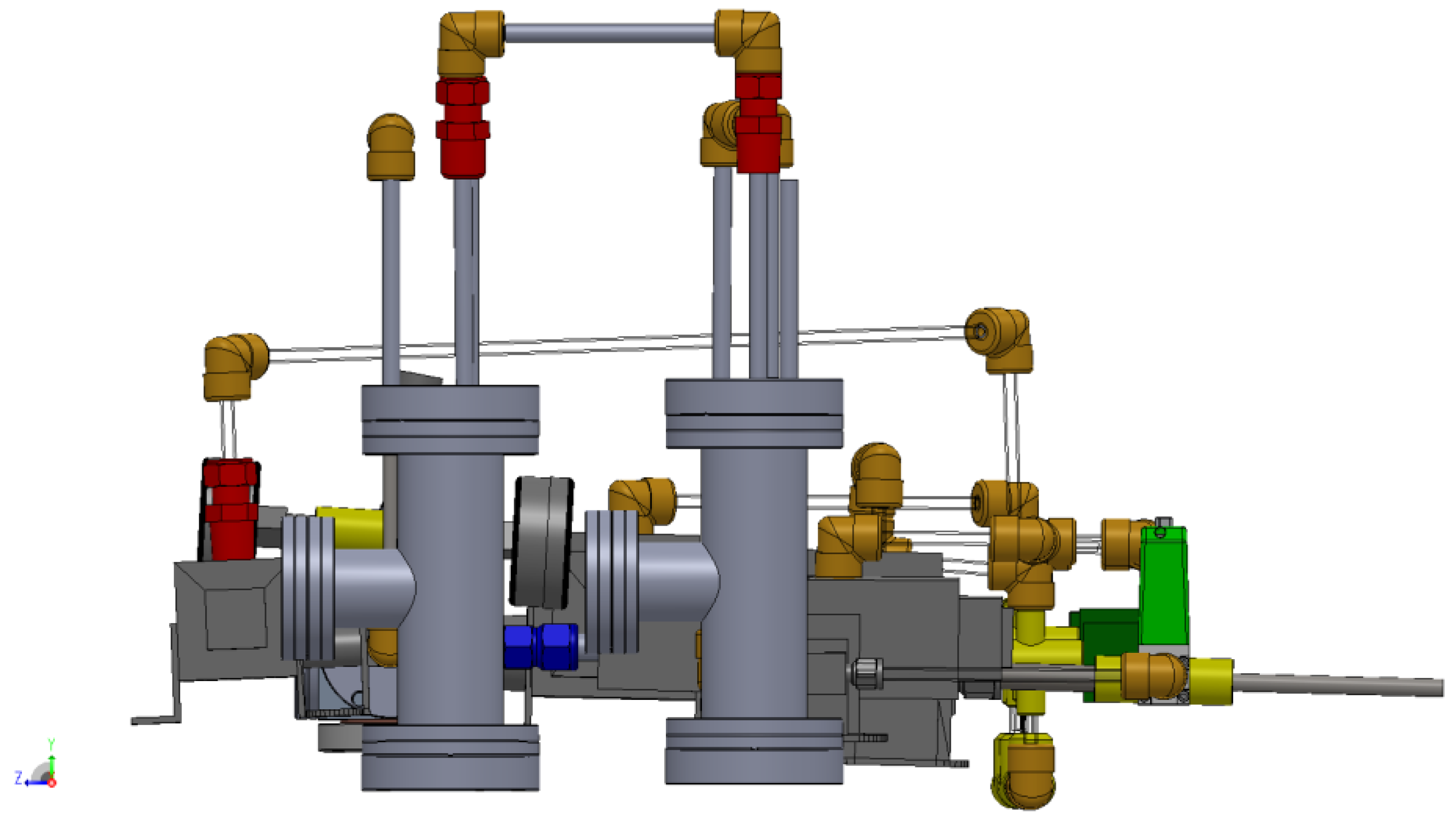
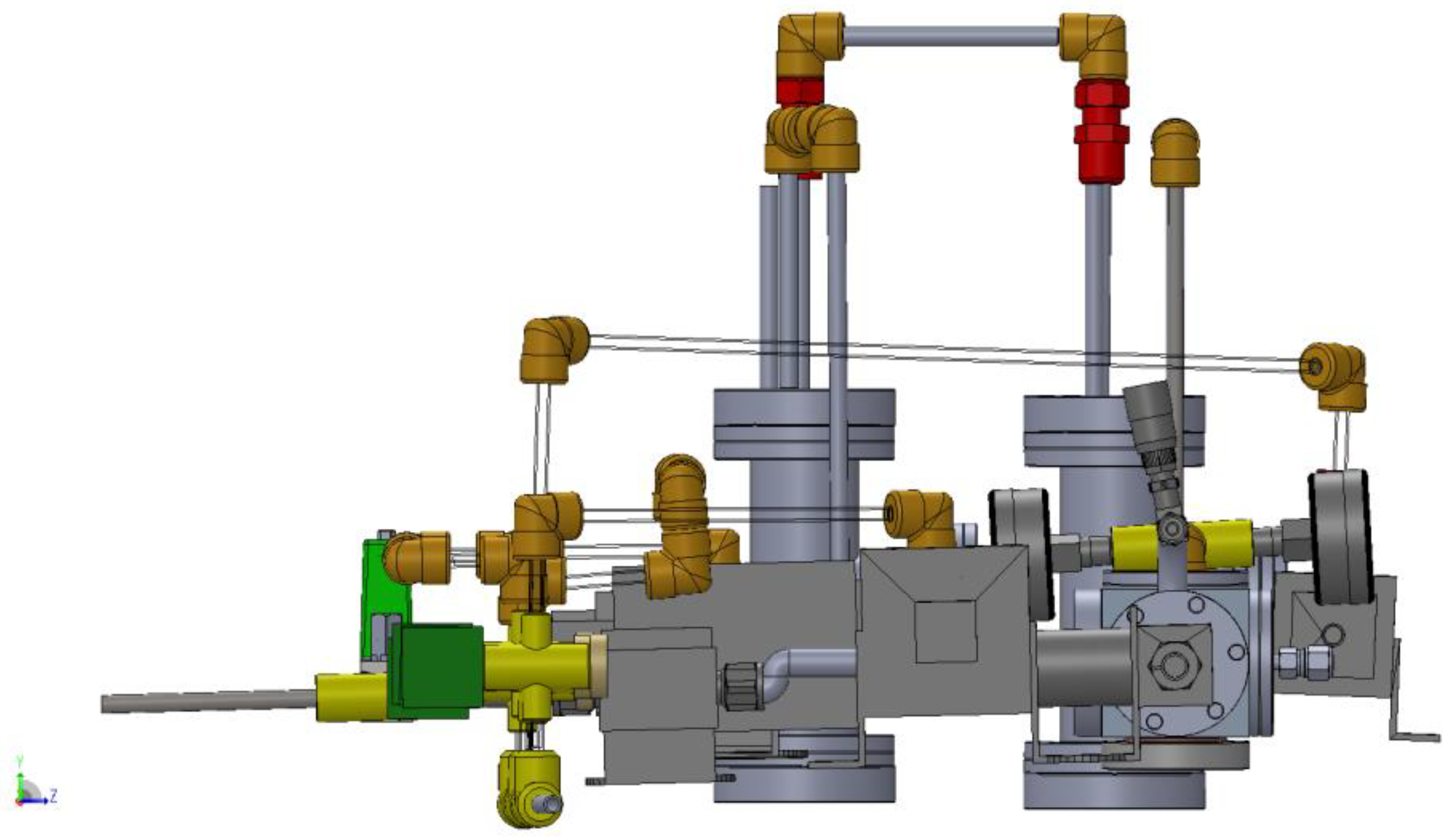
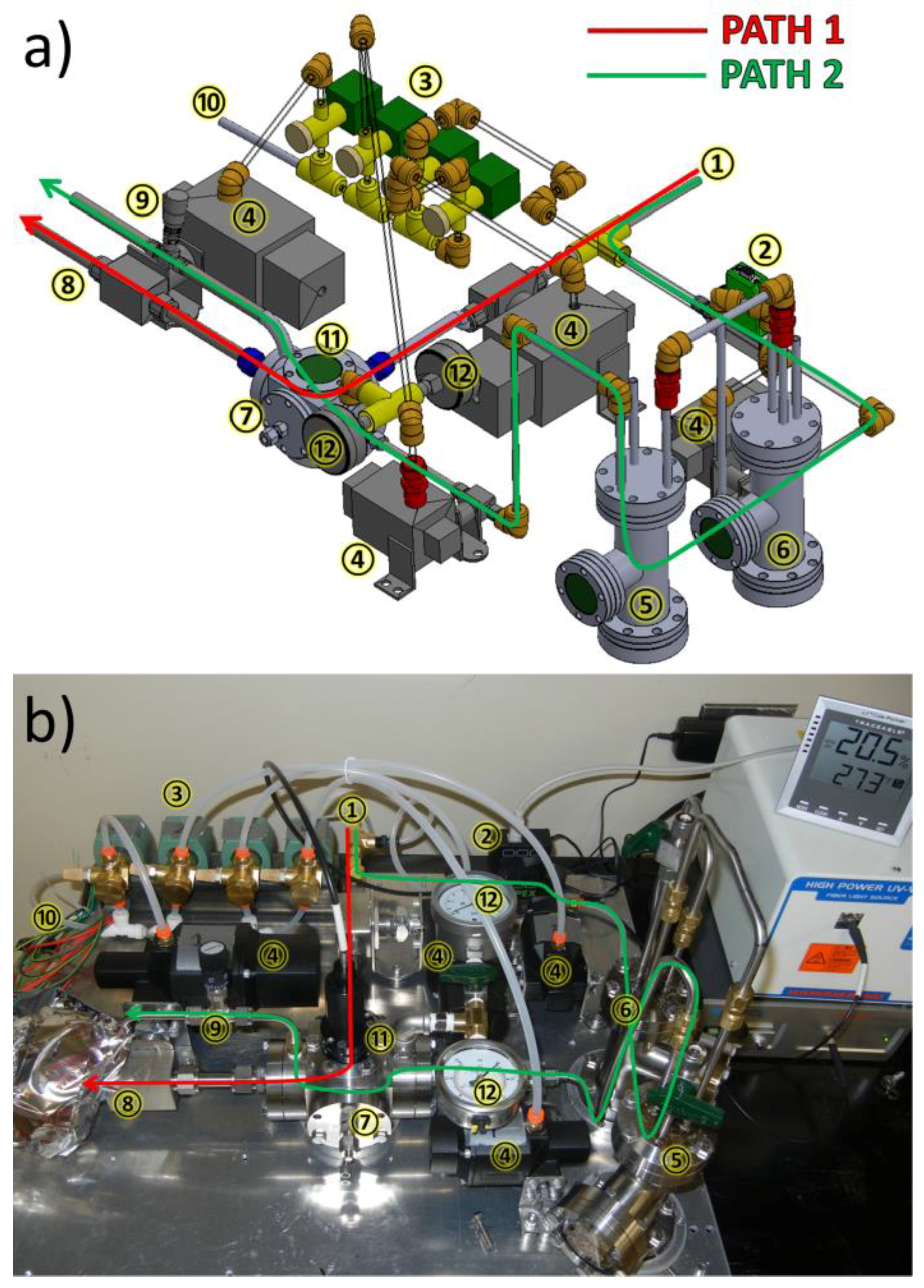
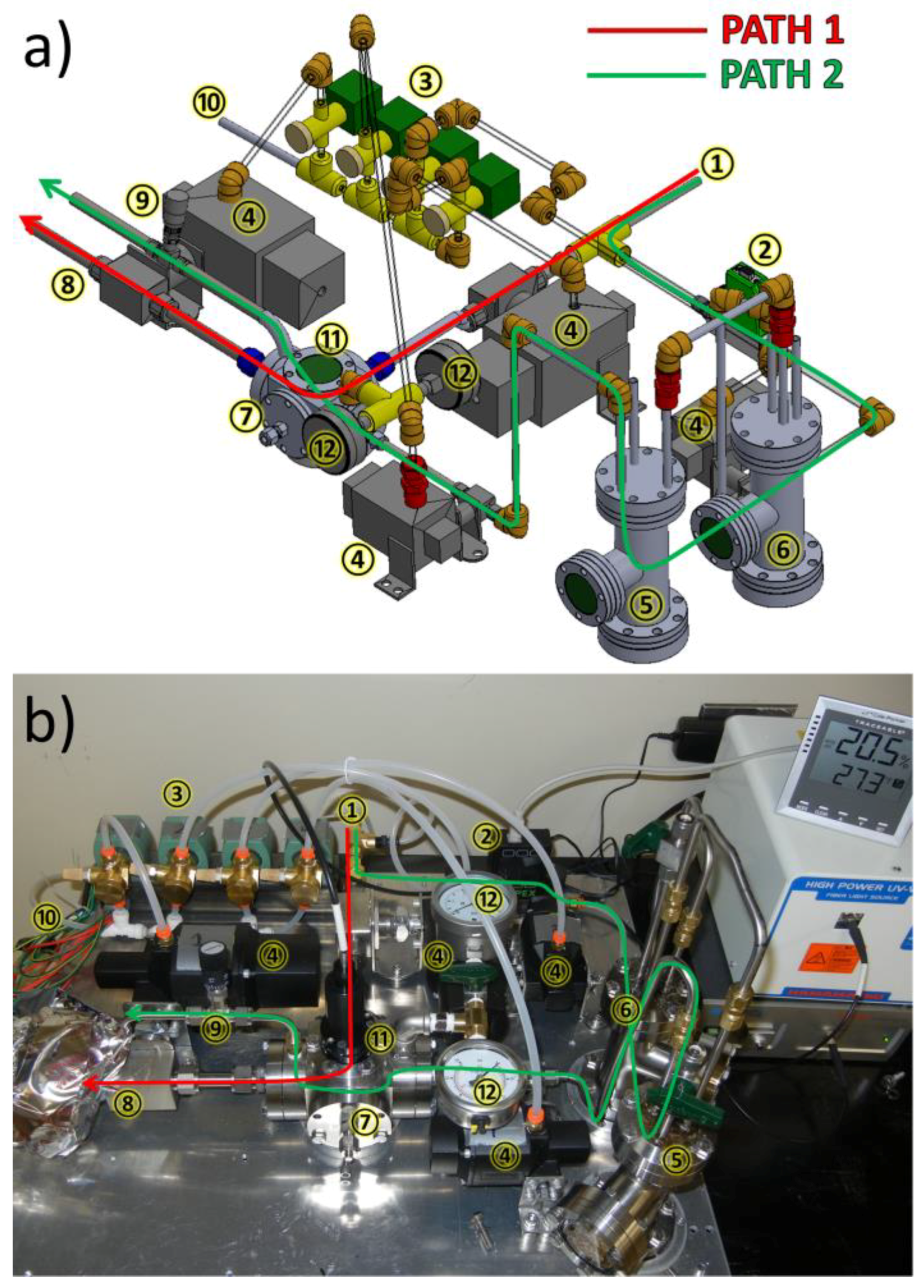
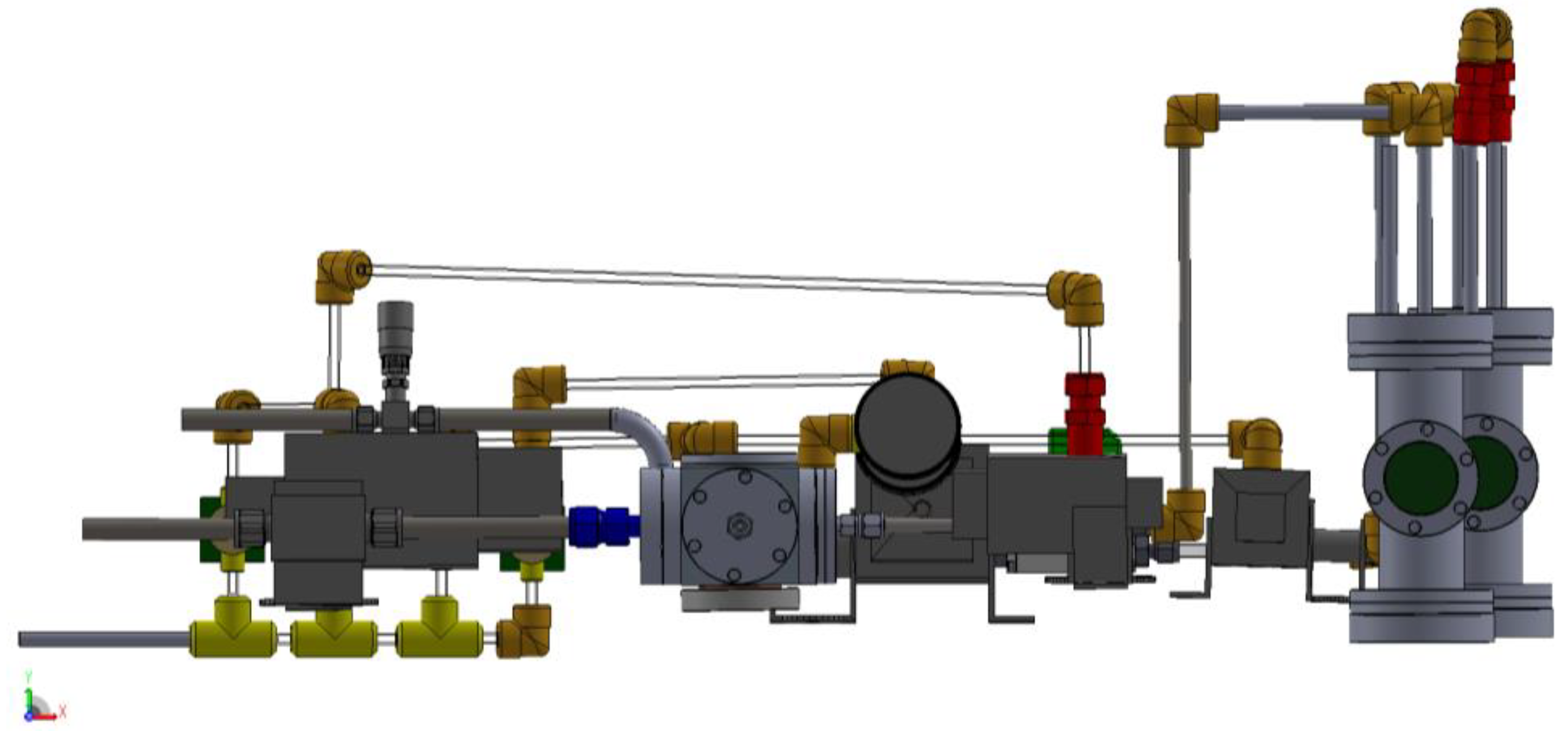
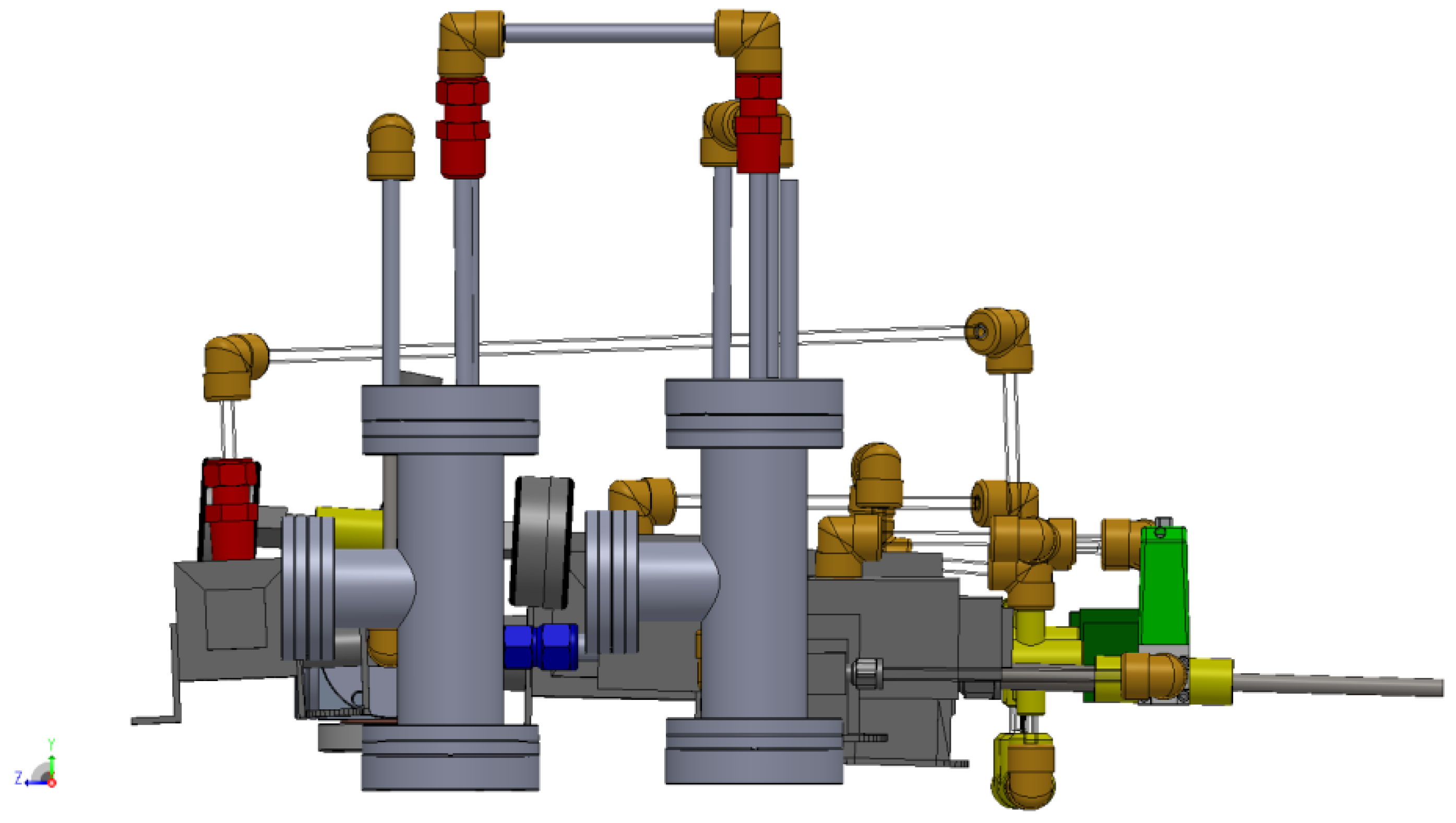
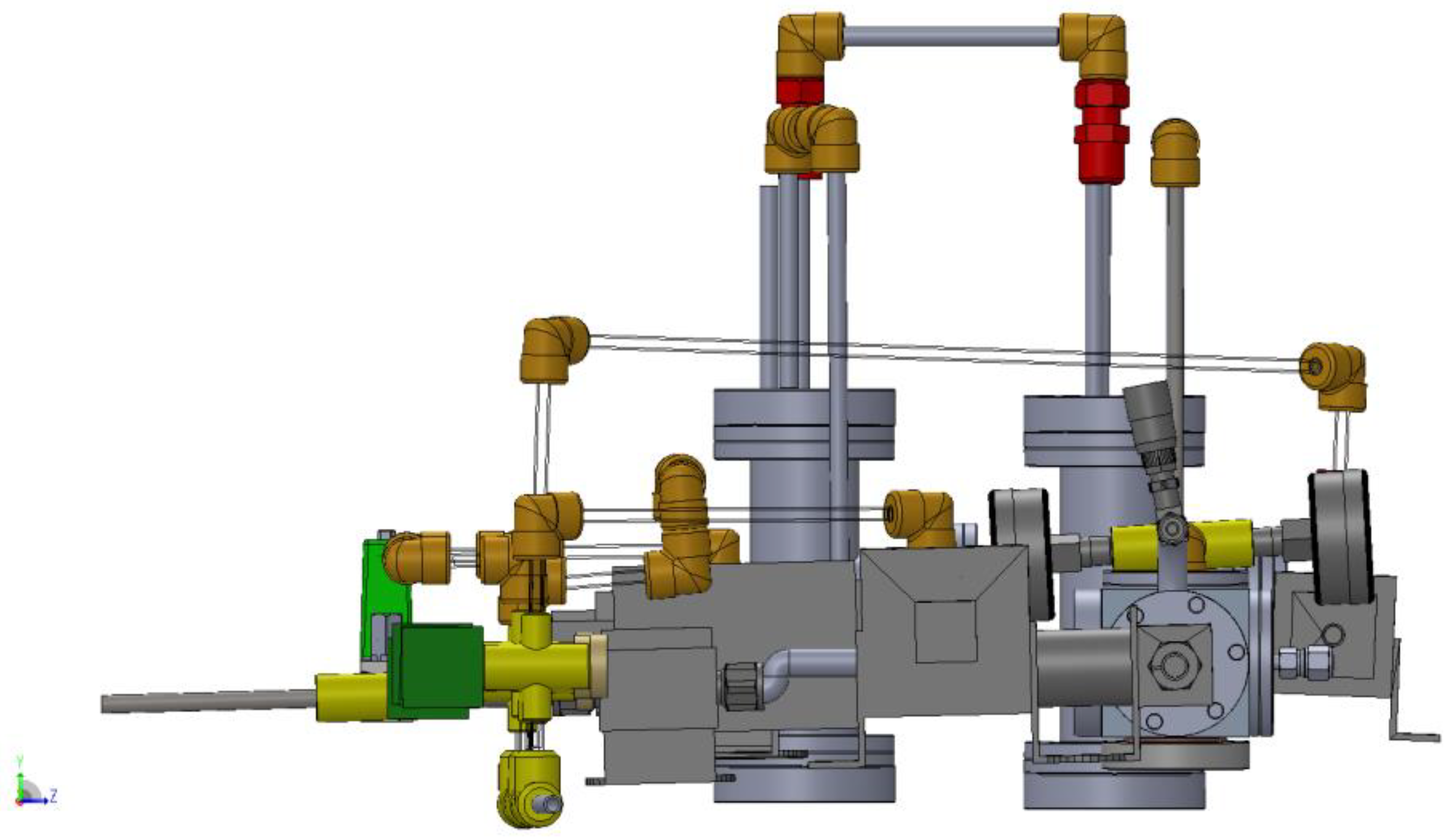
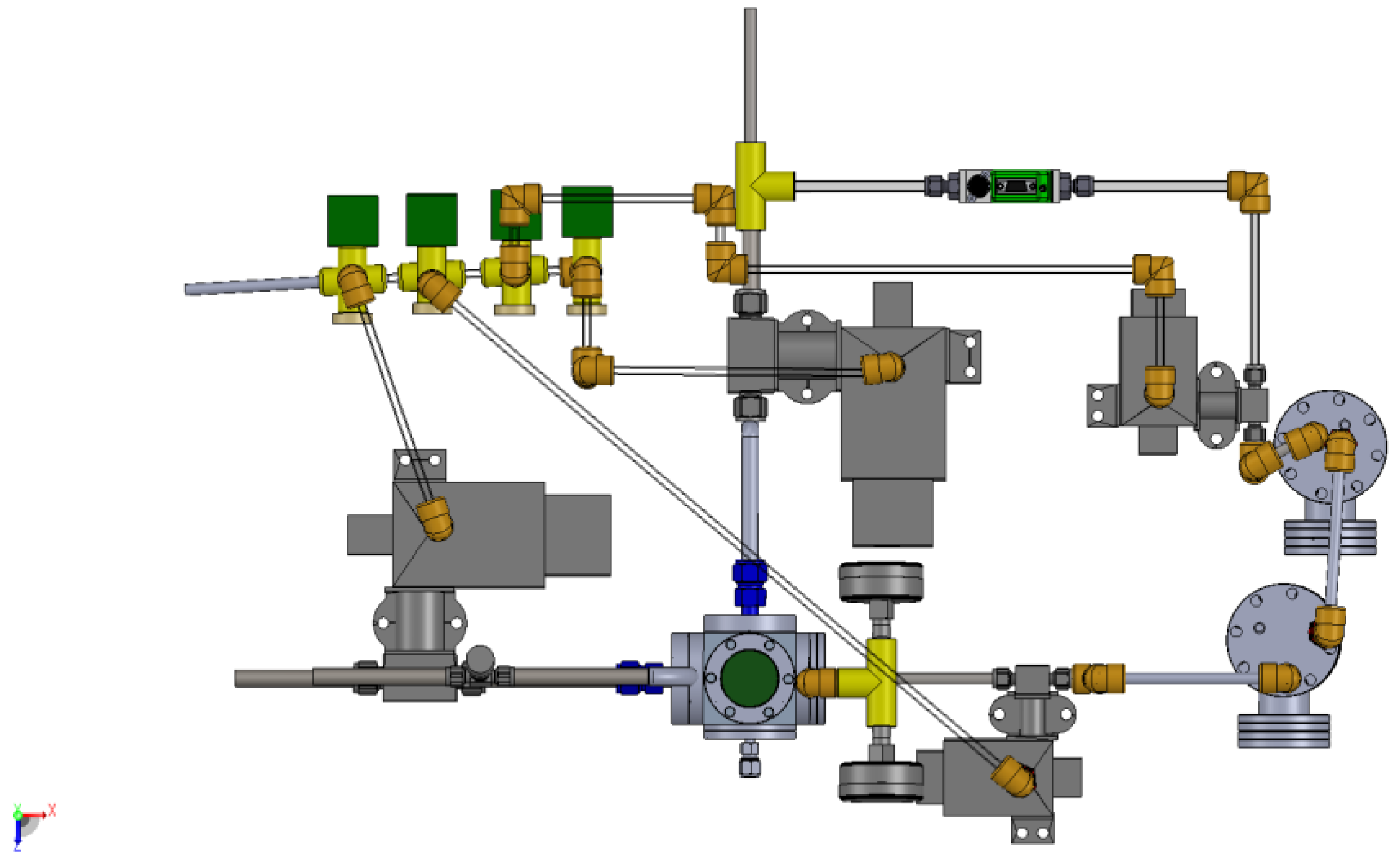
3.1. Design of a Purpose-Built Solvent Vapor Annealing Chamber
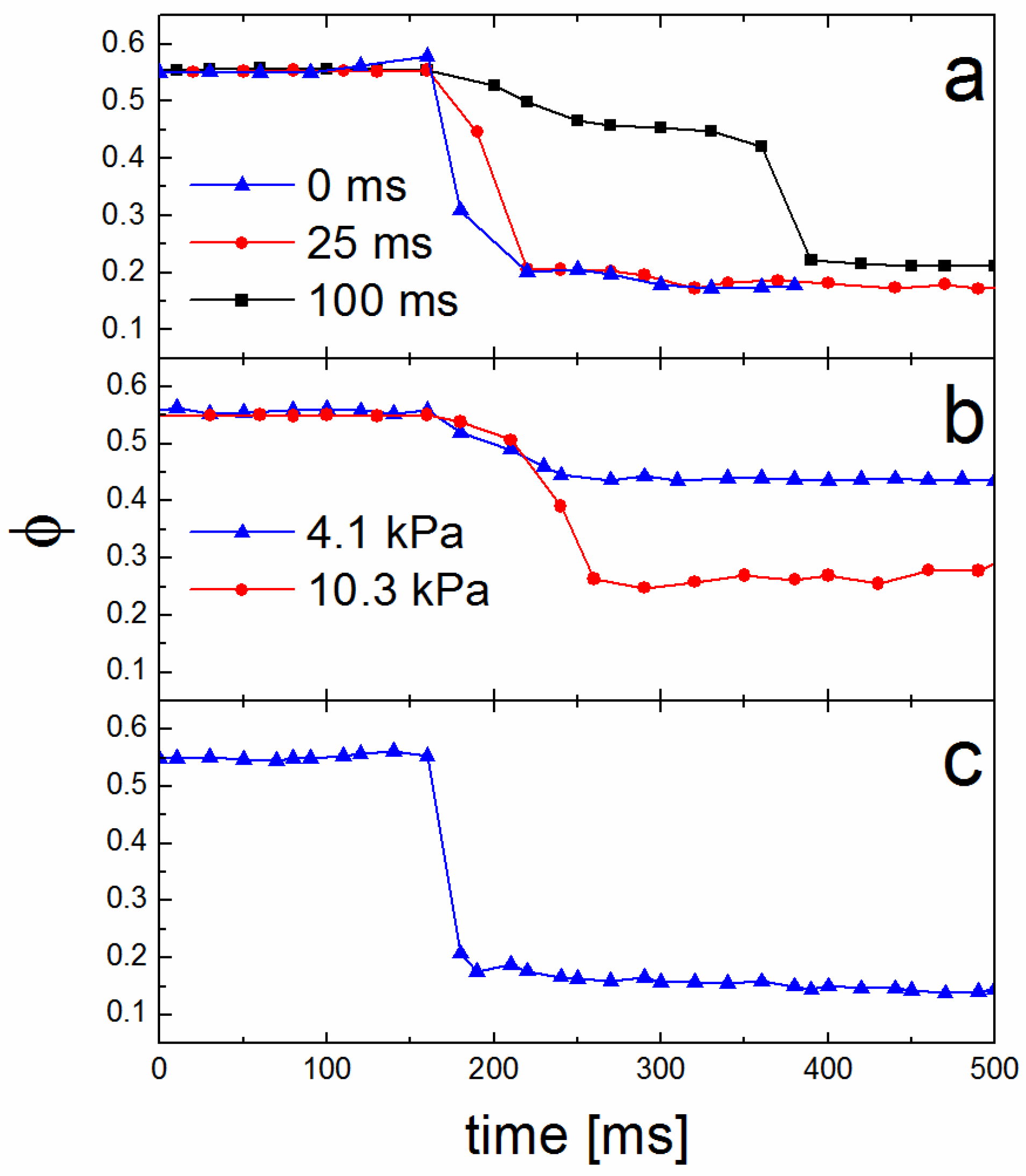
3.2. Theory of Operation
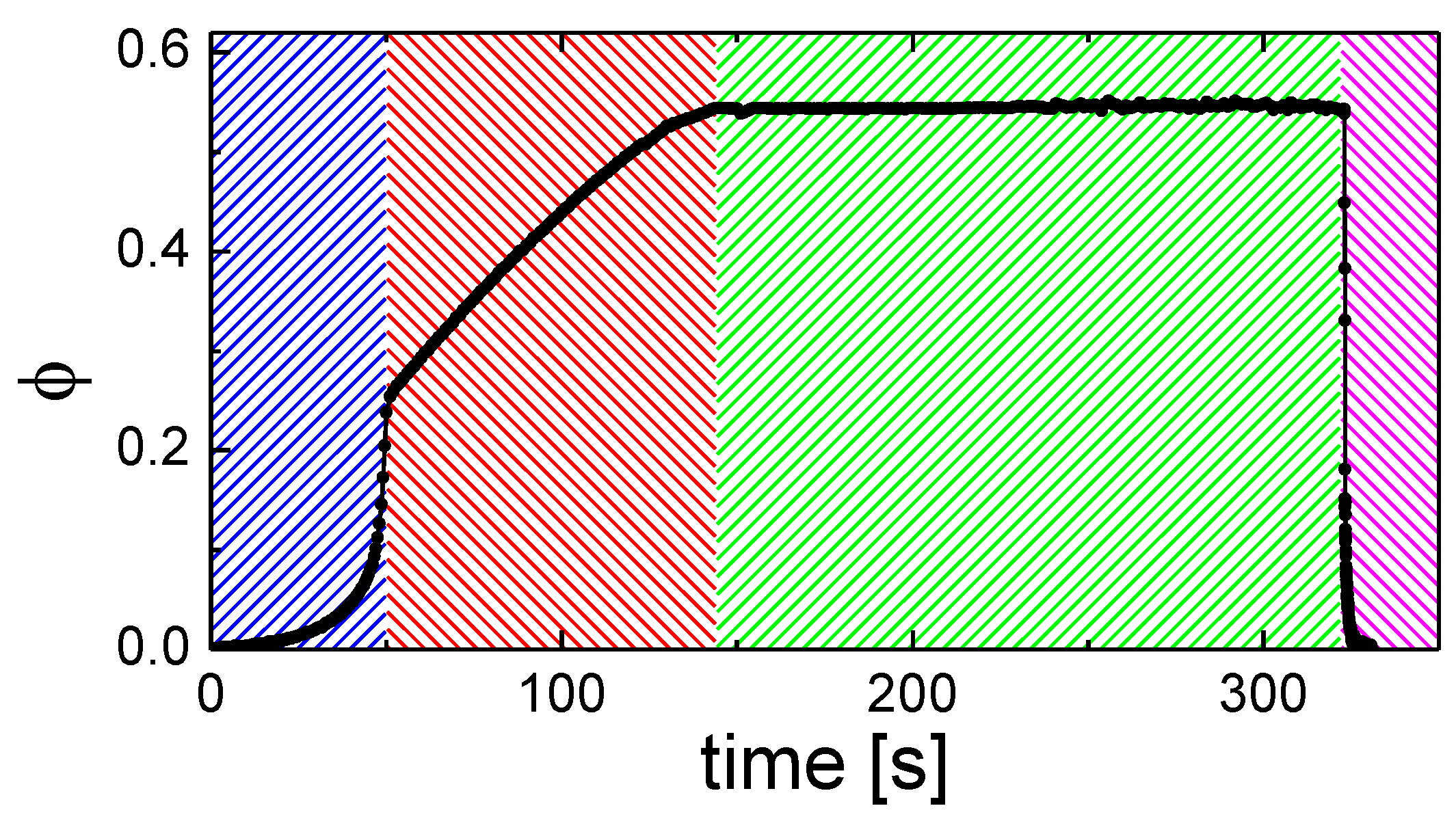
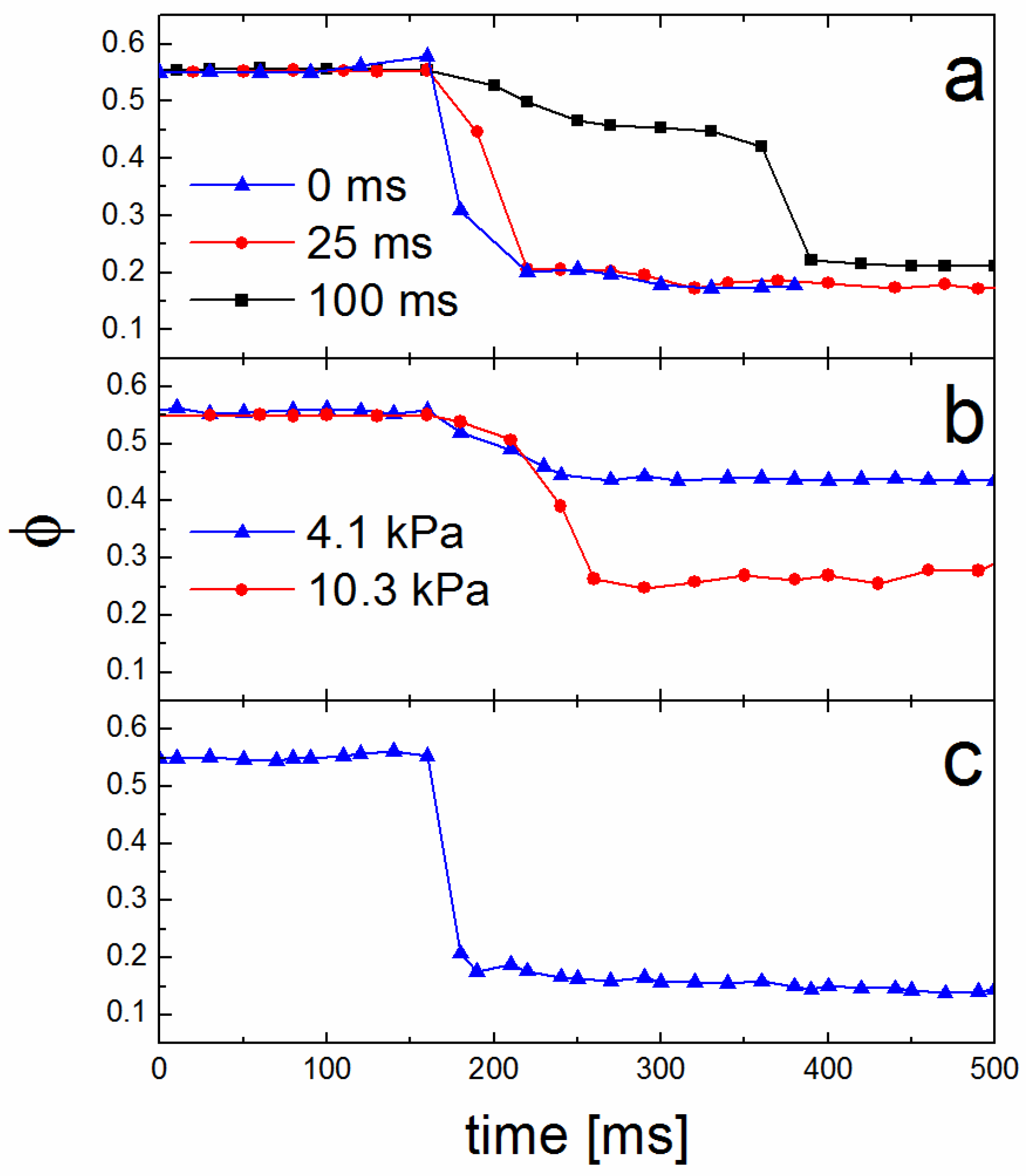
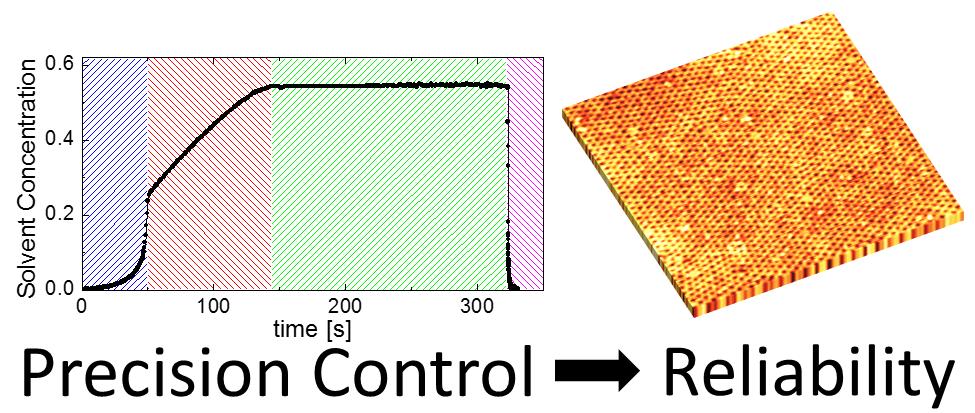
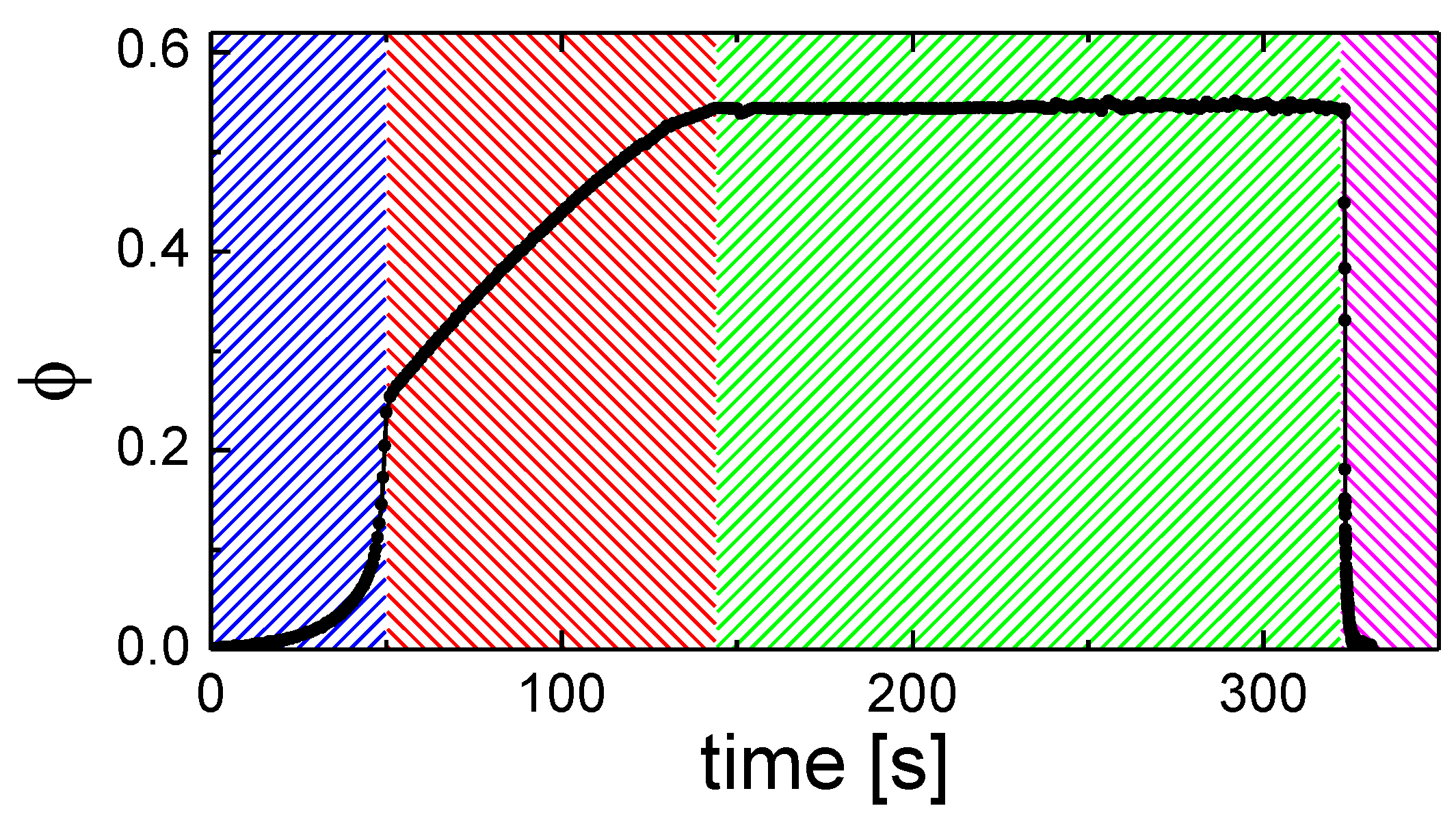
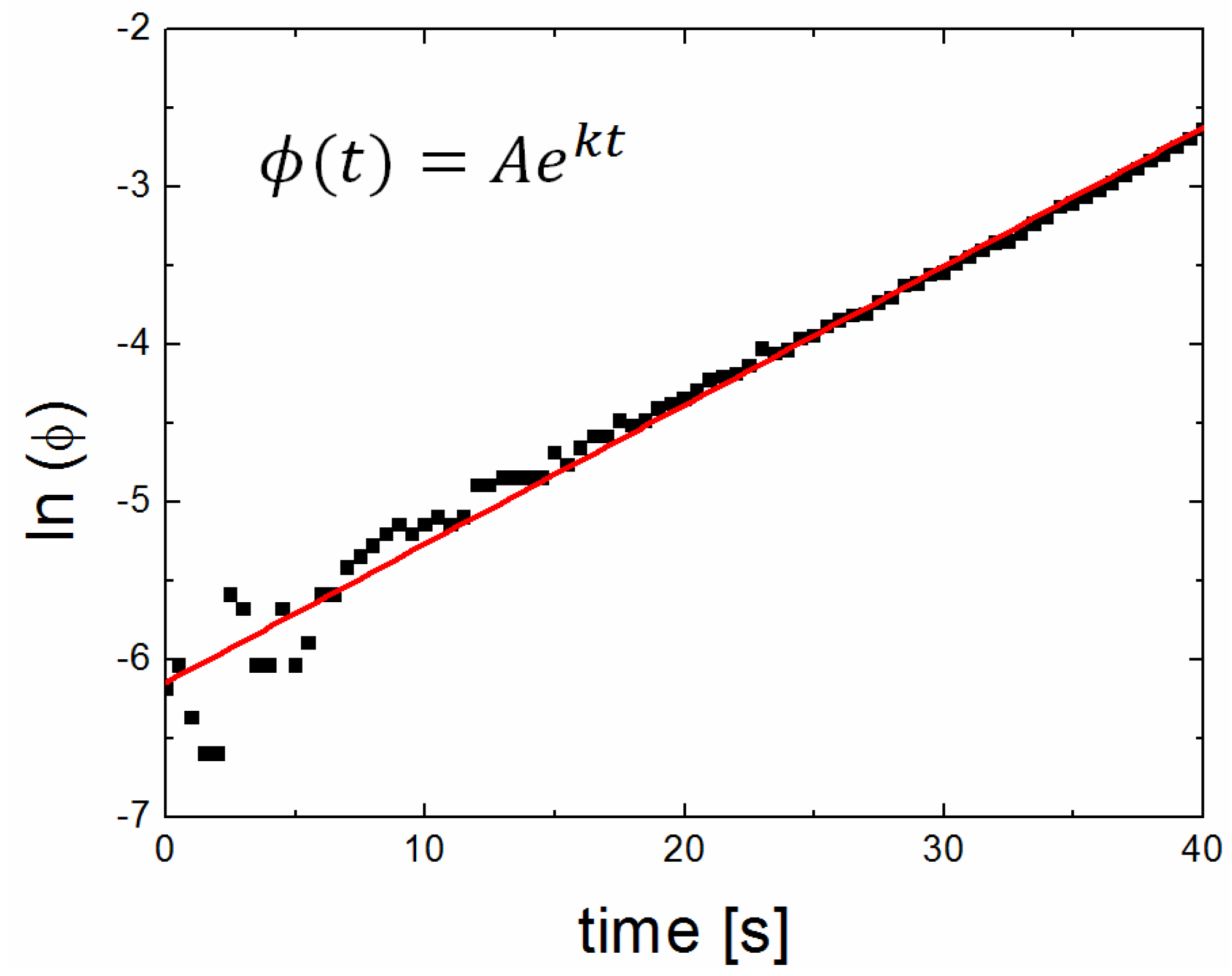
3.3. Solvent Evaporation
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A
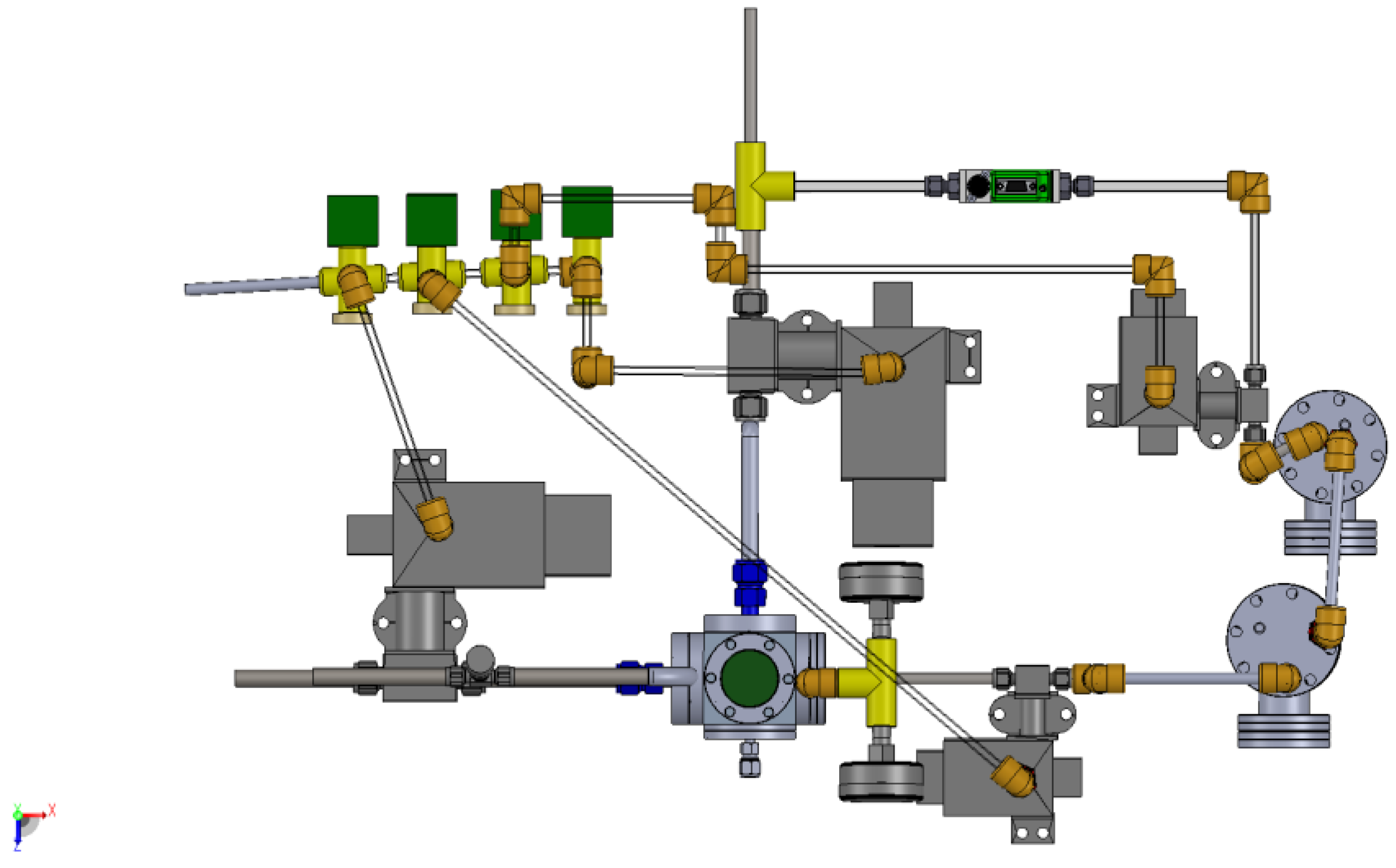




Appendix B
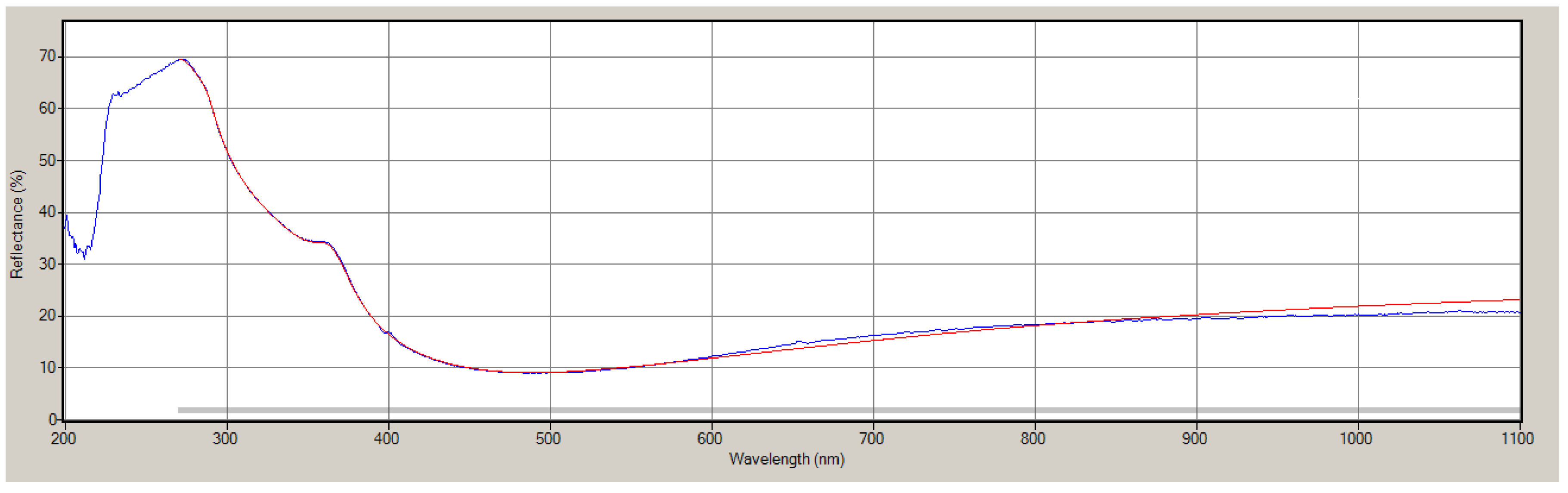
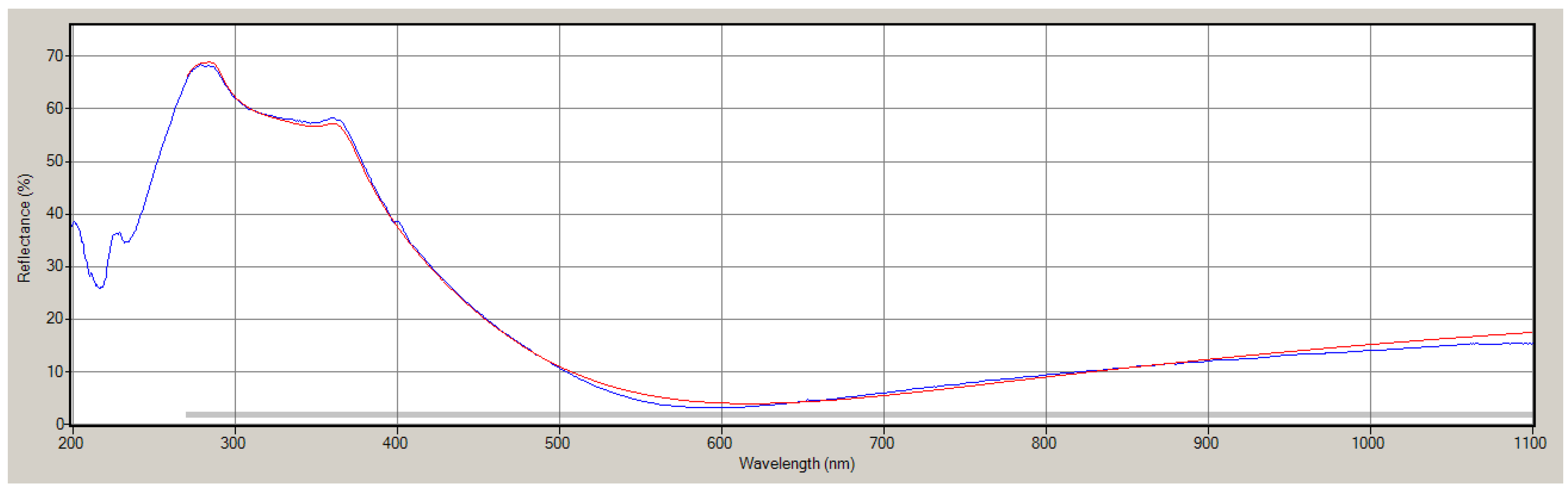
References
- Terris, B.D.; Thomson, T. Nanofabricated and self-assembled magnetic structures as data storage media. J. Phys. D Appl. Phys. 2005, 38, R199–R222. [Google Scholar] [CrossRef]
- Borah, D.; Shaw, M.T.; Rasappa, S.; Farrell, R.A.; O’Mahony, C.; Faulkner, C.M.; Bosea, M.; Gleeson, P.; Holmes, J.D.; Morris, M.A. Plasma etch technologies for the development of ultra-small feature size transistor devices. J. Phys. D Appl. Phys. 2011, 44, 174012. [Google Scholar] [CrossRef]
- Luo, Y.; Montarnal, D.; Kim, S.; Shi, W.; Barteau, K.P.; Pester, C.W.; Hustad, P.D.; Christianson, M.D.; Fredrickson, G.H.; Kramer, E.J.; et al. Poly(dimethylsiloxane-b-methyl methacrylate): A Promising Candidate for Sub-10 nm Patterning. Macromolecules 2015, 48, 3422–3430. [Google Scholar] [CrossRef]
- Bang, J.; Jeong, U.; Ryu, D. Block copolymer nanolithography: Translation of molecular level control to nanoscale patterns. Adv. Mater. 2009, 21, 4769–4792. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.M.; Maher, M.J.; Janes, D.W.; Ellison, C.J.; Willson, C.G. Block copolymer lithography. Macromolecules 2014, 47, 2–12. [Google Scholar] [CrossRef]
- Hamley, I.W. Ordering in thin films of block copolymers: Fundamentals to potential applications. Prog. Polym. Sci. 2009, 34, 1161–1210. [Google Scholar] [CrossRef]
- Black, C.T.; Ruiz, R.; Breyta, G.; Cheng, J.Y.; Colburn, M.E.; Guarini, K.W.; Kim, H.-C.; Zhang, Y. Polymer self assembly in semiconductor microelectronics. IBM J. Res. Dev. 2007, 51, 605–633. [Google Scholar] [CrossRef]
- Kim, H.-C.; Park, S.-M.; Hinsberg, W.D. Block copolymer based nanostructures: Materials, processes, and applications to electronics. Chem. Rev. 2010, 110, 146–177. [Google Scholar] [CrossRef] [PubMed]
- Stoykovich, M.P.; Nealey, P.F. Block copolymers and conventional lithography. Mater. Today 2006, 9, 20–29. [Google Scholar] [CrossRef]
- Guarini, K.W.; Black, C.T.; Yeung, S.H.I. Optimization of diblock copolymer thin film self assembly. Adv. Mater. 2002, 14, 1290–1294. [Google Scholar] [CrossRef]
- International Technology Roadmap for Semiconductors (ITRS). 2013. Available online: https://www.semiconductors.org/main/2013_international_technology_roadmap_for_semiconductors_itrs/ (accessed on 18 April 2018).
- Hamley, I.W. The Physics of Block Copolymers; Oxford University Press: Oxford, UK, 1998; ISBN 0198502184. [Google Scholar]
- Bates, F.S. Polymer-polymer phase behavior. Science 1991, 251, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, S.H.; Russell, T.P.; Satija, S.K.; Majkrzak, C.F. Neutron reflectivity studies of the surface-induced ordering of diblock copolymer films. Phys. Rev. Lett. 1989, 62, 1852–1855. [Google Scholar] [CrossRef] [PubMed]
- Menelle, A.; Russell, T.P.; Anastasiadis, S.H.; Satija, S.K.; Majkrzak, C.F. Ordering of thin diblock copolymer films. Phys. Rev. Lett. 1992, 68, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Fasolka, M.J.M.; Mayes, A.M.A. Block copolymer thin films: Physics and applications. Annu. Rev. Mater. Res. 2003, 31, 323–355. [Google Scholar] [CrossRef]
- Albert, J.N.L.; Epps, T.H., III. Self-assembly of block copolymer thin films. Mater. Today 2010, 13, 24–33. [Google Scholar] [CrossRef]
- Olayo-Valles, R.; Lund, M.S.; Leighton, C.; Hillmyer, M.A. Large area nanolithographic templates by selective etching of chemically stained block copolymer thin films. J. Mater. Chem. 2004, 14, 2729–2731. [Google Scholar] [CrossRef]
- Zhang, X.; Harris, K.D.; Wu, N.L.Y.; Murphy, J.N.; Buriak, J.M. Fast assembly of ordered block copolymer nanostructures through microwave annealing. ACS Nano 2010, 4, 7021–7029. [Google Scholar] [CrossRef] [PubMed]
- Morkved, T.L.; Lu, M.; Urbas, A.M.; Ehrichs, E.E.; Jaeger, H.M.; Mansky, P.; Russell, T.P. Local control of microdomain orientation in diblock copolymer thin films with electric fields. Science 1996, 273, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Thurn-Albrecht, T. Ultrahigh-density nanowire arrays grown in self-assembled diblock copolymer templates. Science 2000, 290, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Wang, R.F.; Olson, D.A.; Rodwogin, M.; Hillmyer, M.A.; Leighton, C. Spontaneous alignment of self-assembled ABC triblock terpolymers for large-area nanolithography. Appl. Phys. Lett. 2008, 93, 133112. [Google Scholar] [CrossRef]
- Kubo, T.; Parker, J.S.; Hillmyer, M.A.; Leighton, C. Characterization of pattern transfer in the fabrication of magnetic nanostructure arrays by block copolymer lithography. Appl. Phys. Lett. 2007, 90, 233113. [Google Scholar] [CrossRef]
- Ruiz, R.; Kang, H.; Detcheverry, F.A.; Dobisz, E.; Kercher, D.S.; Albrecht, T.R.; de Pablo, J.J.; Nealey, P.F. Density multiplication and improved lithography by directed block copolymer assembly. Science 2008, 321, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, C.; Vayer, M.; Morris, M.; Hillmyer, M.A. Solvent vapor annealing of block polymer thin films. Macromolecules 2013, 46, 5399–5415. [Google Scholar] [CrossRef]
- Kelly, J.Y.; Albert, J.N.L.; Howarter, J.A.; Kang, S.; Stafford, C.M.; Epps, T.H.; Fasolka, M.J. Investigation of thermally responsive block copolymer thin film morphologies using gradients. ACS Appl. Mater. Interfaces 2010, 2, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Di, Z.; Posselt, D.; Smilgies, D.-M.; Papadakis, C.M. Structural rearrangements in a lamellar diblock copolymer thin film during treatment with saturated solvent vapor. Macromolecules 2010, 43, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Henderson, K.J.; Jiang, Z.; Strzalka, J.W.; Wang, J.; Shull, K.R. Effects of reactive annealing on the structure of poly(methacrylic acid)-poly(methyl methacrylate) diblock copolymer thin films. Macromolecules 2011, 44, 6525–6531. [Google Scholar] [CrossRef]
- Sinturel, C.; Grosso, D.; Boudot, M.; Amenitsch, H.; Hillmyer, M.A.; Pineau, A.; Vayer, M. Structural transitions in asymmetric poly(styrene)-block-poly(lactide) thin films induced by solvent vapor exposure. ACS Appl. Mater. Interfaces 2014, 6, 12146–12152. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Gunkel, I.; Hexemer, A.; Gu, W.; Russell, T.P. An in situ grazing incidence X-ray scattering study of block copolymer thin films during solvent vapor annealing. Adv. Mater. 2014, 26, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nealey, P.F.; de Pablo, J.J.; Müller, M. Defect removal in the course of directed self-assembly is facilitated in the vicinity of the order-disorder transition. Phys. Rev. Lett. 2014, 113, 168301. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Gunkel, I.; Hexemer, A.; Russell, T.P. Controlling domain spacing and grain size in cylindrical block copolymer thin films by means of thermal and solvent vapor annealing. Macromolecules 2016, 49, 3373–3381. [Google Scholar] [CrossRef]
- Lodge, T.P.; Pudil, B.; Hanley, K.J. The full phase behavior for block copolymers in solvents of varying selectivity. Macromolecules 2002, 35, 4707–4717. [Google Scholar] [CrossRef]
- Baruth, A.; Seo, M.; Lin, C.H.; Walster, K.; Shankar, A.; Hillmyer, M.A.; Leighton, C. Optimization of long-range order in solvent vapor annealed poly(styrene)-block-poly(lactide) thin films for nanolithography. ACS Appl. Mater. Interfaces 2014, 16, 13770–13781. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Polisetty, S.; O’Brien, L.; Baruth, A.; Hillmyer, M.A.; Leighton, C.; Gladfelter, W.L.W.L. Size-tuned ZnO nanocrucible arrays for magnetic nanodot synthesis via atomic layer deposition-assisted block polymer lithography. ACS Nano 2015, 9, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.N.L.; Bogart, T.D.; Lewis, R.L.; Beers, K.L.; Fasolka, M.J.; Hutchison, J.B.; Vogt, B.D.; Epps, T.H. Gradient solvent vapor annealing of block copolymer thin films using a microfluidic mixing device. Nano Lett. 2011, 11, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Epps, T.H., III. Directed block copolymer thin film self-assembly: Emerging trends in nanopattern fabrication. Macromolecules 2013, 46, 7567–7579. [Google Scholar] [CrossRef]
- Baruth, A.; Rodwogin, M.D.; Shankar, A.; Erickson, M.J.; Hillmyer, M.A.; Leighton, C. Non-lift-off block copolymer lithography of 25 nm magnetic nanodot arrays. ACS Appl. Mater. Interfaces 2011, 3, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Berezkin, A.V.; Papadakis, C.M.; Potemkin, I.I. Vertical domain orientation in cylinder-forming diblock copolymer films upon solvent vapor annealing. Macromolecules 2016, 49, 415–424. [Google Scholar] [CrossRef]
- Berezkin, A.V.; Jung, F.; Posselt, D.; Smilgies, D.M.; Papadakis, C.M. Vertical vs. lateral macrophase separation in thin films of block copolymer mixtures: Computer simulations and GISAXS experiments. ACS Appl. Mater. Interfaces 2017, 9, 31291–31301. [Google Scholar] [CrossRef] [PubMed]
- Berezkin, A.V.; Jung, F.; Posselt, D.; Smilgies, D.-M.; Papadakis, C.M. In situ tracking of composition and morphology of a diblock copolymer film with GISAXS during exchange of solvent vapors at elevated temperatures. Adv. Funct. Mater. 2018, 28, 1706226. [Google Scholar] [CrossRef]
- Bang, J.; Kim, B.J.; Stein, G.E.; Russell, T.P.; Li, X.; Wang, J.; Kramer, E.J.; Hawker, C.J. Effect of humidity on the ordering of PEO-based copolymer thin films. Macromolecules 2007, 40, 7019–7025. [Google Scholar] [CrossRef]
- Knoll, A.; Magerle, R.; Krausch, G. Phase behavior in thin films of cylinder-forming ABA block copolymers: Experiments. J. Chem. Phys. 2004, 120, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Zettl, U.; Knoll, A.; Tsarkova, L. Effect of confinement on the mesoscale and macroscopic swelling of thin block copolymer films. Langmuir 2010, 26, 6610–6617. [Google Scholar] [CrossRef] [PubMed]
- Dehmel, R.; Dolan, J.A.; Gu, Y.; Wiesner, U.; Wilkinson, T.D.; Baumberg, J.J.; Steiner, U.; Wilts, B.D.; Gunkel, I. Optical imaging of large gyroid grains in block copolymer templates by confined crystallization. Macromolecules 2017, 50, 6255–6262. [Google Scholar] [CrossRef] [PubMed]
- Gotrik, K.W.; Hannon, A.F.; Son, J.G.; Keller, B.; Alexander-Katz, A.; Ross, C.A. Morphology control in block copolymer films using mixed solvent vapors. ACS Nano 2012, 6, 8052–8059. [Google Scholar] [CrossRef] [PubMed]
- Posselt, D.; Zhang, J.; Smilgies, D.-M.; Berezkin, A.V.; Potemkin, I.I.; Papadakis, C.M. Restructuring in block copolymer thin films: In situ GISAXS investigations during solvent vapor annealing. Prog. Polym. Sci. 2017, 66, 80–115. [Google Scholar] [CrossRef]
- Jung, Y.S.; Ross, C.A. Solvent vapor induced tunability of self assembled block copolymer patterns. Adv. Mater. 2009, 21, 2540–2545. [Google Scholar] [CrossRef]
- Bai, W.; Hannon, A.F.; Gotrik, K.W.; Choi, H.K.; Aissou, K.; Liontos, G.; Ntetsikas, K.; Alexander-katz, A.; Ross, C.A. Thin film morphologies of bulk-gyroid polystyrene-block- polydimethylsiloxane under solvent vapor annealing. Macromolecules 2014, 47, 6000–6008. [Google Scholar] [CrossRef]
- Di, Z.; Posselt, D.; Smilgies, D.-M.; Li, R.; Rauscher, M.; Potemkin, I.I.; Papadakis, C.M. Stepwise swelling of a thin film of lamellae-forming poly(styrene-b-butadiene) in cyclohexane vapor. Macromolecules 2012, 45, 5185–5195. [Google Scholar] [CrossRef]
- Son, J.G.; Gotrik, K.W.; Ross, C.A. High-aspect-ratio perpendicular orientation of PS-b-PDMS thin films under solvent annealing. ACS Macro Lett. 2012, 1, 1279–1284. [Google Scholar] [CrossRef]
- Albert, J.N.L.; Young, W.-S.; Lewis, R.L.; Bogart, T.D.; Smith, J.R.; Epps, T.H. Systematic study on the effect of solvent removal rate on the morphology of solvent vapor annealed ABA triblock copolymer thin films. ACS Nano 2012, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Yager, K.G.; Ross, C.A. In Situ Characterization of the Self-Assembly of a Polystyrene–polydimethylsiloxane block copolymer during solvent vapor annealing. Macromolecules 2015, 48, 8574–8584. [Google Scholar] [CrossRef]
- Müller-Buschbaum, P. GISAXS and GISANS as metrology technique for understanding the 3D morphology of block copolymer thin films. Eur. Polym. J. 2016. [Google Scholar] [CrossRef]
- Kim, S.H.; Misner, M.J.; Xu, T.; Kimura, M.; Russell, T.P. Highly oriented and ordered arrays from block copolymers via solvent evaporation. Adv. Mater. 2004, 16, 226–231. [Google Scholar] [CrossRef]
- Nandan, B.; Vyas, M.K.; Böhme, M.; Stamm, M. Composition-dependent morphological transitions and pathways in switching of fine structure in thin films of block copolymer supramolecular assemblies. Macromolecules 2010, 43, 2463–2473. [Google Scholar] [CrossRef]
- Paradiso, S.P.; Delaney, K.T.; García-Cervera, C.J.; Ceniceros, H.D.; Fredrickson, G.H. Block copolymer self assembly during rapid solvent evaporation: Insights into cylinder growth and stability. ACS Macro Lett. 2014, 3, 16–20. [Google Scholar] [CrossRef]
- Phillip, W.; Hillmyer, M.; Cussler, E. Cylinder orientation mechanism in block copolymer thin films upon solvent evaporation. Macromolecules 2010, 43, 7763–7770. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef] [PubMed]
- Cummins, C.; Mokarian-Tabari, P.; Holmes, J.D.; Morris, M.A. Selective etching of polylactic acid in poly(styrene)-block-poly(d,l)lactide diblock copolymer for nanoscale patterning. J. Appl. Polym. Sci. 2014, 131, 40798. [Google Scholar] [CrossRef]
- Zalusky, A.S.; Olayo-Valles, R.; Wolf, J.H.; Hillmyer, M.A. Ordered nanoporous polymers from polystyrene-polylactide block copolymers. J. Am. Chem. Soc. 2002, 124, 12761–12773. [Google Scholar] [CrossRef] [PubMed]
- Vayer, M.; Hillmyer, M.A.; Dirany, M.; Thevenin, G.; Erre, R.; Sinturel, C. Perpendicular orientation of cylindrical domains upon solvent annealing thin films of polystyrene-b-polylactide. Thin Solid Films 2010, 518, 3710–3715. [Google Scholar] [CrossRef]
- Miller, S.; Fanchini, G.; Lin, Y.-Y.; Li, C.; Chen, C.-W.; Su, W.-F.; Chhowalla, M. Investigation of nanoscale morphological changes in organic photovoltaics during solvent vapor annealing. J. Mater. Chem. 2008, 18, 306–312. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
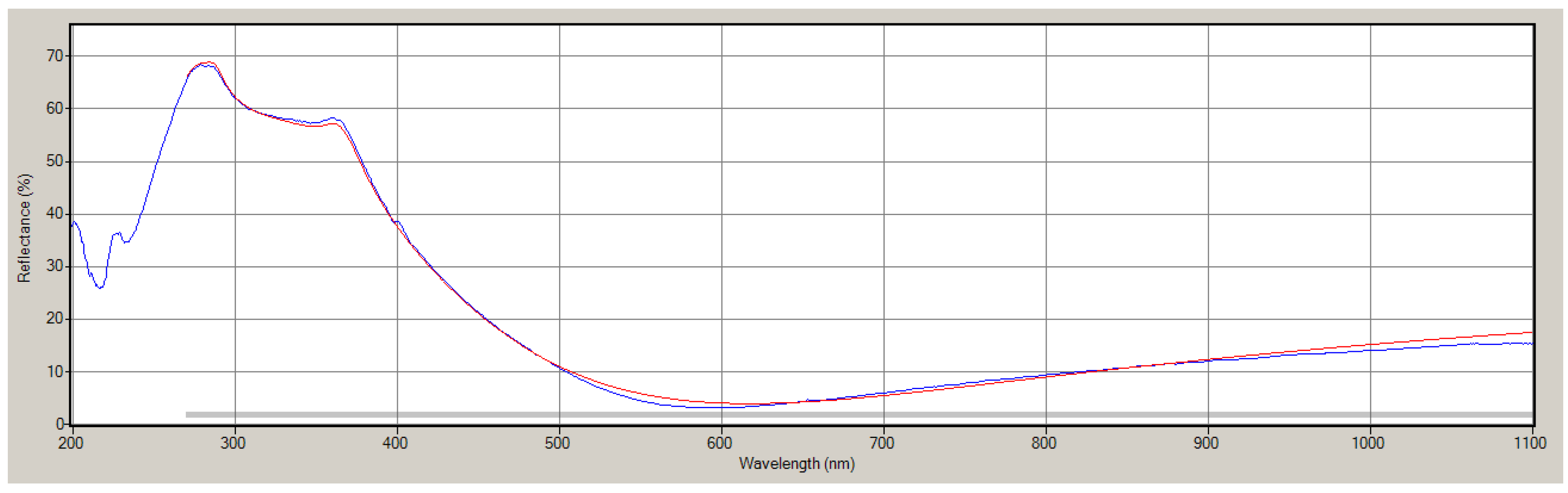{kind=link}
| Variable | Importance | Protocols in Design |
|---|---|---|
| Humidity | Water is a polar solvent, which will modify the solubility. For example, water is PLA selective | Samples are stored in 75 °C oven Samples are hot loaded into chamber Vacuum-grade, copper gaskets and Swagelok seals throughout chamber Molecular sieve-dried solvent (tetrahydrofuran (THF)) Actively purged (dry N2) sample cell Drierite Gas Purifier (−100 °C dewpoint) |
| Solvent Vapor Flow Rate | Flow rate is proportional to solvent uptake in film | Computer-controlled mass flow controller Low-flow metering outlet valve |
| Solvent Concentration | Solvent concentration in film during solvent vapor annealing (SVA) modifies mobility | In situ optical detection of solvent concentration, assuming proportional to film thickness, with 10–20 ms resolution |
| Solvent Evaporation Rate | Evaporation rate in linked to morphology alignment | Computer-controlled pneumatic valves Variable flow N2 purge line |
| Initial Thickness | Role of commensurate thickness on morphology | Films are all spun cast at a constant spin speed from the same solution concentration |
| Vapor Pressure | Vapor pressure during SVA modifies solvent uptake and evaporation | High and low pressure gauges Low-flow metering outlet valve can finely adjust vapor pressure |
| Solvent Selectivity | Solvent selectivity modifies surface energy and polymer-polymer interactions | THF is a relatively neutral solvent for PS and PLA. It has slight PS selectivity |
| Surface Selectivity | Substrate preparation modifies surface energy | HMDS-functionalized Si substrate surface promotes PS (majority block) adhesion |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, G.; Drapes, C.S.; Grant, M.A.; Gnabasik, R.; Wong, J.; Baruth, A. High-Precision Solvent Vapor Annealing for Block Copolymer Thin Films. Micromachines 2018, 9, 271. https://doi.org/10.3390/mi9060271
Nelson G, Drapes CS, Grant MA, Gnabasik R, Wong J, Baruth A. High-Precision Solvent Vapor Annealing for Block Copolymer Thin Films. Micromachines. 2018; 9(6):271. https://doi.org/10.3390/mi9060271
Chicago/Turabian StyleNelson, Gunnar, Chloe S. Drapes, Meagan A. Grant, Ryan Gnabasik, Jeffrey Wong, and Andrew Baruth. 2018. "High-Precision Solvent Vapor Annealing for Block Copolymer Thin Films" Micromachines 9, no. 6: 271. https://doi.org/10.3390/mi9060271
APA StyleNelson, G., Drapes, C. S., Grant, M. A., Gnabasik, R., Wong, J., & Baruth, A. (2018). High-Precision Solvent Vapor Annealing for Block Copolymer Thin Films. Micromachines, 9(6), 271. https://doi.org/10.3390/mi9060271