

A Computational Approach for Graphene Doped with N,P,B Structures as Possible Electrode Materials for Potassium Ion Batteries (PIBs): A DFT Investigation


Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
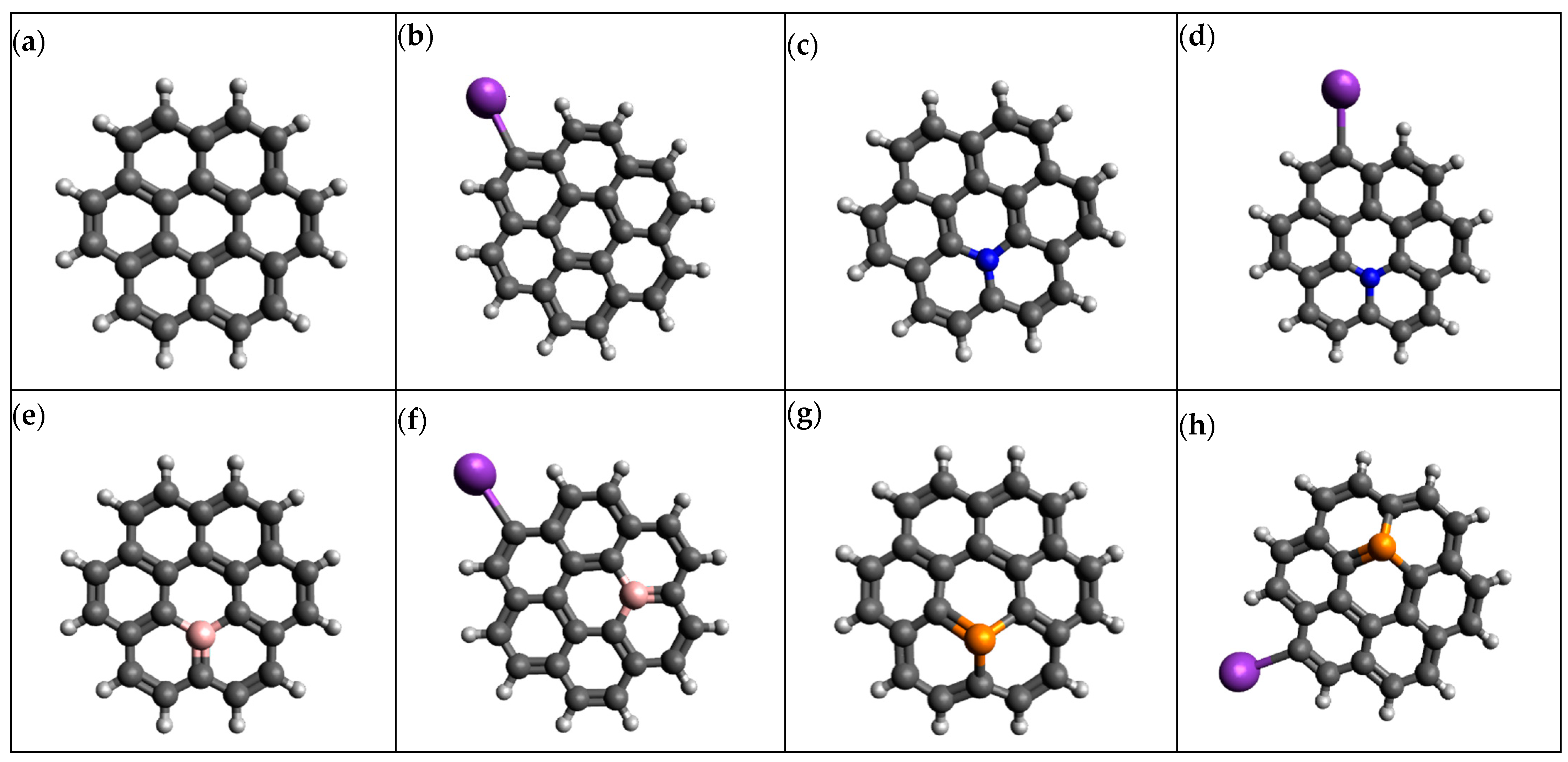
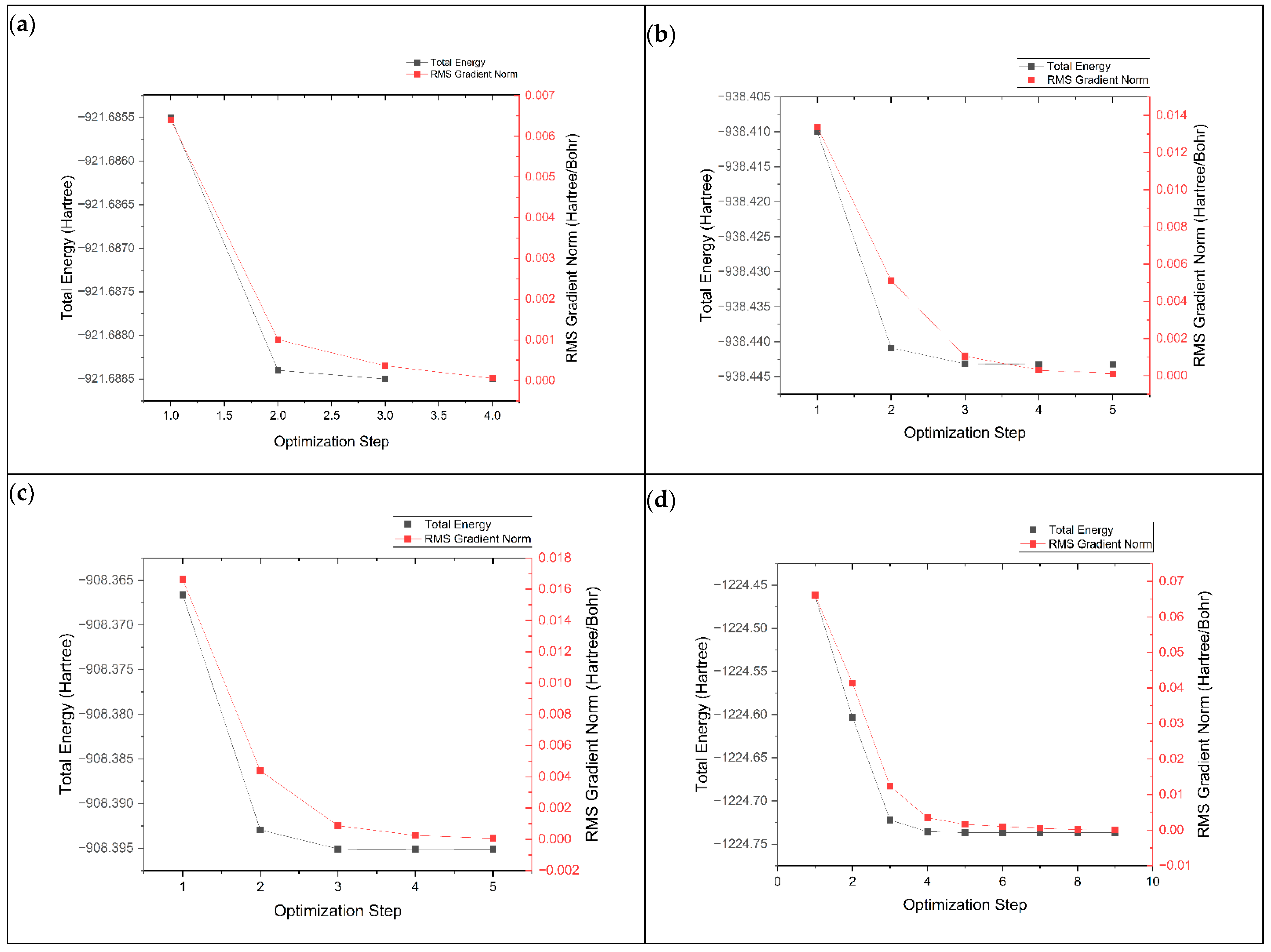
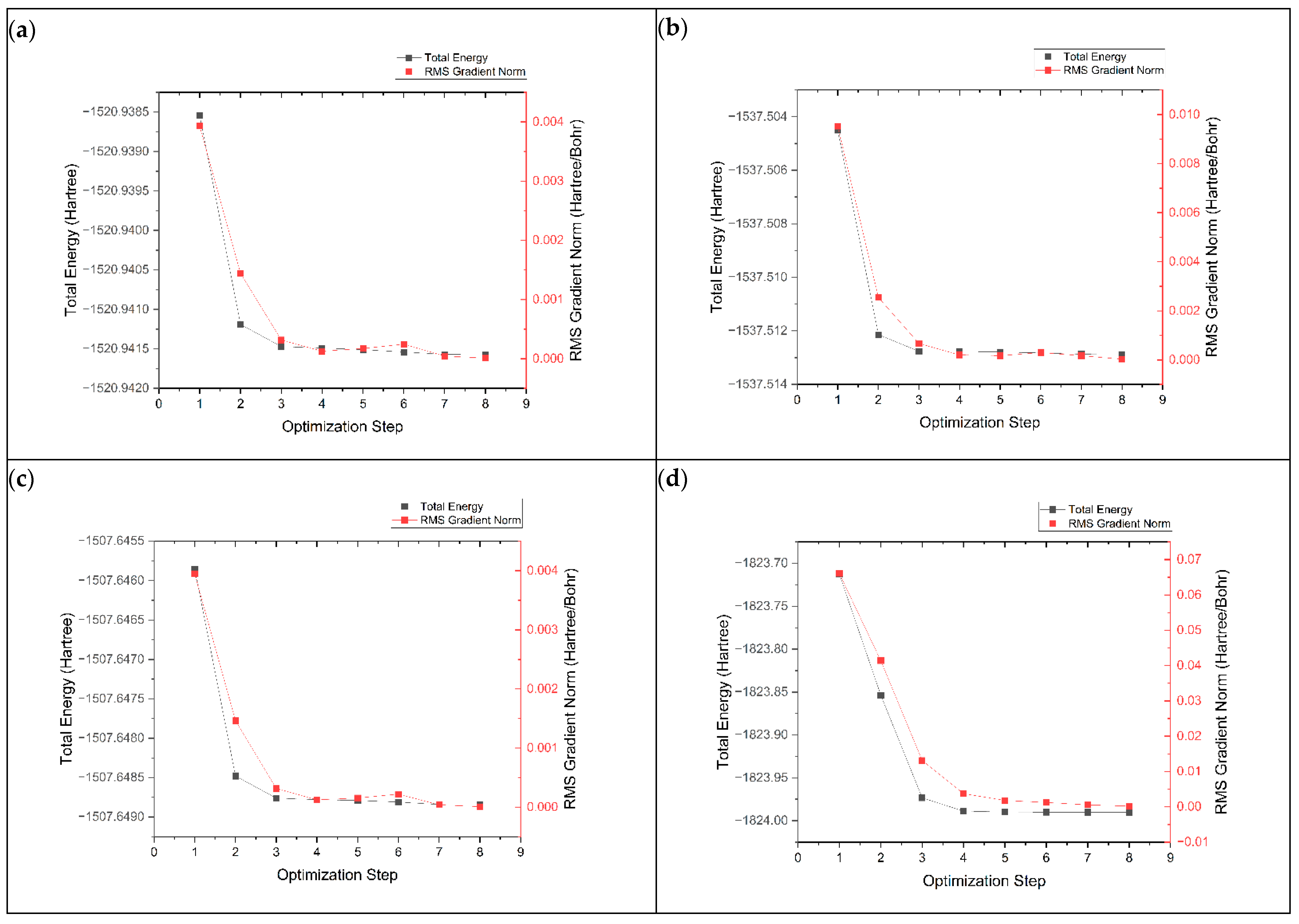
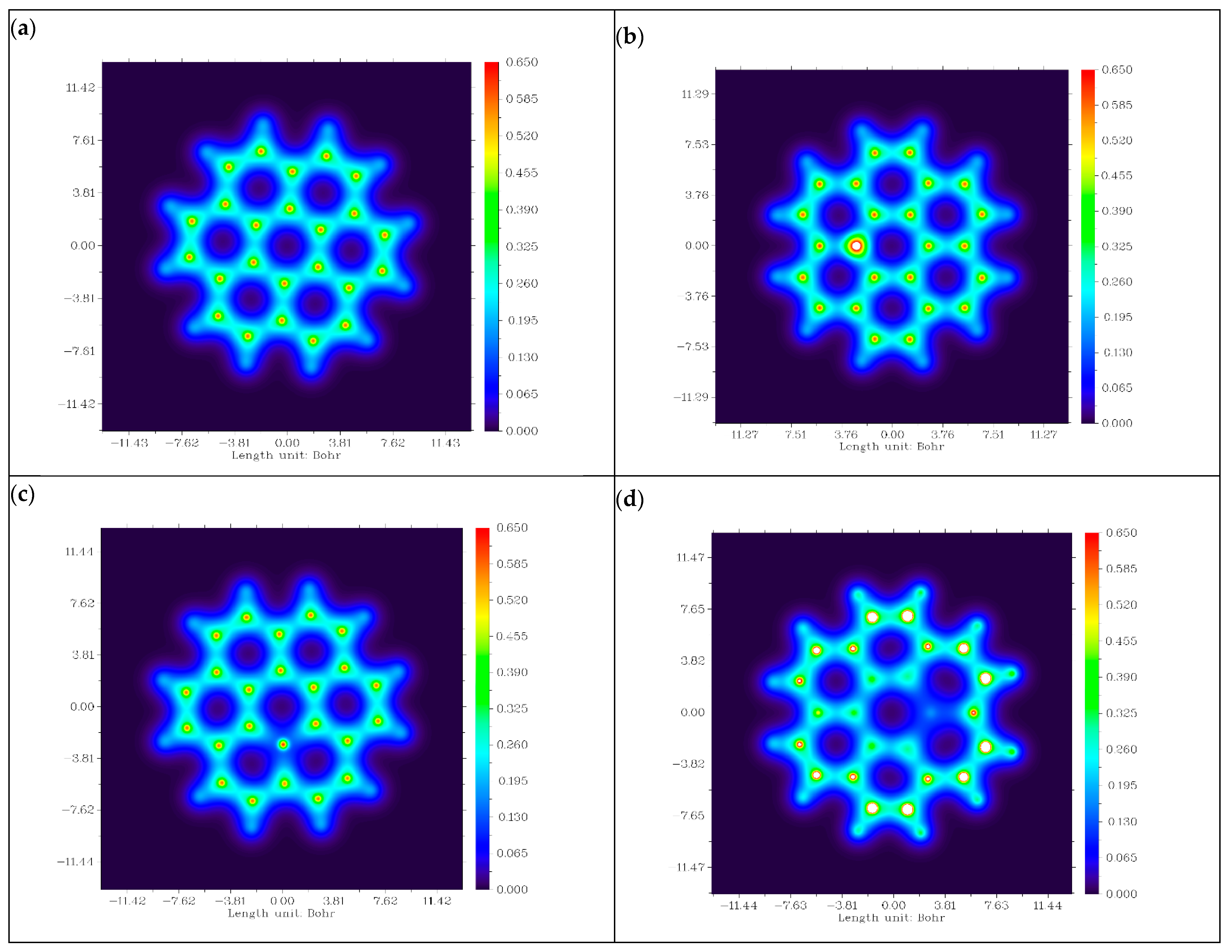
3.1. Structure Enhancement and Geometry Optimization
3.2. Adsorption Energy Calculations
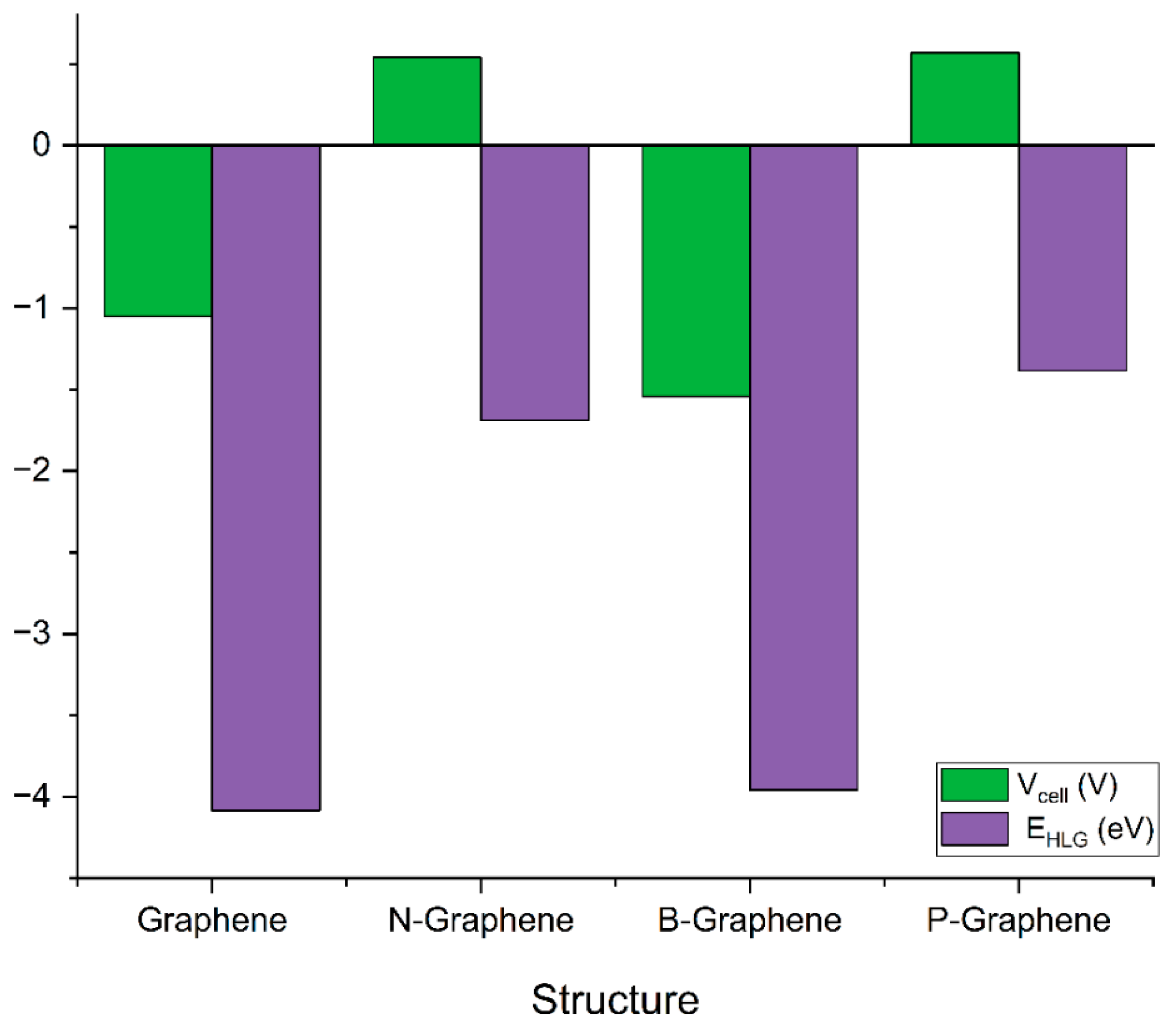
3.3. Cell Voltage and Gibbs Free Energy Change
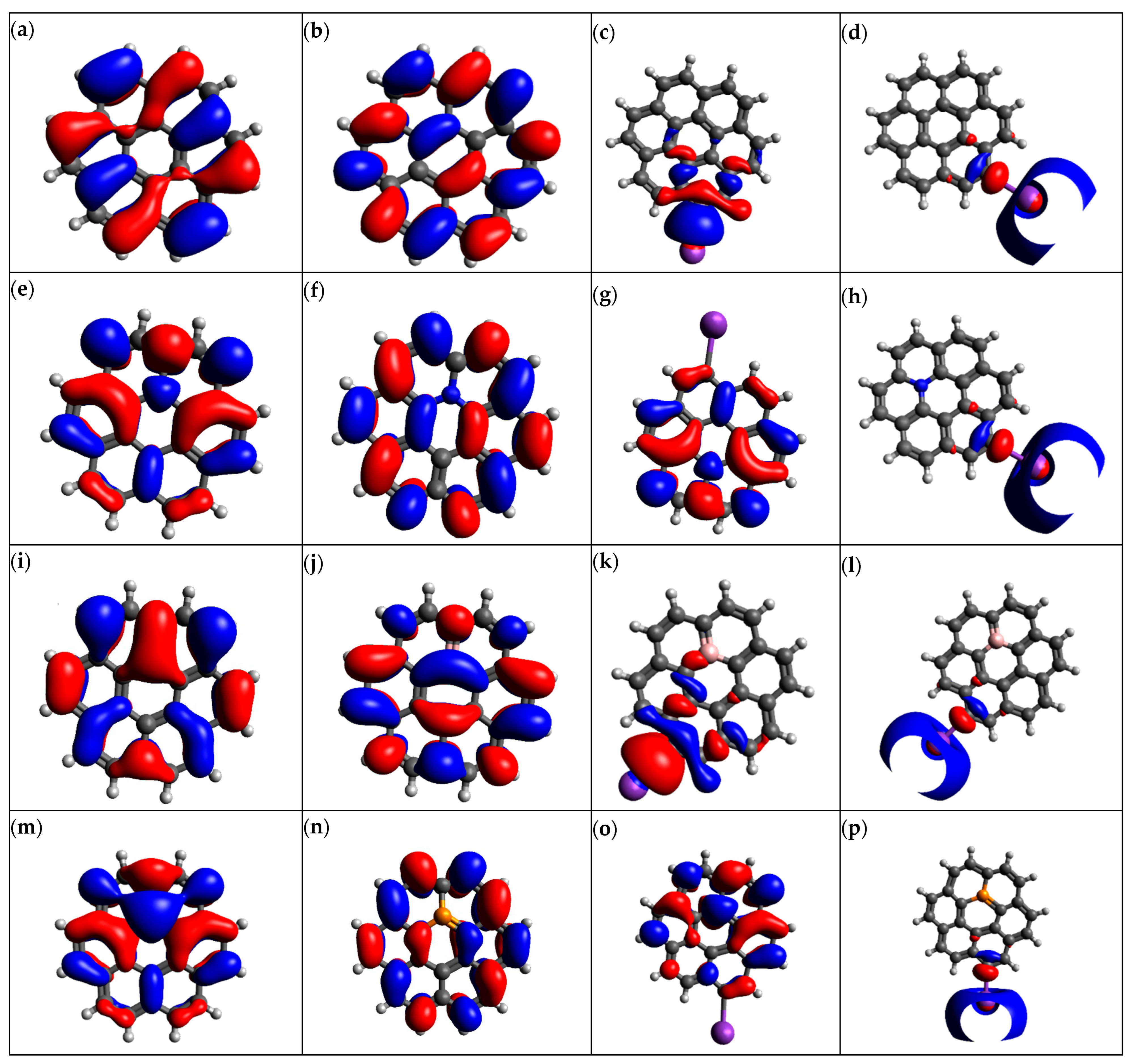
3.4. Molecular Orbital Analysis and Energies
3.5. Electronic Properties
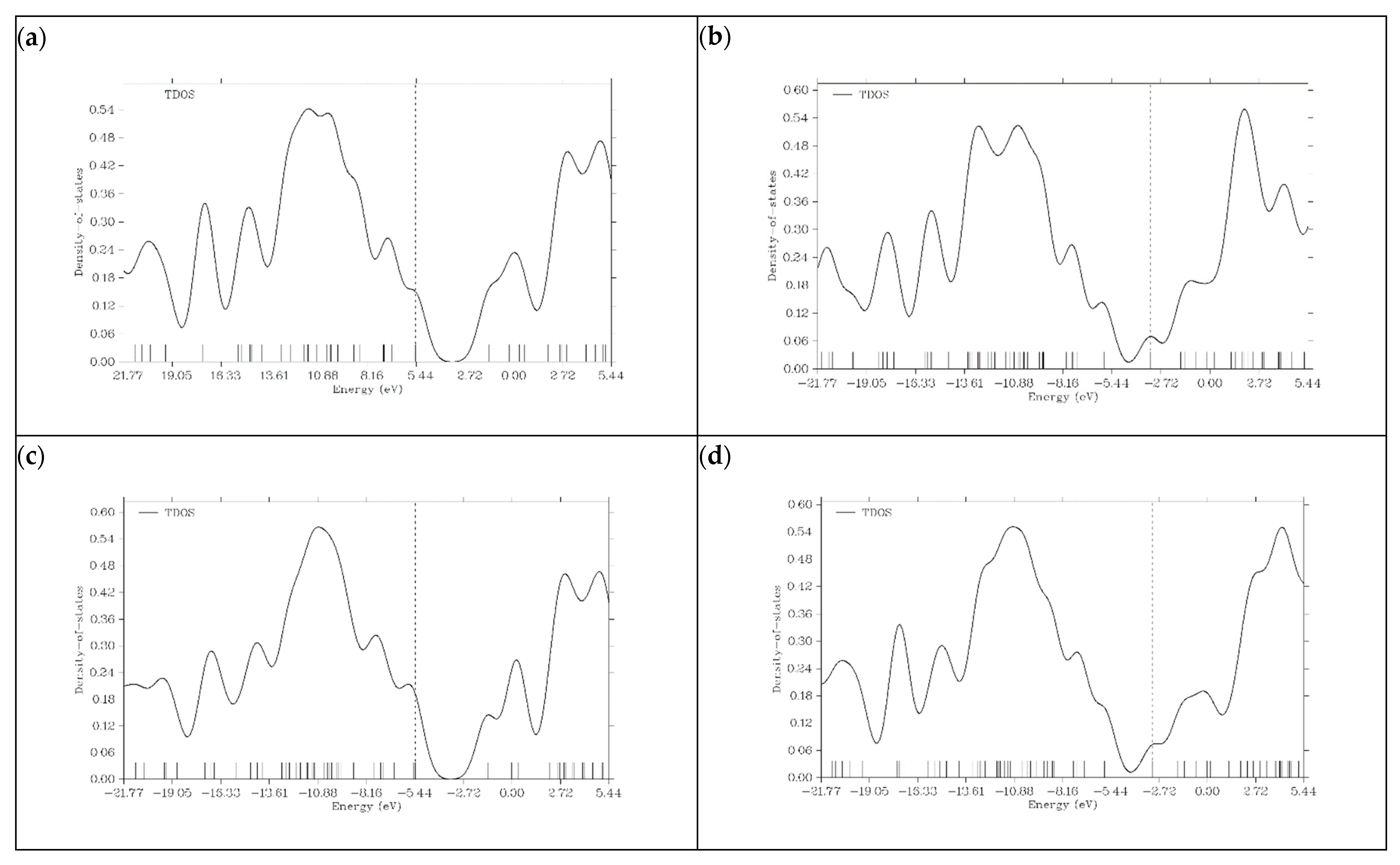
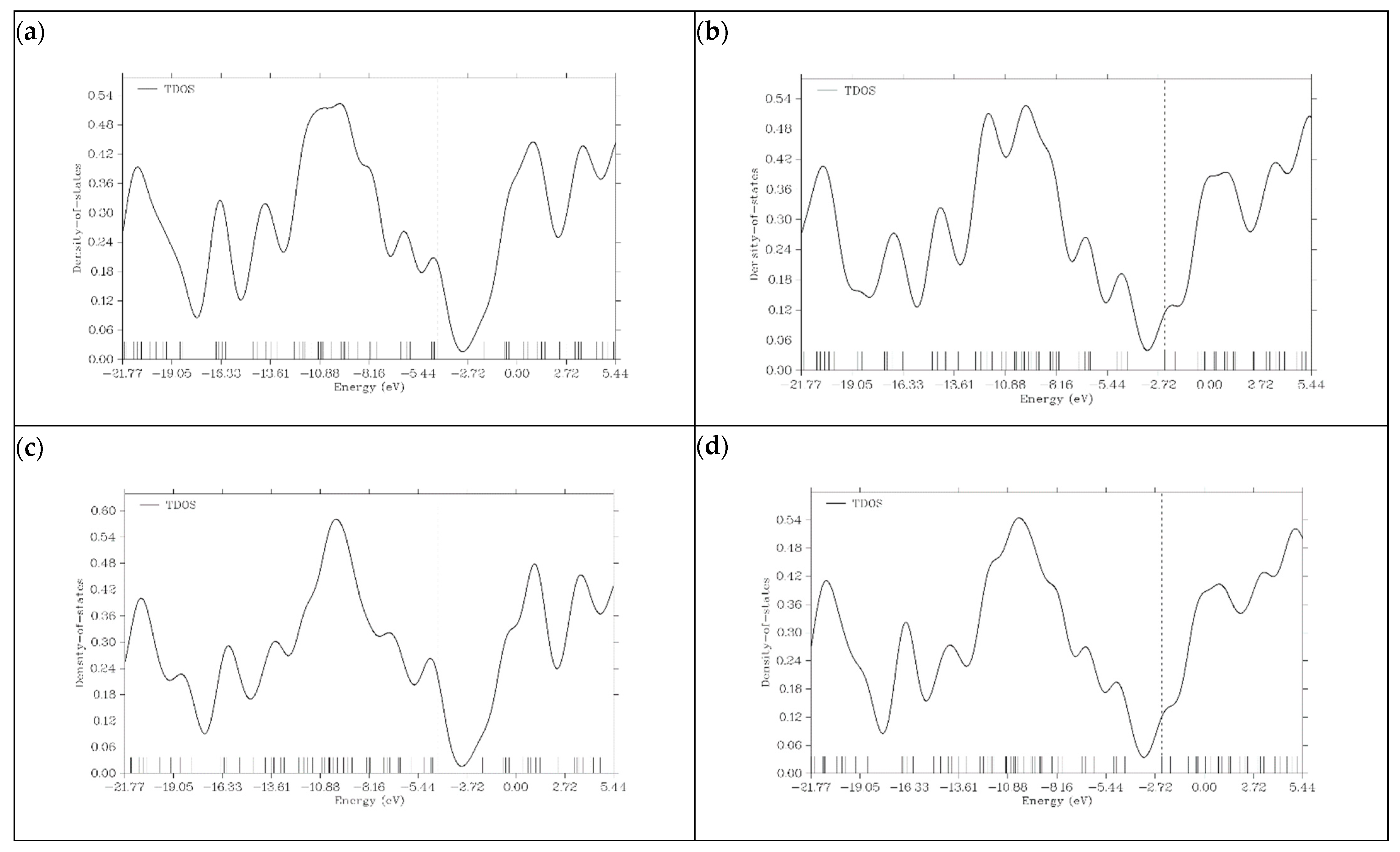
3.6. Density of State (DOS) Plots
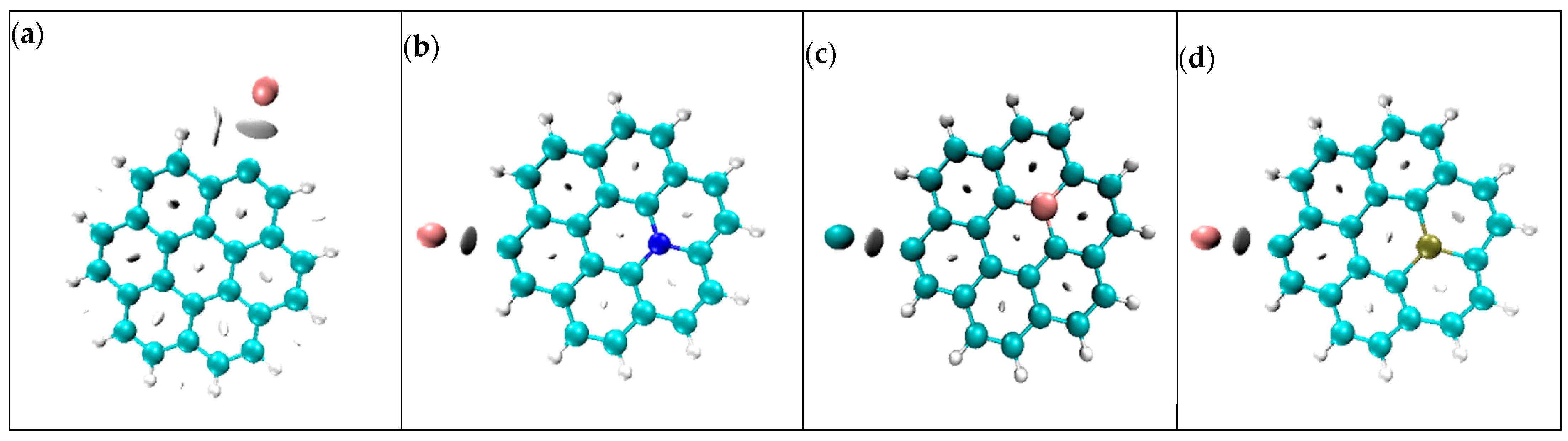
3.7. Non-Covalent Interaction Analysis
3.8. Electron Density Distribution Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lain, M.J.; Kendrick, E. Understanding the limitations of lithium ion batteries at high rates. J. Power Sources 2021, 493, 229690. [Google Scholar] [CrossRef]
- Mitali, J.; Dhinakaran, S.; Mohamad, A.A. Energy storage systems: A review. Energy Storage Sav. 2022, 1, 166–216. [Google Scholar]
- Islam, J.; Anwar, R.; Shareef, M.; Zabed, H.M.; Sahu, J.; Qi, X.; Khandaker, M.U.; Ragauskas, A.; Boukhris, I.; Rahman, R.; et al. Rechargeable metal-metal alkaline batteries: Recent advances, current issues and future research strategies. J. Power Sources 2023, 563, 232777. [Google Scholar]
- Goikolea, E.; Palomares, V.; Wang, S.; de Larramendi, I.R.; Guo, X.; Wang, G.; Rojo, T. Na-ion batteries—Approaching old and new challenges. Adv. Energy Mater. 2020, 10, 2002055. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Tang, Y.; Ji, X.; Jia, C.; Wang, H. Advancements and challenges in potassium ion batteries: A comprehensive review. Adv. Funct. Mater. 2020, 30, 1909486. [Google Scholar] [CrossRef]
- Wu, X.; Qiu, S.; Liu, Y.; Xu, Y.; Jian, Z.; Yang, J.; Ji, X.; Liu, J. The Quest for Stable Potassium-Ion Battery Chemistry. Adv. Mater. 2022, 34, 2106876. [Google Scholar] [CrossRef]
- Zuo, C.; Sun, J.; Li, J.; Zhang, J.; Asundi, A.; Chen, Q. Highresolution transport-of-intensity quantitative phase microscopy with annular illumination. Sci. Rep. 2017, 7, 7622–7654. [Google Scholar] [CrossRef]
- Feng, W.; Feng, N.; Liu, W.; Cui, Y.; Chen, C.; Dong, T.; Liu, S.; Deng, W.; Wang, H.; Jin, Y. Liquid-state templates for constructing B, N, co-doping porous carbons with a boosting of potassium-ion storage performance. Adv. Energy Mater. 2021, 11, 2003215. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, H.; Lei, Y. Recent research progress of anode materials for potassium-ion batteries. Energy Environ. Mater. 2020, 3, 105–120. [Google Scholar] [CrossRef]
- Wasalathilake, K.C.; Li, H.; Xu, L.; Yan, C. Recent advances in graphene based materials as anode materials in sodium-ion batteries. J. Energy Chem. 2020, 42, 91–107. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, L.; Dai, X.; Chen, G.; Liu, G. A record-high ion storage capacity of T-graphene as two-dimensional anode material for Li-ion and Na-ion batteries. Appl. Surf. Sci. 2020, 527, 146849. [Google Scholar] [CrossRef]
- Ahmad, A.; Abahussain, A.A.M.; Nazir, M.H.; Zaidi, S.Z.J. Templating nanostructured aromatic based materials as possible anode electrodes for Na-ion batteries: A computational DFT approach. J. New Mater. Electrochem. Syst. 2024, 27, 38–43. [Google Scholar] [CrossRef]
- Zaidi, S.Z.J.; Hassan, S.; Raza, M.; Walsh, F.C. Transition metal oxide as possible electrode materials for Li-ion batteries: A DFT Analysis. Int. J. Electrochem. Sci. 2021, 16, 210322. [Google Scholar] [CrossRef]
- Zaidi, S.Z.J.; Raza, M.; Hassan, S.; Harito, C.; Walsh, F.C. A DFT Study of Heteroatom Doped-Pyrazine as an Anode in Sodium ion Batteries. J. New Mater. Electrochem. Syst. 2021, 24, 1–8. [Google Scholar] [CrossRef]
- Zaidi, S.Z.J.; Hassan, S.; Raza, M.; Harito, C.; Yuliarto, B.; Walsh, F.C. Conceptualized Simulation for Templating Carbon Based Nano Structures for Li-ion Batteries: A DFT Investigation. J. New Mater. Electrochem. Syst. 2021, 24, 66–72. [Google Scholar] [CrossRef]
- Abahussain, A.A.; Zaidi, S.Z.J.; Nazir, M.H.; Raza, M.; Hassan, M.N.S. A DFT study of Graphene as a drug carrier for gemcitabine anticancer drug. J. New Mater. Electrochem. Syst. 2022, 25, 234–239. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Berenjaghi, H.M.; Mansouri, S.; Beheshtian, J. A DFT study on the potential application of pristine, B and N doped carbon nanocones in potassium-ion batteries. J. Mol. Model. 2021, 27, 168. [Google Scholar] [CrossRef]
- Ferre-Vilaplana, A. Numerical treatment discussion and ab initio computational reinvestigation of physisorption of molecular hydrogen on graphene. J. Chem. Phys. 2005, 122, 104709. [Google Scholar] [CrossRef] [PubMed]
- Jeloaica, L.; Sidis, V. DFT investigation of the adsorption of atomic hydrogen on a cluster-model graphite surface. Chem. Phys. Lett. 1999, 300, 157–162. [Google Scholar] [CrossRef]
- Okamoto, Y.; Miyamoto, Y. Ab initio investigation of physisorption of molecular hydrogen on planar and curved graphenes. J. Phys. Chem. B 2001, 105, 3470–3474. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. van der Waals Potential: An Important Complement to Molecular Electrostatic Potential in Studying Intermolecular Interactions. J. Mol. Model. 2020, 26, 315. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Revealing Molecular Electronic Structure via Analysis of Valence Electron Density. Acta Phys. Chim. Sin. 2018, 34, 503–513. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Ead (Hartree) | Ead (Kcal/mol) |
|---|---|---|
| Graphene | −2.34742 | −1472.26 |
| N-Graphene | −2.16403 | −1357.78 |
| B-Graphene | −2.34810 | −1472.66 |
| P-Graphene | −2.34778 | −1472.48 |
| Structure | ΔG (Hartree) | Vcell (V) |
|---|---|---|
| Graphene | 0.038439 | −1.05 |
| N-Graphene | −0.020056 | 0.54 |
| B-Graphene | 0.056729 | −1.54 |
| P-Graphene | −0.021117 | 0.57 |
| Structure | EHOMO (eV) | ELUMO (eV) | EHLG (eV) |
|---|---|---|---|
| Graphene | −5.4900 | −1.4045 | −4.0854 |
| N-Graphene | −3.3222 | −1.6363 | −1.6860 |
| B-Graphene | −5.4197 | −1.4602 | −3.9576 |
| P-Graphene | −3.0818 | −1.6994 | −1.3832 |
| Structure | EHOMO (eV) | ELUMO (eV) | EHLG (eV) |
|---|---|---|---|
| K@Graphene | −4.3492 | −1.8435 | −2.5057 |
| K@N-Graphene | −2.3847 | −1.8460 | −0.5397 |
| K@B-Graphene | −4.3627 | −1.8538 | −2.5068 |
| K@P-Graphene | −2.3486 | −1.8771 | −0.4719 |
| Structure | μ (eV) | η (eV) | s (eV−1) | ω (eV) |
|---|---|---|---|---|
| Graphene | −3.4473 | 2.0428 | 0.4896 | 2.9085 |
| N-Graphene | −2.4793 | 0.84295 | 1.1865 | 3.6454 |
| B-Graphene | −3.43995 | 1.97975 | 0.5051 | 2.9889 |
| P-Graphene | −2.3906 | 0.6912 | 1.4468 | 4.1346 |
| Structure | EHOMO (eV) | ELUMO (eV) | EHLG (eV) | ΔG (Hartree) | Vcell (V) | Ead (Hartree) |
|---|---|---|---|---|---|---|
| Graphene | −5.4900 | −1.4045 | −4.0854 | 0.038439 | −1.05 | −2.34742 |
| N-Graphene | −3.3222 | −1.6363 | −1.6860 | −0.020056 | 0.54 | −2.16403 |
| B-Graphene | −5.4197 | −1.4602 | −3.9576 | 0.056729 | −1.54 | −2.34810 |
| P-Graphene | −3.0818 | −1.6994 | −1.3832 | −0.021117 | 0.57 | −2.34778 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.; Abahussain, A.A.M.; Nazir, M.H.; Zaidi, S.Z.J. A Computational Approach for Graphene Doped with N,P,B Structures as Possible Electrode Materials for Potassium Ion Batteries (PIBs): A DFT Investigation. Micromachines 2025, 16, 735. https://doi.org/10.3390/mi16070735
Ahmad A, Abahussain AAM, Nazir MH, Zaidi SZJ. A Computational Approach for Graphene Doped with N,P,B Structures as Possible Electrode Materials for Potassium Ion Batteries (PIBs): A DFT Investigation. Micromachines. 2025; 16(7):735. https://doi.org/10.3390/mi16070735
Chicago/Turabian StyleAhmad, A., A. A. M. Abahussain, M. H. Nazir, and S. Z. J. Zaidi. 2025. "A Computational Approach for Graphene Doped with N,P,B Structures as Possible Electrode Materials for Potassium Ion Batteries (PIBs): A DFT Investigation" Micromachines 16, no. 7: 735. https://doi.org/10.3390/mi16070735
APA StyleAhmad, A., Abahussain, A. A. M., Nazir, M. H., & Zaidi, S. Z. J. (2025). A Computational Approach for Graphene Doped with N,P,B Structures as Possible Electrode Materials for Potassium Ion Batteries (PIBs): A DFT Investigation. Micromachines, 16(7), 735. https://doi.org/10.3390/mi16070735