

Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics

Abstract
1. Introduction
2. Advances in Single-Cell RNA Sequencing Technologies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Year | Method | Sample Type | Fit for Rare Cell (Y/N) | Resolution | Ref. |
|---|---|---|---|---|---|---|
| Droplet-based scRNA-seq | 2015 | Drop-seq | Cell suspension | N | Single cell | [23] |
| Droplet-based scRNA-seq | 2015 | inDrop | Cell suspension | N | Single cell | [20] |
| Droplet-based scRNA-seq | 2017 | Chromium 10X | Cell suspension | N | Single cell | [37] |
| Droplet-based scRNA-seq | 2017 | DropNc-seq | Cell suspension | N | Single cell | [24] |
| Droplet-based scRNA-seq | 2023 | SPEAC-seq | Cell suspension | N | Single cell | [38] |
| Microwell-based scRNA-seq | 2015 | Cytoseq | Cell suspension | N | Single cell | [39] |
| Microwell-based scRNA-seq | 2017 | BD Rhapsody | Cell suspension | N | Single cell | [40] |
| Microwell-based scRNA-seq | 2015 | Solid-phase RNA capture | Cell suspension | N | Single cell | [36] |
| Microwell-based scRNA-seq | 2017 | Seq-Well | Cell suspension | N | Single cell | [21] |
| Microwell-based scRNA-seq | 2020 | Seq-Well S3 | Cell suspension | N | Single cell | [41] |
| Microwell-based scRNA-seq | 2018 | scFTD-seq | Cell suspension | N | Single cell | [42] |
| Valve-based scRNA-seq | 2014 | Microfluidic Single-cell whole-transcriptome Sequencing | Cell suspension | N | Single cell | [22] |
| Valve-based scRNA-seq | 2019 | Hydro-Seq | blood | Y | Single cell | [43] |
| Valve-based scRNA-seq | 2020 | Paired-seq | Cell suspension | N | Single cell | [44] |
2.1. Droplet-Based scRNA-Seq Methods
2.2. Microwell-Based scRNA-Seq Methods
2.3. Valve-Based scRNA-Seq Methods
2.4. Single-Cell Multi-Omics Profiling
2.5. Technical Challenges and Future Directions
3. Advances in Spatial Transcriptomics
3.1. Array-Based SRT Methods
3.2. Microchannel-Based SRT Methods
3.3. Spatiotemporal Transcriptome
3.4. Technical Challenges and Future Directions
4. Clinical Applications and Conclusions
4.1. Early Detection and Disease Classification in Oncology
4.2. Cellular Dynamics of the Brain
4.3. Discussion and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Neumann, P.E.; Neumann, E.E. General Histological Woes: Definition and Classification of Tissues. Clin. Anat. 2021, 34, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Tato, C.M.; Furman, D. Systems Immunology: Just Getting Started. Nat. Immunol. 2017, 18, 725–732. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A Molecular Cell Atlas of the Human Lung from Single-Cell RNA Sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef]
- Williams, J.W.; Winkels, H.; Durant, C.P.; Zaitsev, K.; Ghosheh, Y.; Ley, K. Single Cell RNA Sequencing in Atherosclerosis Research. Circ. Res. 2020, 126, 1112–1126. [Google Scholar] [CrossRef]
- Nawy, T. Single-Cell Sequencing. Nat. Methods 2014, 11, 18. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of Spatial Transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays 2020, 42, 1900221. [Google Scholar] [CrossRef]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring Tissue Architecture Using Spatial Transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation Dynamics of Mammary Epithelial Cells Revealed by Single-Cell RNA Sequencing. Nat. Commun. 2017, 8, 2128. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-Cell RNA-Seq Reveals Dynamic Paracrine Control of Cellular Variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Chen, Y.; Vaillant, F.; Jamieson, P.; Gordon, L.; Rios, A.C.; Wilcox, S.; Fu, N.; Liu, K.H.; Jackling, F.C.; et al. Construction of Developmental Lineage Relationships in the Mouse Mammary Gland by Single-Cell RNA Profiling. Nat. Commun. 2017, 8, 1627. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The Origins and the Future of Microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by Shrinking: Empowering Single-Cell “omics” with Microfluidic Devices. Nat. Rev. Genet. 2017, 18, 345–361. [Google Scholar] [CrossRef]
- Tavakoli, H.; Zhou, W.; Ma, L.; Perez, S.; Ibarra, A.; Xu, F.; Zhan, S.; Li, X. Recent Advances in Microfluidic Platforms for Single-Cell Analysis in Cancer Biology, Diagnosis and Therapy. TrAC Trends Anal. Chem. 2019, 117, 13–26. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity Through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint Profiling of Chromatin Accessibility and Gene Expression in Thousands of Single Cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H., II; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, Low-Cost RNA Sequencing of Single Cells at High Throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.M.; Zhang, X.; Cao, C.; Pang, Y.; Wu, X.; Xiong, L.; Yang, L.; Fu, Y.; Zhao, L.; Tang, F.; et al. Microfluidic Single-Cell Whole-Transcriptome Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7048–7053. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively Parallel Single-Nucleus RNA-Seq with DroNc-Seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef]
- Yu, N.; Jin, Z.; Liang, C.; Zhang, J.; Yang, B. Well-ST-Seq: Cost-Effective Spatial Transcriptomics at Cellular Level and High RNA Capture Efficiency. bioRxiv 2023, 546974. [Google Scholar]
- Ahmed, R.; Zaman, T.; Chowdhury, F.; Mraiche, F.; Tariq, M.; Ahmad, I.S.; Hasan, A. Single-Cell RNA Sequencing with Spatial Transcriptomics of Cancer Tissues. Int. J. Mol. Sci. 2022, 23, 3042. [Google Scholar] [CrossRef]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating Single-Cell and Spatial Transcriptomics to Elucidate Intercellular Tissue Dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]
- Jiao, Y.; Gao, L.; Ji, Y.; Liu, W. Recent Advances in Microfluidic Single-Cell Analysis and Its Applications in Drug Development. TrAC Trends Anal. Chem. 2022, 157, 116796. [Google Scholar] [CrossRef]
- Kalisky, T.; Blainey, P.; Quake, S.R. Genomic Analysis at the Single-Cell Level. Annu. Rev. Genet. 2011, 45, 431–445. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The Technological Landscape and Applications of Single-Cell Multi-Omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [PubMed]
- Schmid, L.; Franke, T. SAW-Controlled Drop Size for Flow Focusing. Lab A Chip 2013, 13, 1691–1694. [Google Scholar] [CrossRef] [PubMed]
- Schmid, L.; Franke, T. Acoustic Modulation of Droplet Size in a T-Junction. Appl. Phys. Lett. 2014, 104, 133501. [Google Scholar] [CrossRef]
- Fan, H.C.; Wang, J.; Potanina, A.; Quake, S.R. Whole-Genome Molecular Haplotyping of Single Cells. Nat. Biotechnol. 2011, 29, 51–57. [Google Scholar] [CrossRef]
- Bose, S.; Wan, Z.; Carr, A.; Rizvi, A.H.; Vieira, G.; Pe’er, D.; Sims, P.A. Scalable Microfluidics for Single-Cell RNA Printing and Sequencing. Genome Biol. 2015, 16, 120. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, F. SPEAC-Seq: A New Method to Investigate Astrocyte-Microglia Crosstalk. Brain-X 2023, 1, e22. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P.A. Combinatorial Labeling of Single Cells for Gene Expression Cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of Functionally Integrated Human Forebrain Spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef]
- Hughes, T.K.; Wadsworth, M.H.; Gierahn, T.M.; Do, T.; Weiss, D.; Andrade, P.R.; Ma, F.; Silva, B.J.d.A.; Shao, S.; Tsoi, L.C.; et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity 2020, 53, 878–894.e7. [Google Scholar] [CrossRef] [PubMed]
- Dura, B.; Choi, J.-Y.; Zhang, K.; Damsky, W.; Thakral, D.; Bosenberg, M.; Craft, J.; Fan, R. scFTD-Seq: Freeze-Thaw Lysis Based, Portable Approach Toward Highly Distributed Single-Cell 3′ mRNA Profiling. Nucleic Acids Res. 2019, 47, e16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Chen, Y.-C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.-T.; Lee, W.; Hao, Z.; Sahoo, S.; Min Kang, H.; et al. Hydro-Seq Enables Contamination-Free High-Throughput Single-Cell RNA-Sequencing for Circulating Tumor Cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zou, Y.; Xu, X.; Zhang, X.; Gao, M.; Song, J.; Huang, P.; Chen, Q.; Zhu, Z.; Lin, W.; et al. Highly Parallel and Efficient Single Cell mRNA Sequencing with Paired Picoliter Chambers. Nat. Commun. 2020, 11, 2118. [Google Scholar] [CrossRef]
- Grün, D.; Kester, L.; van Oudenaarden, A. Validation of Noise Models for Single-Cell Transcriptomics. Nat. Methods 2014, 11, 637–640. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Abate, A.R.; Chen, C.-H.; Agresti, J.J.; Weitz, D.A. Beating Poisson Encapsulation Statistics Using Close-Packed Ordering. Lab Chip 2009, 9, 2628–2631. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef]
- Meng, Y.; Ye, F.; Nie, P.; Zhao, Q.; An, L.; Wang, W.; Qu, S.; Shen, Z.; Cao, Z.; Zhang, X.; et al. Immunosuppressive CD10+ALPL+ Neutrophils Promote Resistance to Anti-PD-1 Therapy in HCC by Mediating Irreversible Exhaustion of T Cells. J. Hepatol. 2023, 79, 1435–1449. [Google Scholar] [CrossRef]
- Yu, J.; Xiao, K.; Chen, X.; Deng, L.; Zhang, L.; Li, Y.; Gao, A.; Gao, J.; Wu, C.; Yang, X.; et al. Neuron-Derived Neuropeptide Y Fine-Tunes the Splenic Immune Responses. Neuron 2022, 110, 1327–1339.e6. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-Length mRNA-Seq from Single-Cell Levels of RNA and Individual Circulating Tumor Cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.A.; Chou, H.-P.; Thorsen, T.; Scherer, A.; Quake, S.R. Monolithic Microfabricated Valves and Pumps by Multilayer Soft Lithography. Science 2000, 288, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Kim, J.; Ni, M.; Wei, Y.; Okamoto, H.; Lee, J.; Adler, C.; Cavino, K.; Murphy, A.J.; Yancopoulos, G.D.; et al. Use of the Fluidigm C1 Platform for RNA Sequencing of Single Mouse Pancreatic Islet Cells. Proc. Natl. Acad. Sci. USA 2016, 113, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-Cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Saligrama, N.; Buenrostro, J.D.; Wei, Y.; Wu, B.; Rubin, A.J.; Granja, J.M.; Lareau, C.A.; Li, R.; Qi, Y.; et al. Transcript-Indexed ATAC-Seq for Precision Immune Profiling. Nat. Med. 2018, 24, 580–590. [Google Scholar] [CrossRef]
- Yang, Y.; Swennenhuis, J.F.; Rho, H.S.; Le Gac, S.; Terstappen, L.W.M.M. Parallel Single Cancer Cell Whole Genome Amplification Using Button-Valve Assisted Mixing in Nanoliter Chambers. PLoS ONE 2014, 9, e107958. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, P.; Lei, S.; Deng, F.; Xiao, G.G.; Liu, Y.; Chen, X.; Li, L.; Wu, S.; Chen, Y.; et al. How Is mRNA Expression Predictive for Protein Expression? A Correlation Study on Human Circulating Monocytes. Acta Biochim. Biophys. Sin. 2008, 40, 426–436. [Google Scholar] [CrossRef]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and Protein in Complex Biological Samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-Omics Approaches to Disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- He, L.; Wang, W.; Dang, K.; Ge, Q.; Zhao, X. Integration of Single-cell Transcriptome and Proteome Technologies: Toward Spatial Resolution Levels. View 2023, 4, 20230040. [Google Scholar] [CrossRef]
- Kreitmaier, P.; Katsoula, G.; Zeggini, E. Insights from Multi-Omics Integration in Complex Disease Primary Tissues. Trends Genet. 2023, 39, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Vistain, L.F.; Tay, S. Single-Cell Proteomics. Trends Biochem. Sci. 2021, 46, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Nan, J.; Xu, H.; Wang, L.; Niu, J.; Zhang, J.; Yang, B. Neural Network Enables High Accuracy for Hepatitis B Surface Antigen Detection with a Plasmonic Platform. Nano Lett. 2024, 24, 8784–8792. [Google Scholar] [CrossRef] [PubMed]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-Throughput Single-Cell ChIP-Seq Identifies Heterogeneity of Chromatin States in Breast Cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, Multimodal Profiling of Chromatin Accessibility, Gene Expression and Protein Levels in Single Cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of Multiplex Immunohistochemistry/Immunofluorescence Techniques in the Era of Cancer Immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef]
- Park, S.-M.; Lee, J.Y.; Hong, S.; Lee, S.H.; Dimov, I.K.; Lee, H.; Suh, S.; Pan, Q.; Li, K.; Wu, A.M.; et al. Dual Transcript and Protein Quantification in a Massive Single Cell Array. Lab Chip 2016, 16, 3682–3688. [Google Scholar] [CrossRef]
- Frei, A.P.; Bava, F.-A.; Zunder, E.R.; Hsieh, E.W.Y.; Chen, S.-Y.; Nolan, G.P.; Gherardini, P.F. Highly Multiplexed Simultaneous Detection of RNAs and Proteins in Single Cells. Nat. Methods 2016, 13, 269–275. [Google Scholar] [CrossRef]
- Cadot, S.; Valle, C.; Tosolini, M.; Pont, F.; Largeaud, L.; Laurent, C.; Fournie, J.J.; Ysebaert, L.; Quillet-Mary, A. Longitudinal CITE-Seq Profiling of Chronic Lymphocytic Leukemia during Ibrutinib Treatment: Evolution of Leukemic and Immune Cells at Relapse. Biomark. Res. 2020, 8, 72. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-Cell Profiling of Myeloid Cells in Glioblastoma across Species and Disease Stage Reveals Macrophage Competition and Specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-J.; Du, H.; Khabibullin, D.; Zarei, M.; Wei, K.; Freeman, G.J.; Kwiatkowski, D.J.; Henske, E.P. mTORC1 Upregulates B7-H3/CD276 to Inhibit Antitumor T Cells and Drive Tumor Immune Evasion. Nat. Commun. 2023, 14, 1214. [Google Scholar] [CrossRef] [PubMed]
- Delley, C.L.; Liu, L.; Sarhan, M.F.; Abate, A.R. Combined Aptamer and Transcriptome Sequencing of Single Cells. Sci. Rep. 2018, 8, 2919. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.F.; Parks, B.; Kathiria, A.S.; Ober-Reynolds, B.; Goronzy, J.J.; Greenleaf, W.J. NEAT-Seq: Simultaneous Profiling of Intra-Nuclear Proteins, Chromatin Accessibility and Gene Expression in Single Cells. Nat. Methods 2022, 19, 547–553. [Google Scholar] [CrossRef]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef]
- Participants in the 1st Human Cell Atlas Jamboree; Lun, A.T.L.; Riesenfeld, S.; Andrews, T.; Dao, T.P.; Gomes, T.; Marioni, J.C. EmptyDrops: Distinguishing Cells from Empty Droplets in Droplet-Based Single-Cell RNA Sequencing Data. Genome Biol. 2019, 20, 63. [Google Scholar] [CrossRef]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef]
- Bressan, D.; Battistoni, G.; Hannon, G.J. The Dawn of Spatial Omics. Science 2023, 381, eabq4964. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Ding, J.; Ahangari, F.; Espinoza, C.R.; Chhabra, D.; Nicola, T.; Yan, X.; Lal, C.V.; Hagood, J.S.; Kaminski, N.; Bar-Joseph, Z.; et al. Integrating Multiomics Longitudinal Data to Reconstruct Networks Underlying Lung Development. Available online: https://journals.physiology.org/doi/epdf/10.1152/ajplung.00554.2018 (accessed on 29 October 2024).
- Du, J.; Yang, Y.-C.; An, Z.-J.; Zhang, M.-H.; Fu, X.-H.; Huang, Z.-F.; Yuan, Y.; Hou, J. Advances in Spatial Transcriptomics and Related Data Analysis Strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Canozo, F.J.G.; Zuo, Z.; Martin, J.F.; Samee, M.A.H. Cell-Type Modeling in Spatial Transcriptomics Data Elucidates Spatially Variable Colocalization and Communication Between Cell-Types in Mouse Brain. Cell Syst. 2022, 13, 58–70.e5. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, V.; Capellera-Garcia, S.; Ma, F.; Fares, I.; Liebscher, S.; Ng, E.S.; Ekstrand, S.; Aguadé-Gorgorió, J.; Vavilina, A.; Lefaudeux, D.; et al. Mapping Human Haematopoietic Stem Cells from Haemogenic Endothelium to Birth. Nature 2022, 604, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal Transcriptomic Atlas of Mouse Organogenesis Using DNA Nanoball-Patterned Arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef]
- 10x Genomics Acquires Spatial Transcriptomics. Available online: https://www.genomeweb.com/sequencing/10x-genomics-acquires-spatial-transcriptomics (accessed on 13 November 2024).
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-Definition Spatial Transcriptomics for in Situ Tissue Profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
- Ben-Chetrit, N.; Niu, X.; Swett, A.D.; Sotelo, J.; Jiao, M.S.; Stewart, C.M.; Potenski, C.; Mielinis, P.; Roelli, P.; Stoeckius, M.; et al. Integration of Whole Transcriptome Spatial Profiling with Protein Markers. Nat. Biotechnol. 2023, 41, 788–793. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef]
- Liu, Y.; Enninful, A.; Deng, Y.; Fan, R. Spatial Transcriptome Sequencing of FFPE Tissues at Cellular Level. bioRxiv 2020, 338475. [Google Scholar]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial Profiling of Chromatin Accessibility in Mouse and Human Tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially Resolved Chromatin Modification Profiling at the Cellular Level. Science 2022, 375, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; DiStasio, M.; Su, G.; Asashima, H.; Enninful, A.; Qin, X.; Deng, Y.; Nam, J.; Gao, F.; Bordignon, P.; et al. High-Plex Protein and Whole Transcriptome Co-Mapping at Cellular Resolution with Spatial CITE-Seq. Nat. Biotechnol. 2023, 41, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Deng, Y.; Kukanja, P.; Agirre, E.; Bartosovic, M.; Dong, M.; Ma, C.; Ma, S.; Su, G.; Bao, S.; et al. Spatial Epigenome-Transcriptome Co-Profiling of Mammalian Tissues. Nature 2023, 616, 113–122. [Google Scholar] [CrossRef]
- Liu, C.; Li, R.; Li, Y.; Lin, X.; Zhao, K.; Liu, Q.; Wang, S.; Yang, X.; Shi, X.; Ma, Y.; et al. Spatiotemporal Mapping of Gene Expression Landscapes and Developmental Trajectories During Zebrafish Embryogenesis. Dev. Cell 2022, 57, 1284–1298.e5. [Google Scholar] [CrossRef]
- Shi, Y.; Huang, L.; Dong, H.; Yang, M.; Ding, W.; Zhou, X.; Lu, T.; Liu, Z.; Zhou, X.; Wang, M.; et al. Decoding the Spatiotemporal Regulation of Transcription Factors During Human Spinal Cord Development. Cell Res. 2024, 34, 193–213. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Papadopoulos, Z.; Dykstra, T.; Brase, L.; Farias, F.G.; Wall, M.; Jiang, H.; Kodira, C.D.; de Lima, K.A.; Herz, J.; et al. Meningeal Lymphatics Affect Microglia Responses and Anti-Aβ Immunotherapy. Nature 2021, 593, 255–260. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Wang, C.; Yang, M.; He, Z.; Wang, F.; Zhang, Y.; Li, R.; Gong, Y.; Wang, B.; et al. Spatiotemporal Transcriptome Atlas Reveals the Regional Specification of the Developing Human Brain. Cell 2023, 186, 5892–5909.e22. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; Regier, M.C.; Barkan, E.; Franks, J.M.; Packer, J.S.; Grosjean, P.; Duran, M.; Saxton, S.; Ladd, J.J.; Spielmann, M.; et al. Embryo-Scale, Single-Cell Spatial Transcriptomics. Science 2021, 373, 111–117. [Google Scholar] [CrossRef]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323.e30. [Google Scholar] [CrossRef] [PubMed]
- Lohoff, T.; Ghazanfar, S.; Missarova, A.; Koulena, N.; Pierson, N.; Griffiths, J.A.; Bardot, E.S.; Eng, C.-H.L.; Tyser, R.C.V.; Argelaguet, R.; et al. Integration of Spatial and Single-Cell Transcriptomic Data Elucidates Mouse Organogenesis. Nat. Biotechnol. 2022, 40, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal Analysis of Human Intestinal Development at Single-Cell Resolution. Cell 2021, 184, 810–826.e23. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Lorenzi, V.; Mazzeo, C.I.; Alves-Lopes, J.P.; Roberts, K.; Sancho-Serra, C.; Engelbert, J.; Marečková, M.; Gruhn, W.H.; Botting, R.A.; et al. Single-Cell Roadmap of Human Gonadal Development. Nature 2022, 607, 540–547. [Google Scholar] [CrossRef]
- Zhao, L.; Song, W.; Chen, Y.-G. Mesenchymal-Epithelial Interaction Regulates Gastrointestinal Tract Development in Mouse Embryos. Cell Rep. 2022, 40, 111053. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly Sensitive Spatial Transcriptomics at Near-Cellular Resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef]
- Wu, B.; Shentu, X.; Nan, H.; Guo, P.; Hao, S.; Xu, J.; Shangguan, S.; Cui, L.; Cen, J.; Deng, Q.; et al. A Spatiotemporal Atlas of Cholestatic Injury and Repair in Mice. Nat. Genet. 2024, 56, 938–952. [Google Scholar] [CrossRef]
- Zhang, B.; He, P.; Lawrence, J.E.G.; Wang, S.; Tuck, E.; Williams, B.A.; Roberts, K.; Kleshchevnikov, V.; Mamanova, L.; Bolt, L.; et al. A Human Embryonic Limb Cell Atlas Resolved in Space and Time. Nature 2023, 635, 668–678. [Google Scholar] [CrossRef]
- Laurenti, E.; Göttgens, B. From Haematopoietic Stem Cells to Complex Differentiation Landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-Cell and Spatial Analysis Reveal Interaction of FAP+ Fibroblasts and SPP1+ Macrophages in Colorectal Cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial Proteogenomics Reveals Distinct and Evolutionarily Conserved Hepatic Macrophage Niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Shi, Q.; Zhang, Y.; Liang, G.; Kang, Z.; Huang, B.; Ma, D.; Wang, L.; Jiao, J.; Fang, X.; et al. Identification of HSC/MPP Expansion Units in Fetal Liver by Single-Cell Spatiotemporal Transcriptomics. Cell Res. 2022, 32, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Egozi, A.; Halpern, K.B.; Farack, L.; Rotem, H.; Itzkovitz, S. Zonation of Pancreatic Acinar Cells in Diabetic Mice. Cell Rep. 2020, 32, 108043. [Google Scholar] [CrossRef]
- Cho, C.-S.; Xi, J.; Si, Y.; Park, S.-R.; Hsu, J.-E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic Examination of Spatial Transcriptome Using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef]
- Zhang, L. Clinical and Translational Values of Spatial Transcriptomics. Signal Transduct. Target. Ther. 2022, 7, 111. [Google Scholar] [CrossRef]
- Wang, D.C.; Wang, X. Clinical Significance of Spatiotemporal Transcriptional Bursting and Control. Clin. Transl. Med. 2021, 11, e518. [Google Scholar] [CrossRef]
- Pickard, K.; Stephenson, E.; Mitchell, A.; Jardine, L.; Bacon, C.M. Location, Location, Location: Mapping the Lymphoma Tumor Microenvironment Using Spatial Transcriptomics. Front. Oncol. 2023, 13, 1258245. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, Y.; Wang, X.; Fan, J.; Gao, Q. Spatial Omics: Navigating to the Golden Era of Cancer Research. Clin. Transl. Med. 2022, 12, e696. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, M.; Sun, W.; Tang, X.; Zhou, Y.; Zhang, J.; Yang, B. A Hemoglobin Bionics-Based System for Combating Antibiotic Resistance in Chronic Diabetic Wounds via Iron Homeostasis Regulation. Adv. Mater. 2024, 36, 2405002. [Google Scholar] [CrossRef]
- Zhao, T.; Chiang, Z.D.; Morriss, J.W.; LaFave, L.M.; Murray, E.M.; Del Priore, I.; Meli, K.; Lareau, C.A.; Nadaf, N.M.; Li, J.; et al. Spatial Genomics Enables Multi-Modal Study of Clonal Heterogeneity in Tissues. Nature 2022, 601, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.T.A.; Schram, A.M.; Hyman, D.M.; Taylor, B.S. Discovery Through Clinical Sequencing in Oncology. Nat. Cancer 2020, 1, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.; He, M.; Berglund, E.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Kvastad, L.; Andersson, A.; Bergenstråhle, L.; Bergenstråhle, J.; et al. Spatially Resolved Clonal Copy Number Alterations in Benign and Malignant Tissue. Nature 2022, 608, 360–367. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e16. [Google Scholar] [CrossRef]
- Granja, J.M.; Klemm, S.; McGinnis, L.M.; Kathiria, A.S.; Mezger, A.; Corces, M.R.; Parks, B.; Gars, E.; Liedtke, M.; Zheng, G.X.Y.; et al. Single-Cell Multiomic Analysis Identifies Regulatory Programs in Mixed-Phenotype Acute Leukemia. Nat. Biotechnol. 2019, 37, 1458–1465. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Cheng, M.; Jiang, Y.; Xu, J.; Mentis, A.-F.A.; Wang, S.; Zheng, H.; Sahu, S.K.; Liu, L.; Xu, X. Spatially Resolved Transcriptomics: A Comprehensive Review of Their Technological Advances, Applications, and Challenges. J. Genet. Genom. 2023, 50, 625–640. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Lau, B.T.; Kim, H.S.; Sathe, A.; Grimes, S.M.; Ji, H.P.; Zhang, N.R. Integrative Single-Cell Analysis of Allele-Specific Copy Number Alterations and Chromatin Accessibility in Cancer. Nat. Biotechnol. 2021, 39, 1259–1269. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, B.; Yang, Z.; Liu, X.; Huai, Q.; Guo, L.; Xue, X.; Tan, F.; Li, Y.; Xue, Q.; et al. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Sequencing Reveals Tissue Architecture in Esophageal Squamous Cell Carcinoma. EBioMedicine 2022, 84, 104281. [Google Scholar] [CrossRef]
- Ou, Z.; Lin, S.; Qiu, J.; Ding, W.; Ren, P.; Chen, D.; Wang, J.; Tong, Y.; Wu, D.; Chen, A.; et al. Single-Nucleus RNA Sequencing and Spatial Transcriptomics Reveal the Immunological Microenvironment of Cervical Squamous Cell Carcinoma. Adv. Sci. 2022, 9, e2203040. [Google Scholar] [CrossRef] [PubMed]
- Habibey, R.; Arias, J.E.R.; Striebel, J.; Busskamp, V. Microfluidics for Neuronal Cell and Circuit Engineering. Chem. Rev. 2022, 122, 14842–14880. [Google Scholar] [CrossRef] [PubMed]
- Rostom, R.; Svensson, V.; Teichmann, S.A.; Kar, G. Computational Approaches for Interpreting scRNA-Seq Data. FEBS Lett. 2017, 591, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative Single-Cell Analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Gal, E.; London, M.; Globerson, A.; Ramaswamy, S.; Reimann, M.W.; Muller, E.; Markram, H.; Segev, I. Rich Cell-Type-Specific Network Topology in Neocortical Microcircuitry. Nat. Neurosci. 2017, 20, 1004–1013. [Google Scholar] [CrossRef]
- Schafflick, D.; Xu, C.A.; Hartlehnert, M.; Cole, M.; Schulte-Mecklenbeck, A.; Lautwein, T.; Wolbert, J.; Heming, M.; Meuth, S.G.; Kuhlmann, T.; et al. Integrated Single Cell Analysis of Blood and Cerebrospinal Fluid Leukocytes in Multiple Sclerosis. Nat. Commun. 2020, 11, 247. [Google Scholar] [CrossRef]
- Morabito, S.; Miyoshi, E.; Michael, N.; Shahin, S.; Martini, A.C.; Head, E.; Silva, J.; Leavy, K.; Perez-Rosendahl, M.; Swarup, V. Single-Nucleus Chromatin Accessibility and Transcriptomic Characterization of Alzheimer’s Disease. Nat. Genet. 2021, 53, 1143–1155. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally Expanded CD8 T Cells Patrol the Cerebrospinal Fluid in Alzheimer’s Disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Sporns, O.; Tononi, G.; Kötter, R. The Human Connectome: A Structural Description of the Human Brain. PLoS Comput. Biol. 2005, 1, e42. [Google Scholar] [CrossRef]
- Fornito, A.; Arnatkevičiūtė, A.; Fulcher, B.D. Bridging the Gap between Connectome and Transcriptome. Trends Cogn. Sci. 2019, 23, 34–50. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-Wide Atlas of Gene Expression in the Adult Mouse Brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Bernard, A.; Lau, C.; Overly, C.C.; Dong, H.-W.; Kuan, C.; Pathak, S.; Sunkin, S.M.; Dang, C.; Bohland, J.W.; et al. An Anatomic Gene Expression Atlas of the Adult Mouse Brain. Nat. Neurosci. 2009, 12, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An Anatomically Comprehensive Atlas of the Adult Human Brain Transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef]
- Gravina, S.; Dong, X.; Yu, B.; Vijg, J. Single-Cell Genome-Wide Bisulfite Sequencing Uncovers Extensive Heterogeneity in the Mouse Liver Methylome. Genome Biol. 2016, 17, 150. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-Cell RNA Sequencing to Explore Immune Cell Heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Scott, D.W.; Gascoyne, R.D. The Tumour Microenvironment in B Cell Lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef]
- Stewart, B.J.; Fergie, M.; Young, M.D.; Jones, C.; Sachdeva, A.; Blain, A.; Bacon, C.M.; Rand, V.; Ferdinand, J.R.; James, K.R.; et al. Spatial and Molecular Profiling of the Mononuclear Phagocyte Network in Classic Hodgkin Lymphoma. Blood 2023, 141, 2343–2358. [Google Scholar] [CrossRef]
- He, Z.; Maynard, A.; Jain, A.; Gerber, T.; Petri, R.; Lin, H.-C.; Santel, M.; Ly, K.; Dupré, J.-S.; Sidow, L.; et al. Lineage Recording in Human Cerebral Organoids. Nat. Methods 2022, 19, 90–99. [Google Scholar] [CrossRef]
- Peixoto, L.; Risso, D.; Poplawski, S.G.; Wimmer, M.E.; Speed, T.P.; Wood, M.A.; Abel, T. How Data Analysis Affects Power, Reproducibility and Biological Insight of RNA-Seq Studies in Complex Datasets. Nucleic Acids Res. 2015, 43, 7664–7674. [Google Scholar] [CrossRef]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Toth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-Cell Spatial Reconstruction Reveals Global Division of Labour in the Mammalian Liver. Nature 2017, 542, 352–356. [Google Scholar] [CrossRef]
- Madsen, R.; Lundstedt, T.; Trygg, J. Chemometrics in Metabolomics—A Review in Human Disease Diagnosis. Anal. Chim. Acta 2010, 659, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Steuer, R. Review: On the Analysis and Interpretation of Correlations in Metabolomic Data. Brief. Bioinform. 2006, 7, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.R.; Palsson, B.Ø. The Model Organism as a System: Integrating “omics” Data Sets. Nat. Rev. Mol. Cell Biol. 2006, 7, 198–210. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Ren, X.; Zhong, G.; Zhang, Q.; Zhang, L.; Sun, Y.; Zhang, Z. Reconstruction of Cell Spatial Organization from Single-Cell RNA Sequencing Data Based on Ligand-Receptor Mediated Self-Assembly. Cell Res. 2020, 30, 763–778. [Google Scholar] [CrossRef]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-Scale Spatial Gene Expression in the Human Dorsolateral Prefrontal Cortex. Nat. Neurosci. 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene Expression Recovery for Single-Cell RNA Sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef]
- Li, W.V.; Li, J.J. An Accurate and Robust Imputation Method scImpute for Single-Cell RNA-Seq Data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef]
- Pham, D.; Tan, X.; Balderson, B.; Xu, J.; Grice, L.F.; Yoon, S.; Willis, E.F.; Tran, M.; Lam, P.Y.; Raghubar, A.; et al. Robust Mapping of Spatiotemporal Trajectories and Cell–Cell Interactions in Healthy and Diseased Tissues. Nat. Commun. 2023, 14, 7739. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Qing, T.; Zheng, Y.; Jin, L.; Shi, L. Advances in Single-Cell RNA Sequencing and Its Applications in Cancer Research. Oncotarget 2017, 8, 53763–53779. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Ritchie, G.R.S.; Roumeliotis, T.I.; Jayasuriya, R.L.; Clark, M.J.; Brooks, R.A.; Binch, A.L.A.; Shah, K.M.; Coyle, R.; Pardo, M.; et al. Integrative Epigenomics, Transcriptomics and Proteomics of Patient Chondrocytes Reveal Genes and Pathways Involved in Osteoarthritis. Sci. Rep. 2017, 7, 8935. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Southam, L.; Roumeliotis, T.I.; Clark, M.J.; Jayasuriya, R.L.; Swift, D.; Shah, K.M.; Butterfield, N.C.; Brooks, R.A.; McCaskie, A.W.; et al. A Molecular Quantitative Trait Locus Map for Osteoarthritis. Nat. Commun. 2021, 12, 1309. [Google Scholar] [CrossRef]
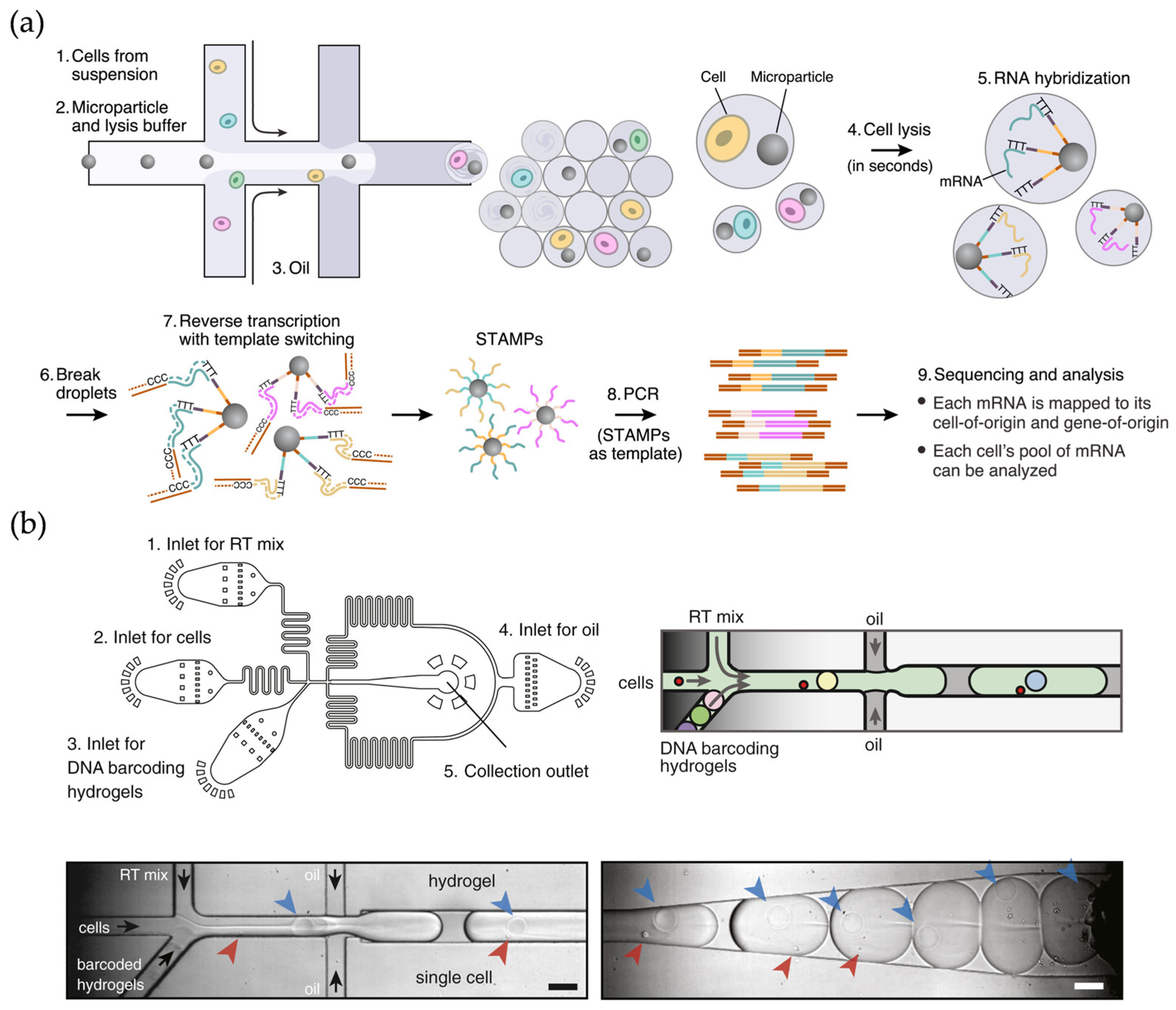
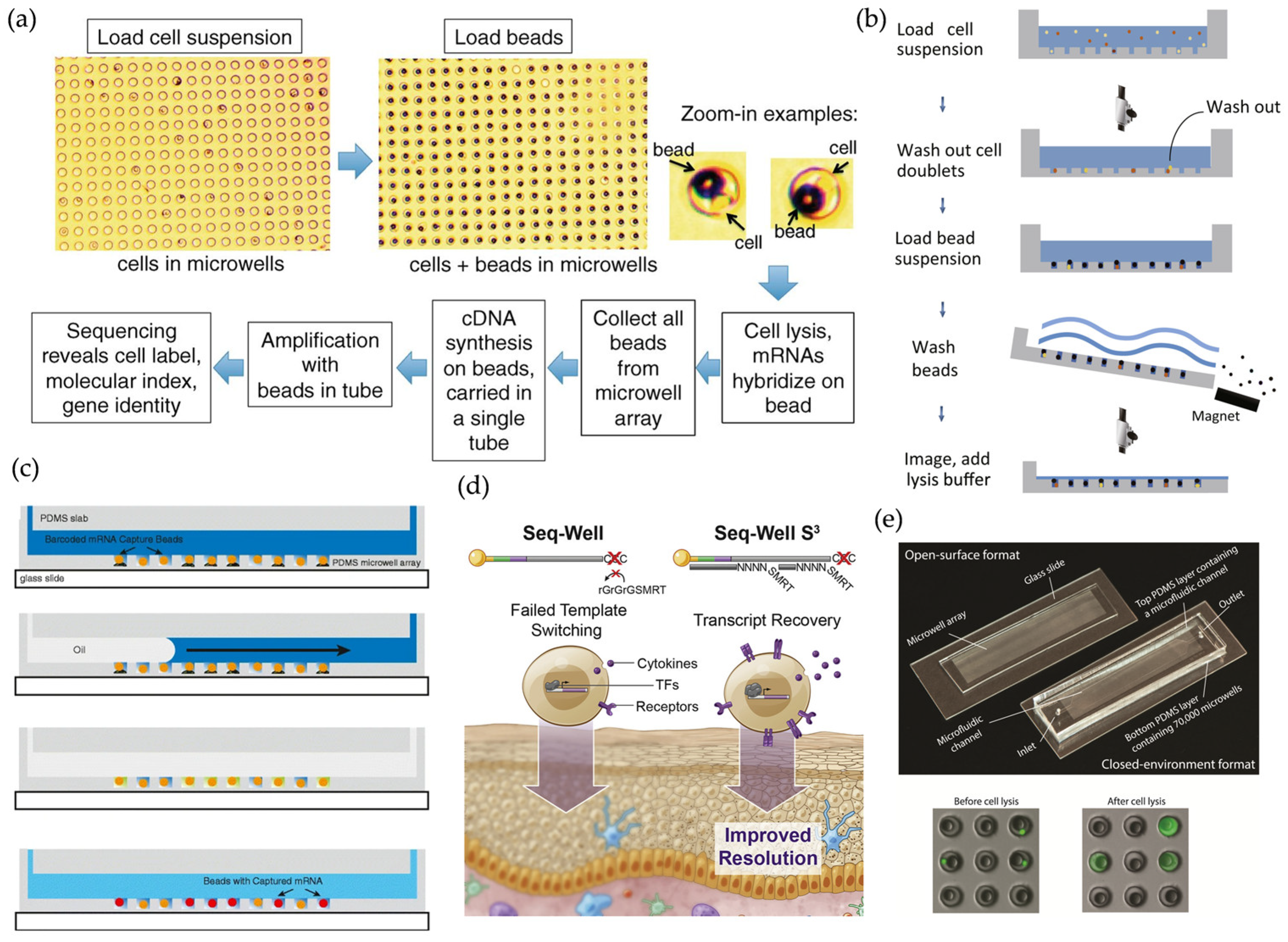
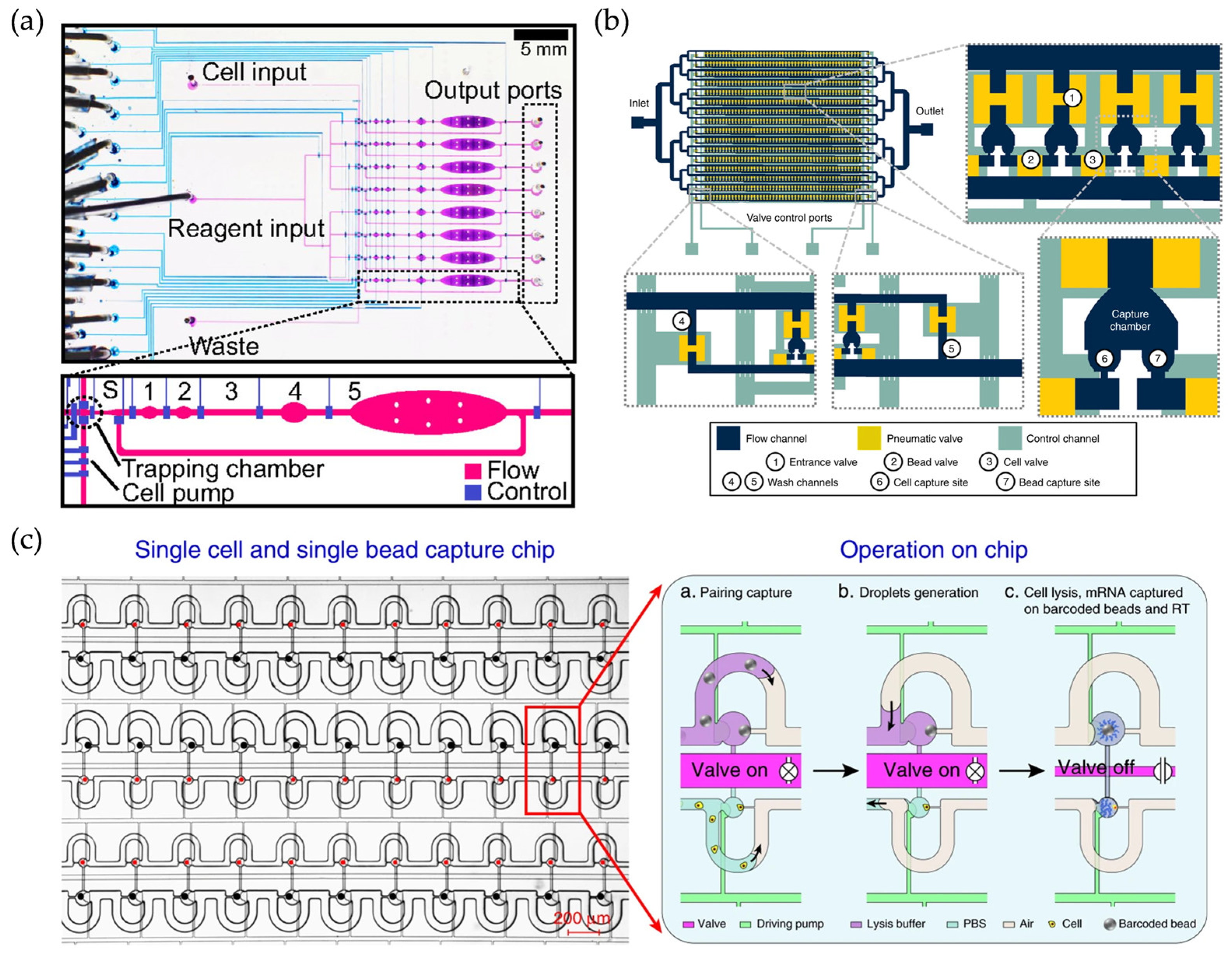
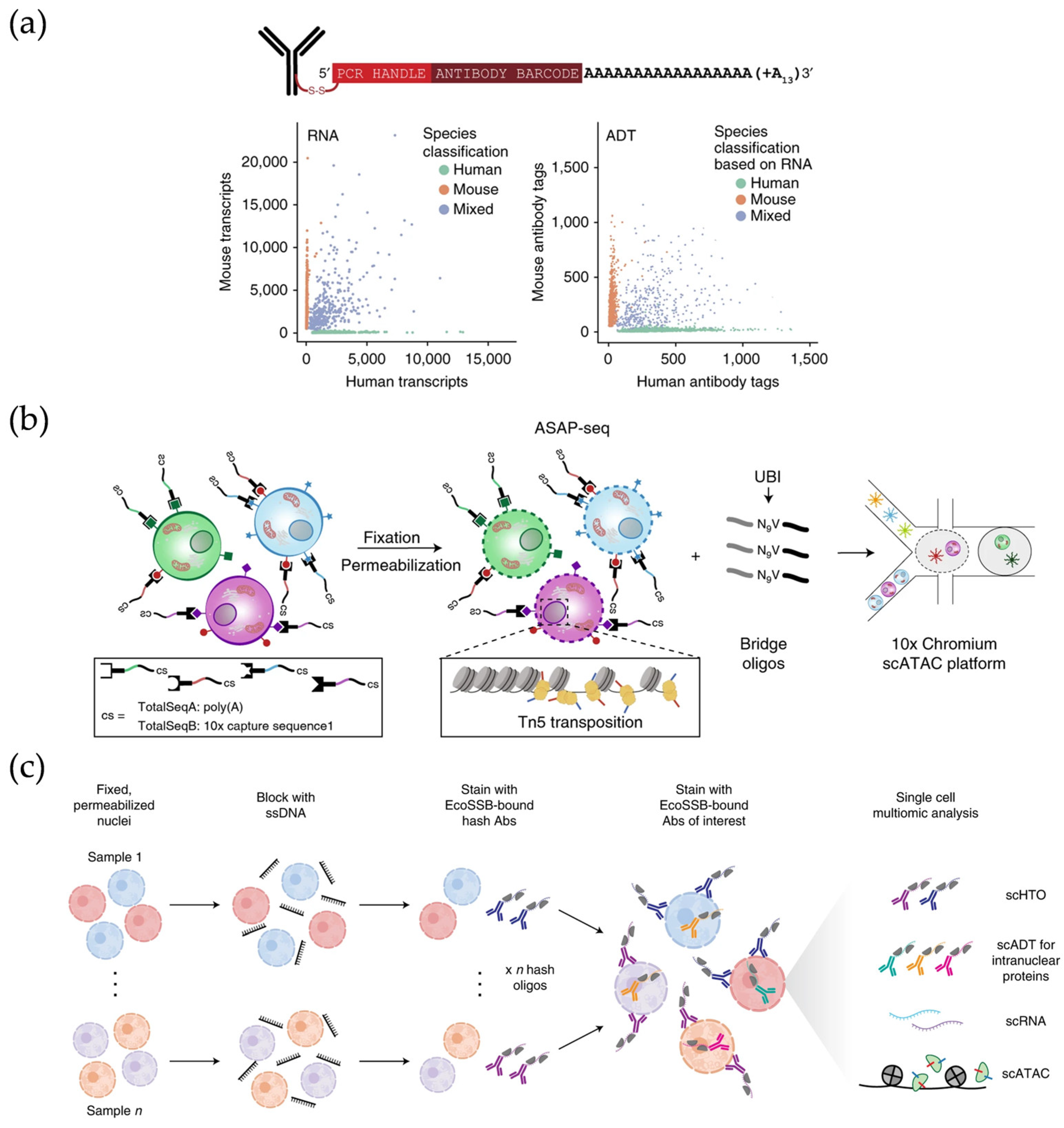

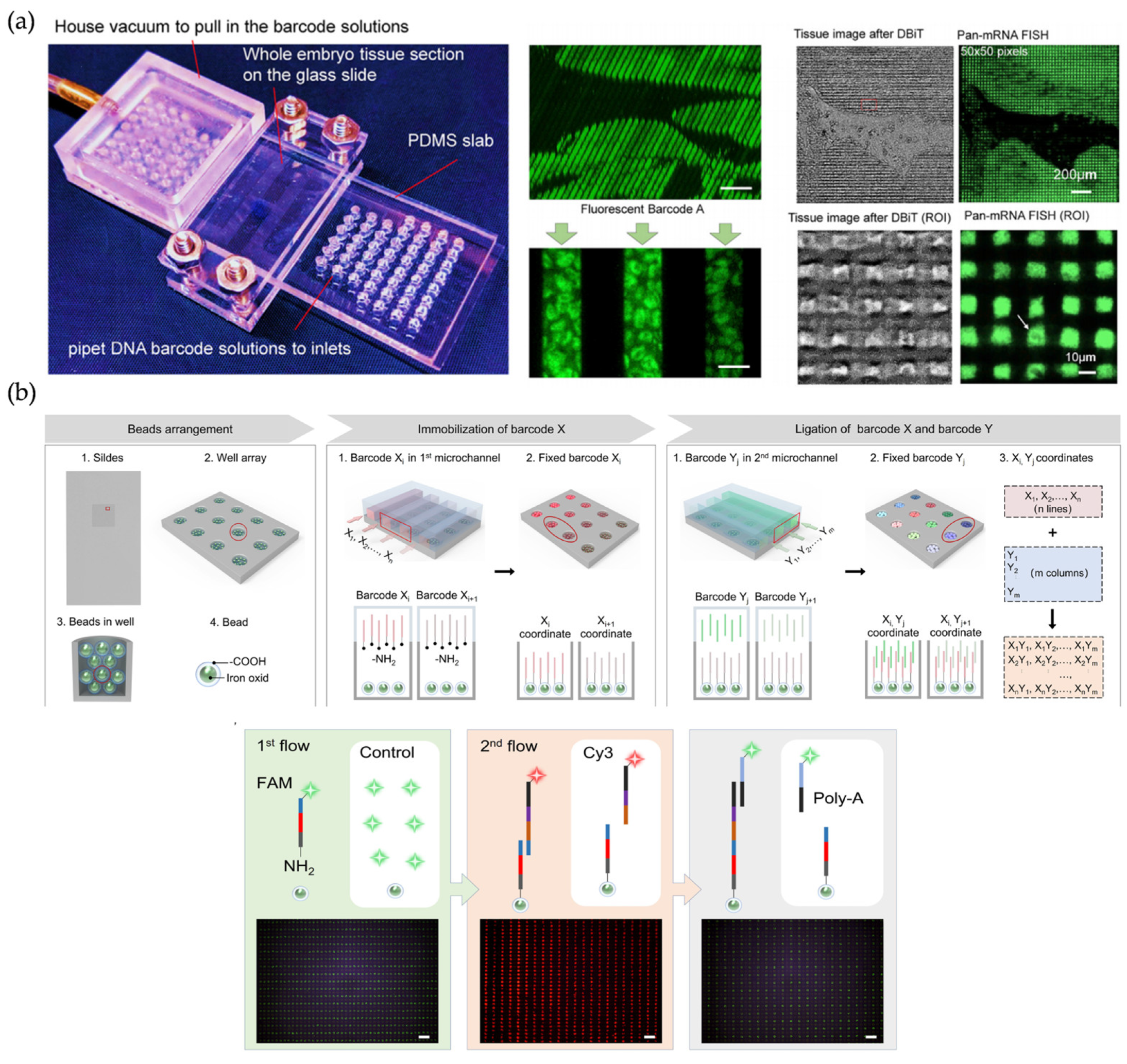

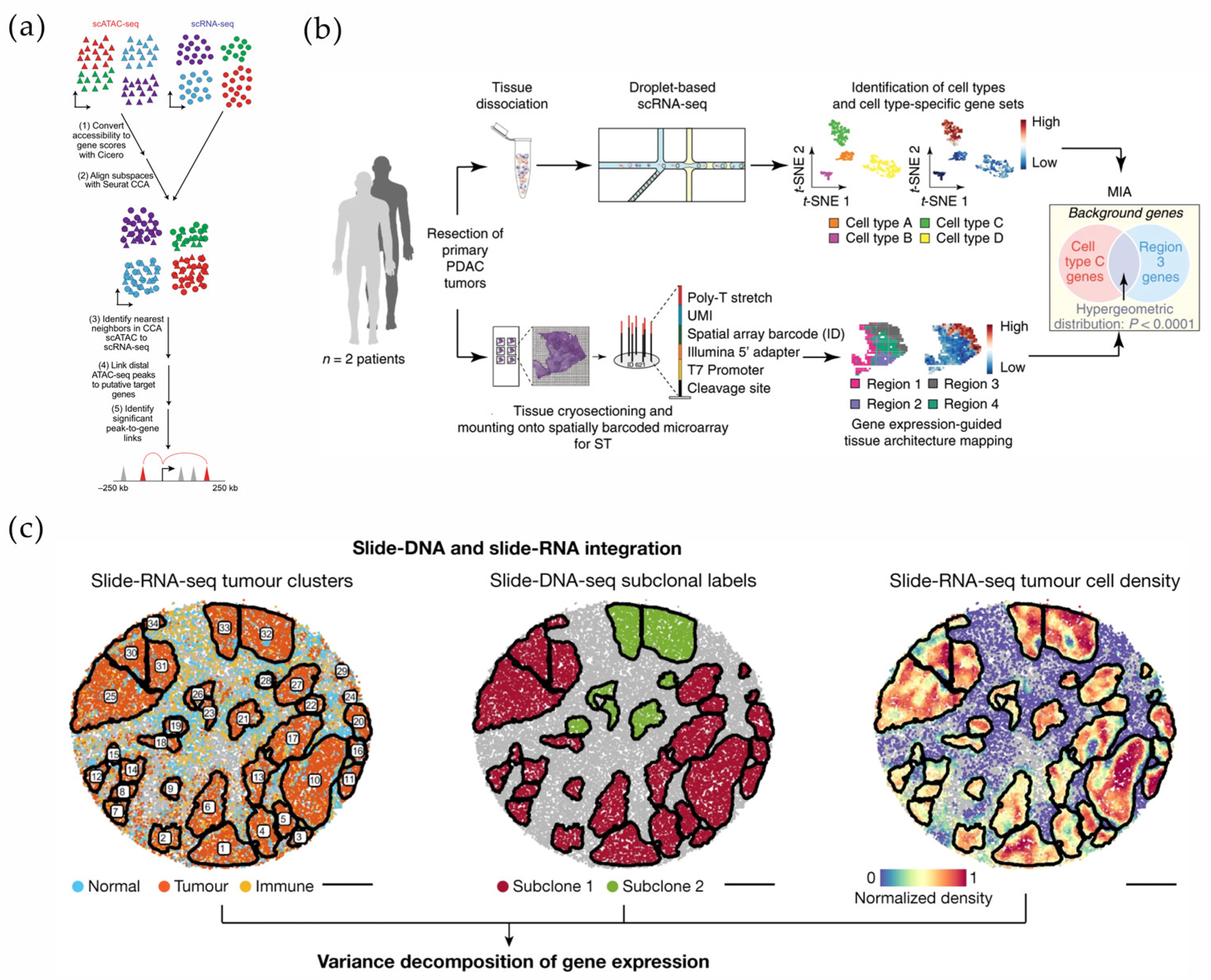
| Technology | Year | Method | Sample Type | Array Substrate | Resolution | Ref. |
|---|---|---|---|---|---|---|
| Array-based SRT | 2016 | ST | FF | Glass | 100 μm | [79] |
| Array-based SRT | 2019 | Visium | FF | Glass | 55 μm | [86] |
| Array-based SRT | 2019 | Slide-Seq | FF | Glass | 10 μm | [81] |
| Array-based SRT | 2019 | HDST | FF | Silicon | 2 μm | [79] |
| Microchannel-based SRT | 2020 | DBiT-seq | FF/FFPE | Microfluidic Channel divided tissue | 10–50 μm | [82,83] |
| Microchannel-based SRT | 2023 | Well-ST-seq | FF | PDMS | 10–50 μm | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Yu, N.; Zhang, J.; Yang, B. Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics. Micromachines 2025, 16, 426. https://doi.org/10.3390/mi16040426
Sun Y, Yu N, Zhang J, Yang B. Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics. Micromachines. 2025; 16(4):426. https://doi.org/10.3390/mi16040426
Chicago/Turabian StyleSun, Yueqiu, Nianzuo Yu, Junhu Zhang, and Bai Yang. 2025. "Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics" Micromachines 16, no. 4: 426. https://doi.org/10.3390/mi16040426
APA StyleSun, Y., Yu, N., Zhang, J., & Yang, B. (2025). Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics. Micromachines, 16(4), 426. https://doi.org/10.3390/mi16040426