

Droplets for Gene Editing Using CRISPR-Cas9 and Clonal Selection Improvement Using Hydrogels



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Design and Fabrication
2.2. Modified HiPSC Line
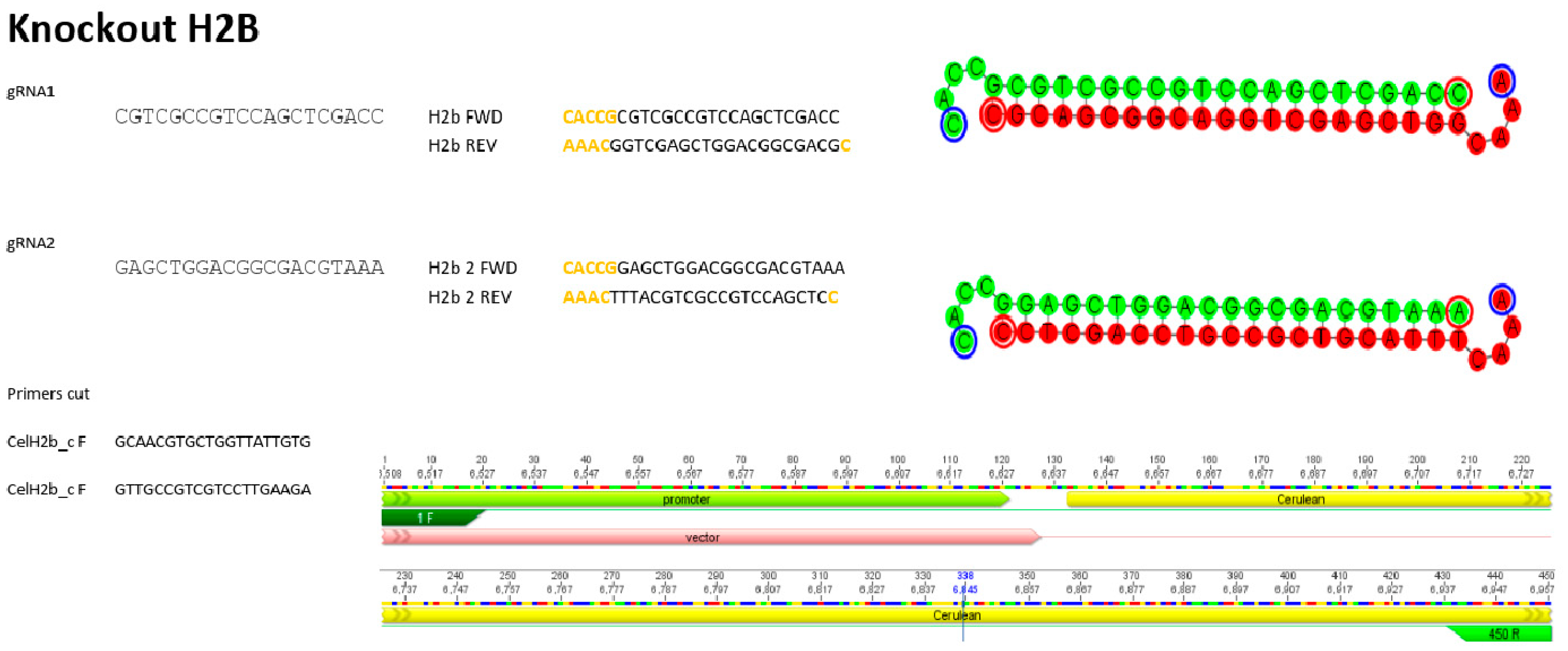
2.3. Design of the Guides and Flanking Primers
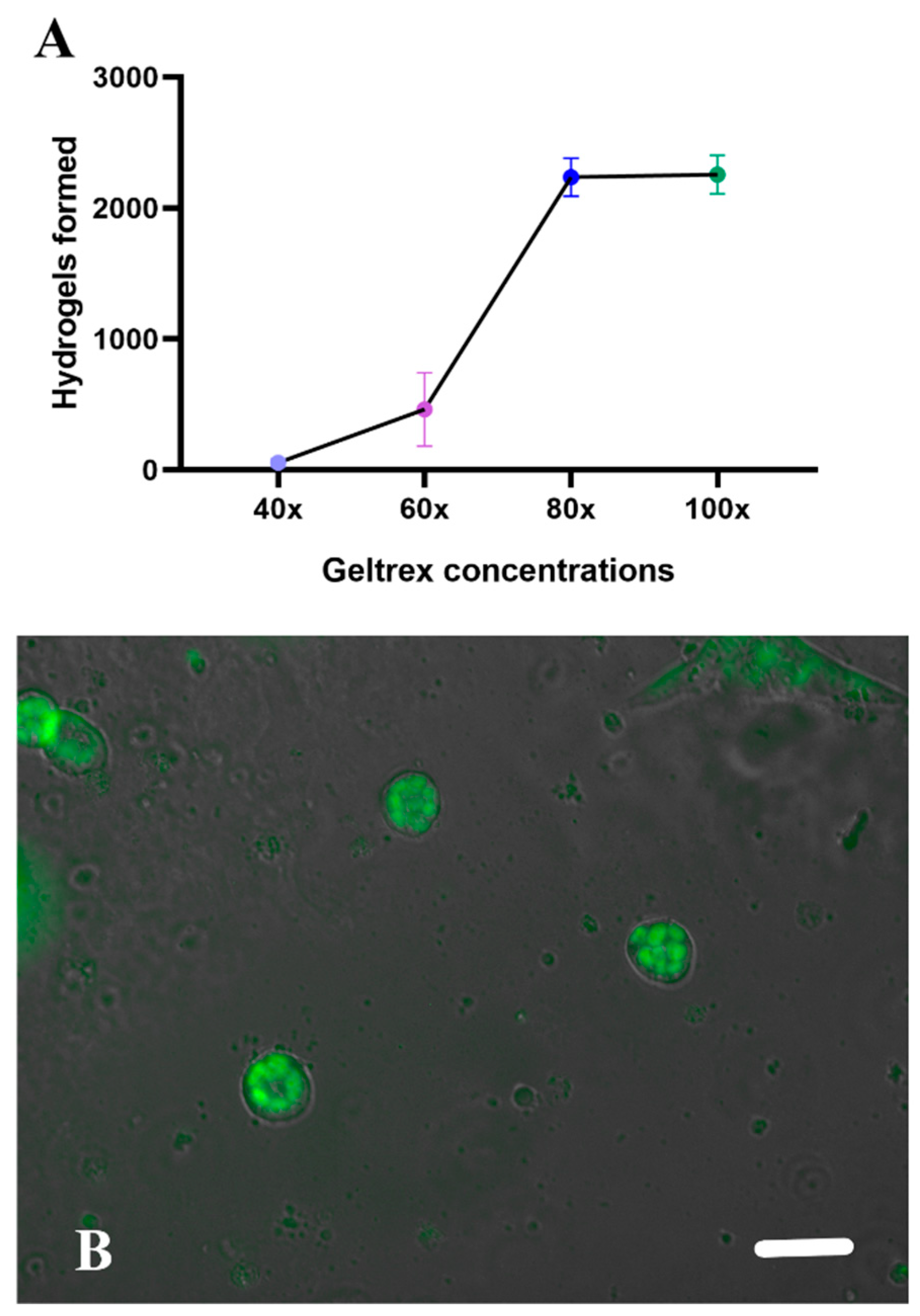
2.4. Production of Droplets and Hydrogels
2.5. Image Analysis
3. Results
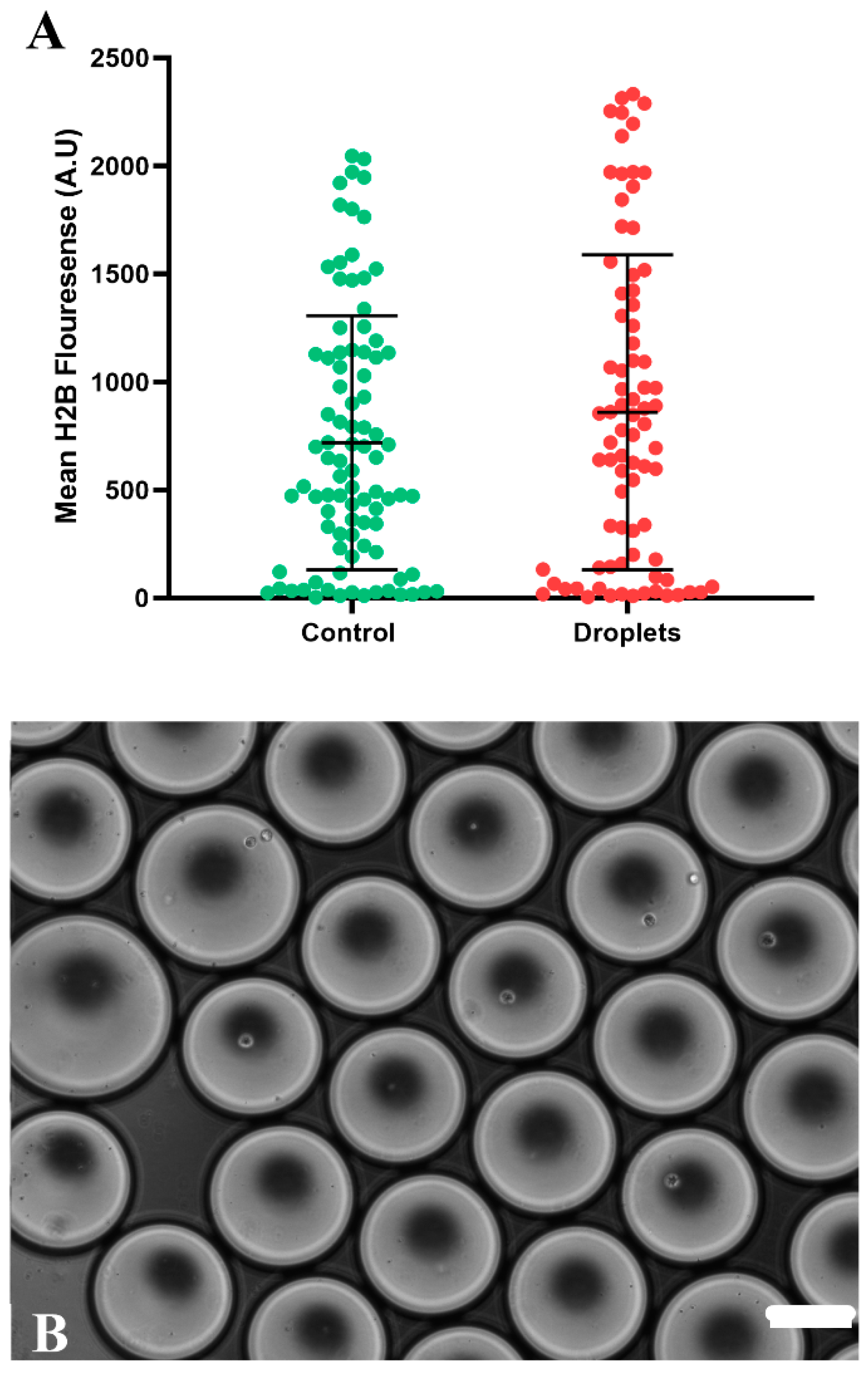
3.1. Editing Efficiency by the CRISPR-Cas9 Method in Droplets
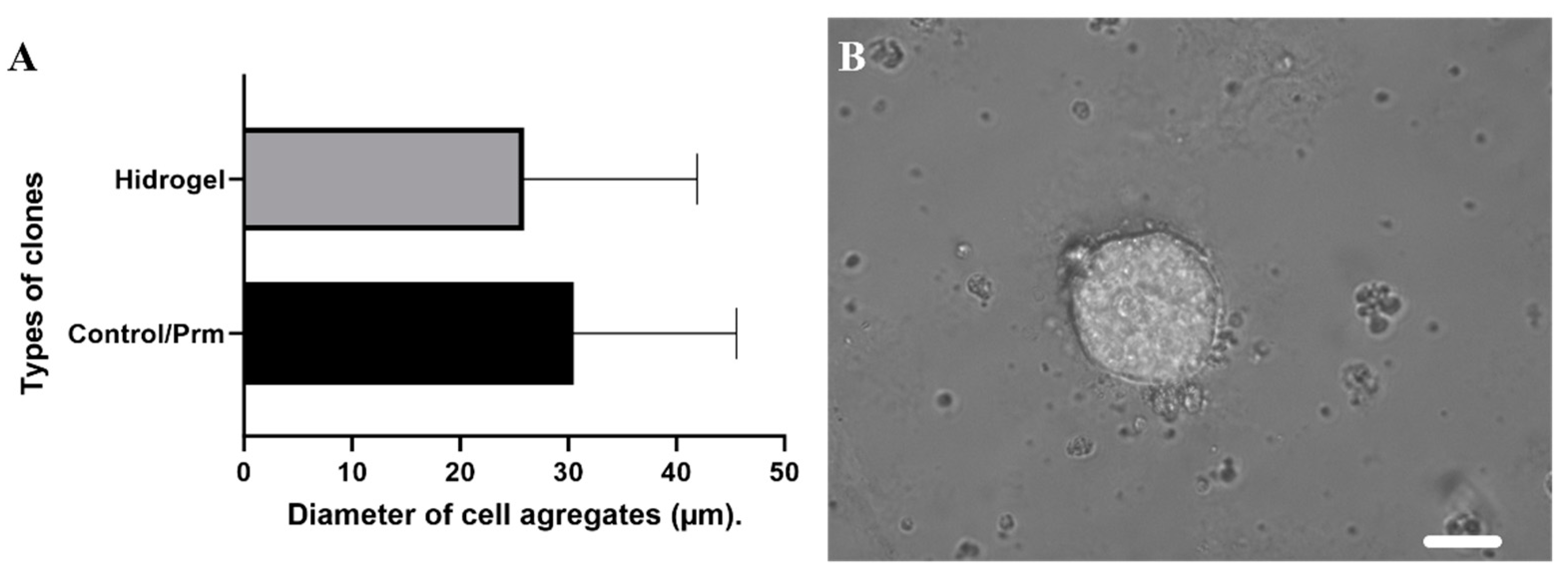
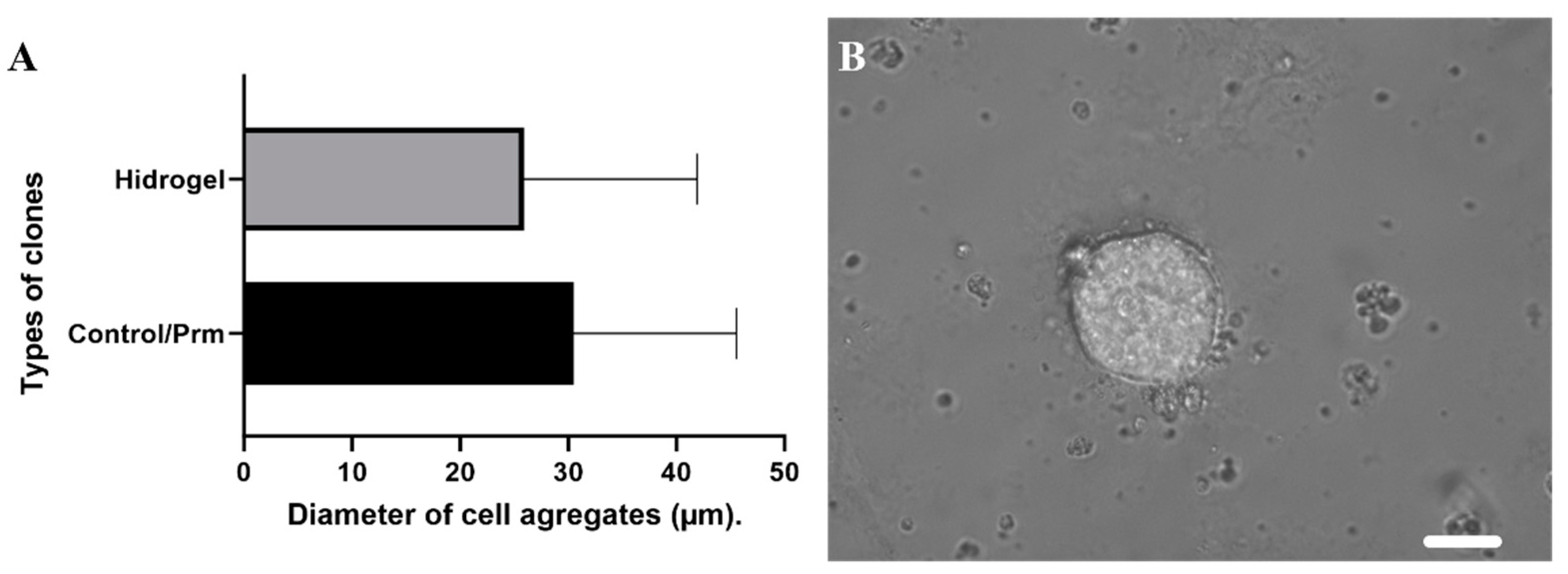
3.2. Clonal Selection Using Extracellular Matrix Hydrogels
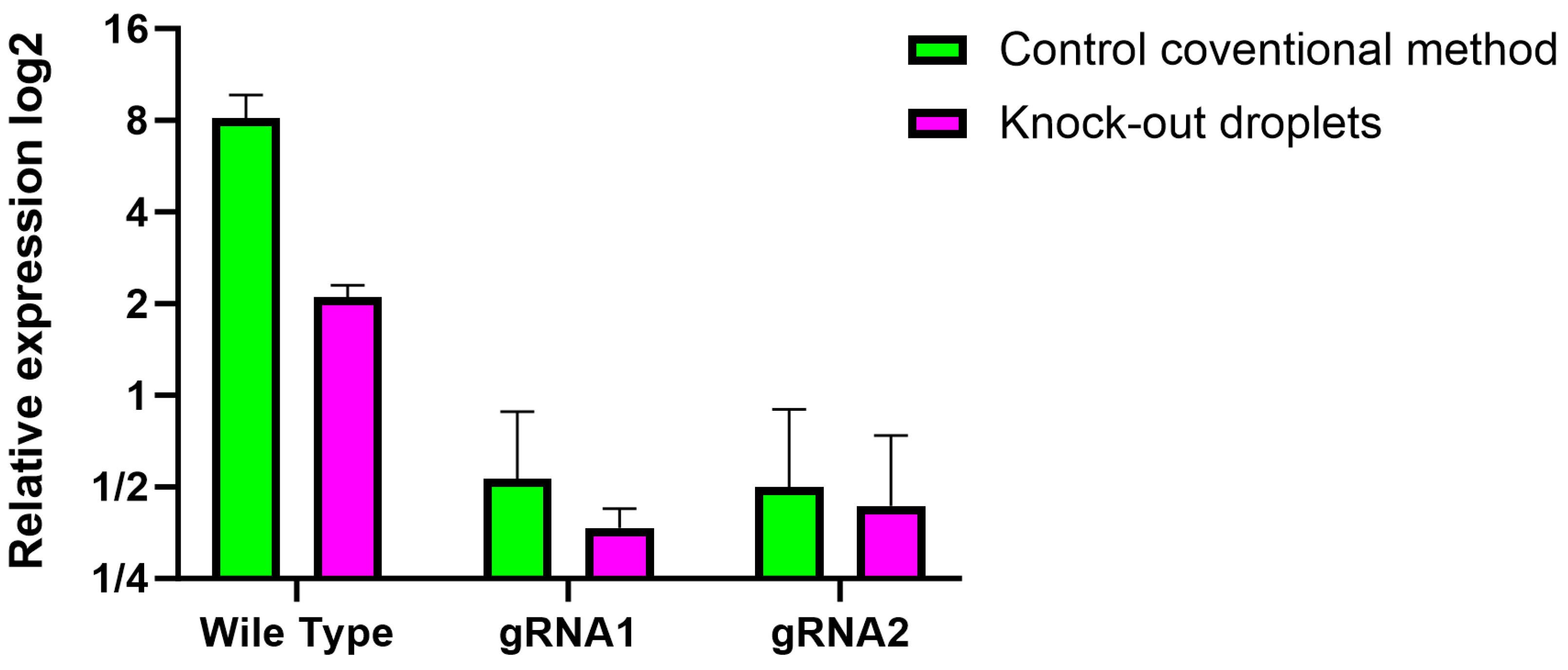
3.3. Molecular Verification of the Knockout of H2B–mCerulean
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef]
- Bire, S.; Ley, D.; Casteret, S.; Mermod, N.; Bigot, Y.; Rouleux-Bonnin, F. Optimization of the piggyBac transposon using mRNA and insulators: Toward a more reliable gene delivery system. PLoS ONE 2013, 8, e82559. [Google Scholar] [CrossRef]
- Mansouri, S.; Lavigne, P.; Corsi, K.; Benderdour, M.; Beaumont, E.; Fernandes, J.C. Chitosan-DNA nanoparticles as non-viral vectors in gene therapy: Strategies to improve transfection efficacy. Eur. J. Pharm. Biopharm. 2004, 57, 1–8. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, J.; Cheng, M.; Liao, X.; Peng, S. Review of CRISPR/Cas9 sgRNA Design Tools. In Interdisciplinary Sciences: Computational Life Sciences; Springer: Berlin/Heidelberg, Germany, 2018; Volume 10, pp. 455–465. [Google Scholar]
- Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. CRISPR–Cas9 gRNA efficiency prediction: An overview of predictive tools and the role of deep learning. Nucleic Acid Res. 2022, 50, 3616–3637. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Akoulitchev, A.; Sharon, B.; Wolf, Y.I. Evolution and classification of CRISPR-Cas systems. Microbiology 2011, 9, 326–336. [Google Scholar]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided DNA silencing systems in bacteria and archaea. Nature 2012, 482, 361–368. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F.; Lander, E.S. Double nicking by RNA-guided CRISPR Cas9 for efficient genome editing. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar]
- Doudna, J.A.; Charpentier, E. Genome editing. N. Engl. J. Med. 2014, 371, 2678–2687. [Google Scholar]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Xing, Y.; Liu, J.; Guo, X.; Liu, H.; Zeng, W.; Wang, Y.; Zhang, C.; Lu, Y.; He, D.; Ma, S.; et al. Engineering organoid microfluidic system for biomedical and health engineering: A review. Chin. J. Chem. Eng. Mater. China 2021, 30, 244–254. [Google Scholar] [CrossRef]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biol. Targets Ther. 2021, 15, 353–361. [Google Scholar]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef]
- Burridge, P.W.; Keller, G.; Gold, J.D.; Wu, J.C. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 2012, 10, 16–28. [Google Scholar] [CrossRef]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef]
- Headen, D.M.; García, J.R.; García, A.J. Parallel droplet microfluidics for high throughput cell encapsulation and synthetic microgel generation. Microsyst. Nanoeng. 2018, 4, 17076. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Baroud, C.N.; Gallaire, F.; Dangla, R. Dynamics of microfluidic droplets. Lab A Chip R. Soc. Chem. 2010, 10, 2032–2045. [Google Scholar] [CrossRef]
- Ishida, T.; McLaughlin, D.; McLaughlin, D.; Tanaka, Y. First-come-first-store microfluidic device of droplets using hydrophobic passive microvalves. Sens. Actuators B Chem. 2018, 254, 1005–1010. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; McCarroll, S.A. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Zhang, F. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef]
- Chen, F.; Zhan, Y.; Geng, T.; Lian, H.; Xu, P.; Lu, C. Chemical transfection of cells in picoliter aqueous droplets in fluorocarbon oil. Anal. Chem. 2011, 83, 8816–8820. [Google Scholar] [CrossRef]
- Wang, J.H.; Lee, G.B. Formation of Tunable, Emulsion Micro-Droplets Utilizing Flow-Focusing Channels and a Normally-Closed Micro-Valve. Micromachines 2013, 4, 306–320. [Google Scholar] [CrossRef]
- Pérez-sosa, C.; Peñaherrera-pazmiño, A.B.; Rosero, G.; Bourguignon, N.; Aravelli, A.; Bhansali, S.; Pérez, M.S.; Lerner, B. Novel Reproducible Manufacturing and Reversible Sealing Method for Microfluidic Devices. Micromachines 2022, 13, 650. [Google Scholar] [CrossRef]
- Olmos, C.M.; Vaca, A.; Rosero, G.; Peñaherrera, A.; Perez, C.; de Sá Carneiro, I.; Vizuete, K.; Arroyo, C.R.; Debut, A.; Pérez, M.S.; et al. Epoxy resin mold and PDMS microfluidic devices through photopolymer flexographic printing plate. Sens. Actuators B Chem. 2019, 288, 742–748. [Google Scholar] [CrossRef]
- Pérez-Sosa, C.; Sanluis-Verdes, A.; Waisman, A.; Lombardi, A.; Rosero, G.; Greca, A.L.; Lerner, B. Single cell transfection of human-induced pluripotent stem cells using a droplet-based microfluidic system. R. Soc. Open Sci. 2022, 9, 211510. [Google Scholar] [CrossRef] [PubMed]
- Joo, B.; Hur, J.; Kim, G.B.; Yun, S.G.; Chung, A.J. Highly Efficient Transfection of Human Primary T Lymphocytes Using Droplet-Enabled Mechanoporation. ACS Nano 2021, 15, 12888–12898. [Google Scholar] [CrossRef]
- Peñaherrera, A.; Payés, C.; Sierra-Rodero, M.; Vega, M.; Rosero, G.; Lerner, B.; Helguera, G.; Pérez, M.S. Evaluation of cell culture in microfluidic chips for application in monoclonal antibody production. Microelectron. Eng. 2016, 158, 126–129. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Sosa, C.; Pérez, M.S.; Vallejo-Janeta, A.P.; Bhansali, S.; Miriuka, S.; Lerner, B. Droplets for Gene Editing Using CRISPR-Cas9 and Clonal Selection Improvement Using Hydrogels. Micromachines 2024, 15, 413. https://doi.org/10.3390/mi15030413
Pérez-Sosa C, Pérez MS, Vallejo-Janeta AP, Bhansali S, Miriuka S, Lerner B. Droplets for Gene Editing Using CRISPR-Cas9 and Clonal Selection Improvement Using Hydrogels. Micromachines. 2024; 15(3):413. https://doi.org/10.3390/mi15030413
Chicago/Turabian StylePérez-Sosa, Camilo, Maximiliano S. Pérez, Alexander Paolo Vallejo-Janeta, Shekhar Bhansali, Santiago Miriuka, and Betiana Lerner. 2024. "Droplets for Gene Editing Using CRISPR-Cas9 and Clonal Selection Improvement Using Hydrogels" Micromachines 15, no. 3: 413. https://doi.org/10.3390/mi15030413
APA StylePérez-Sosa, C., Pérez, M. S., Vallejo-Janeta, A. P., Bhansali, S., Miriuka, S., & Lerner, B. (2024). Droplets for Gene Editing Using CRISPR-Cas9 and Clonal Selection Improvement Using Hydrogels. Micromachines, 15(3), 413. https://doi.org/10.3390/mi15030413