Tissue Chips and Microphysiological Systems for Disease Modeling and Drug Testing


Abstract
1. Introduction
2. Cardiac Tissue Chips
2.1. Function
2.2. Cell Types and Extracellular Matrix
2.3. Cellular Organization
2.4. Physical Stresses, Fluid Flow, and Electrical Signals
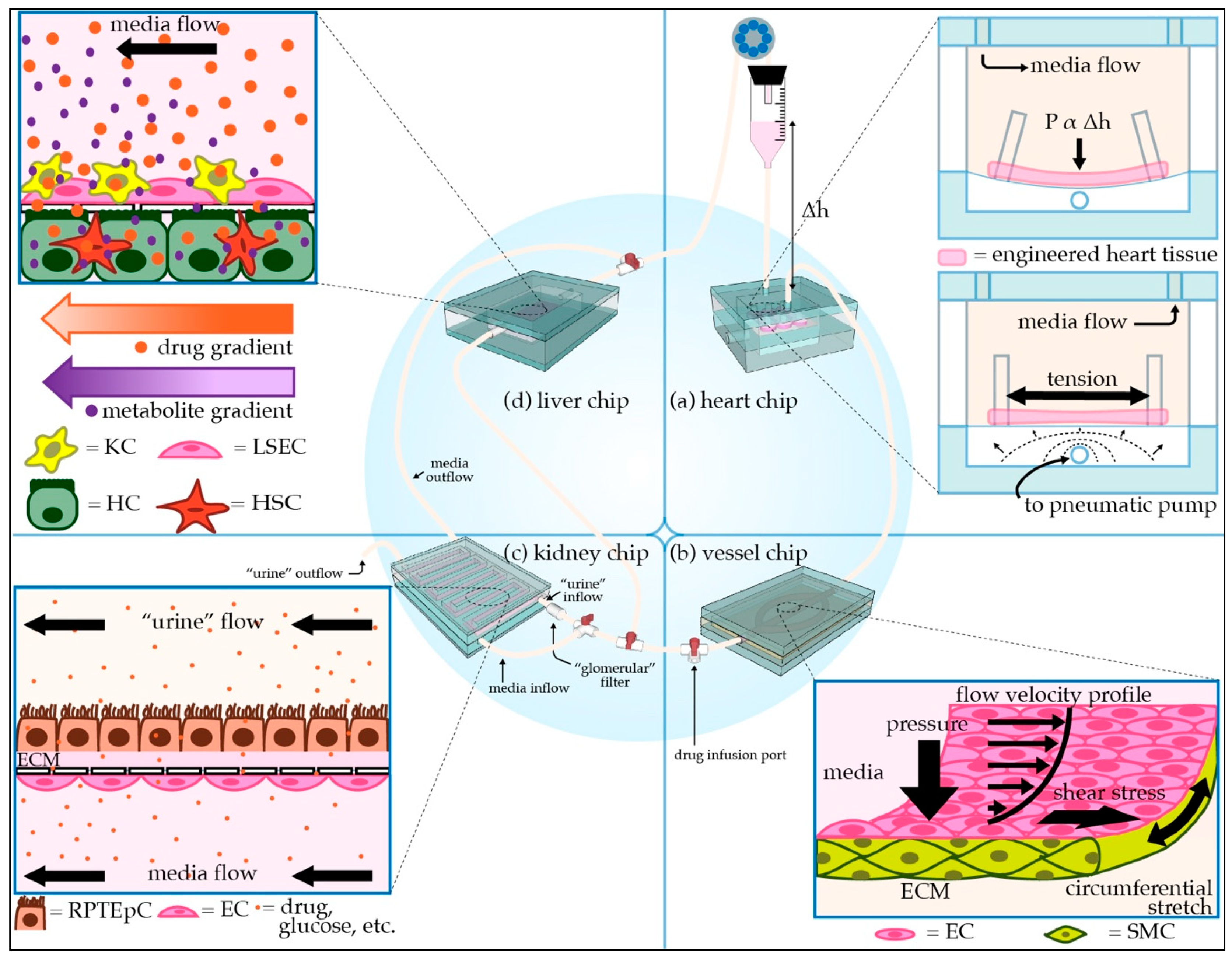
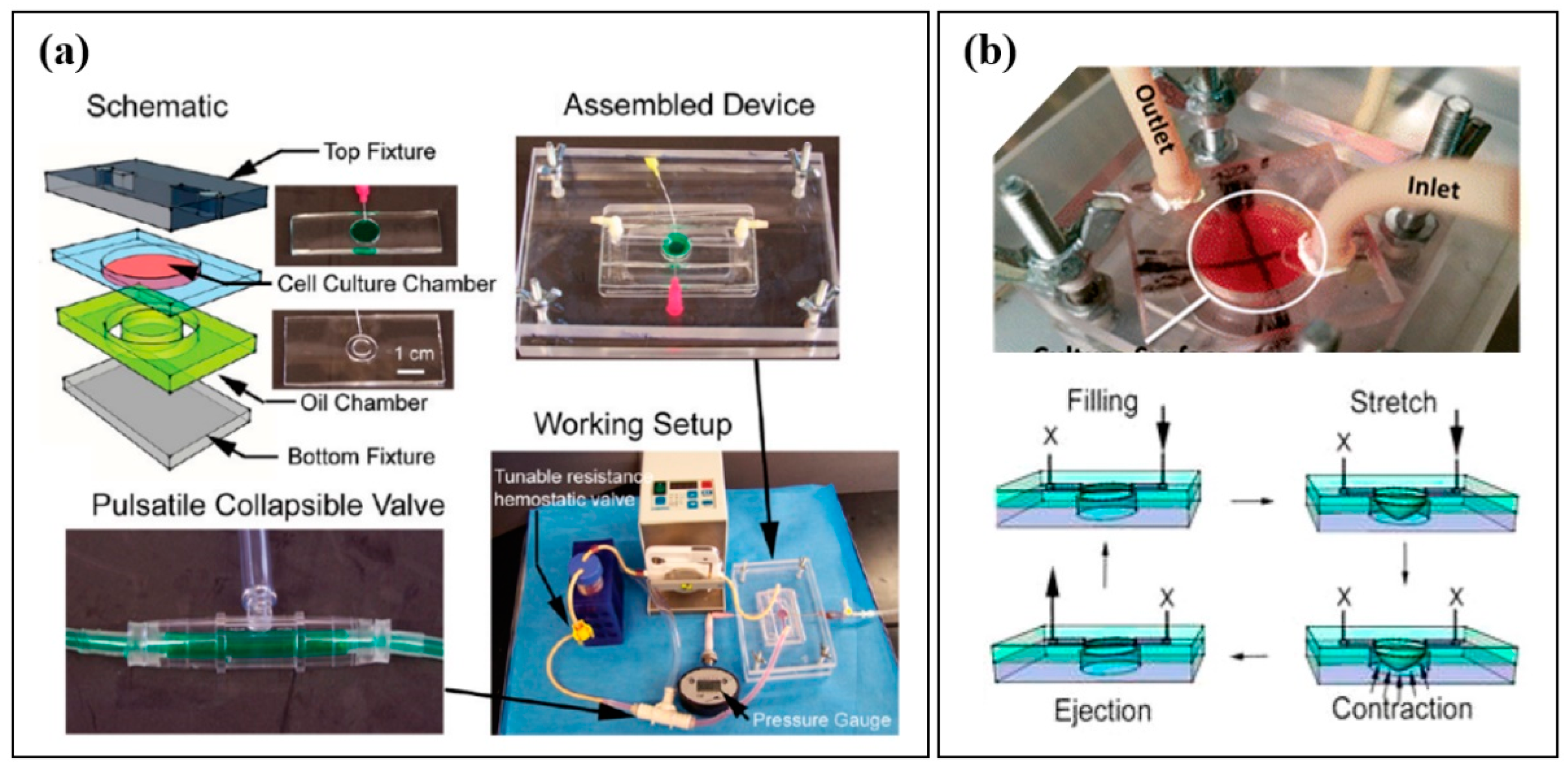
2.5. Examples of Cardiac Tissue Chips
2.6. Limitations
3. Vascular Tissue Chips
3.1. Function
3.2. Cell Types and Extracellular Matrix
3.3. Cellular Organization
3.4. Physical Stresses and Fluid Flow
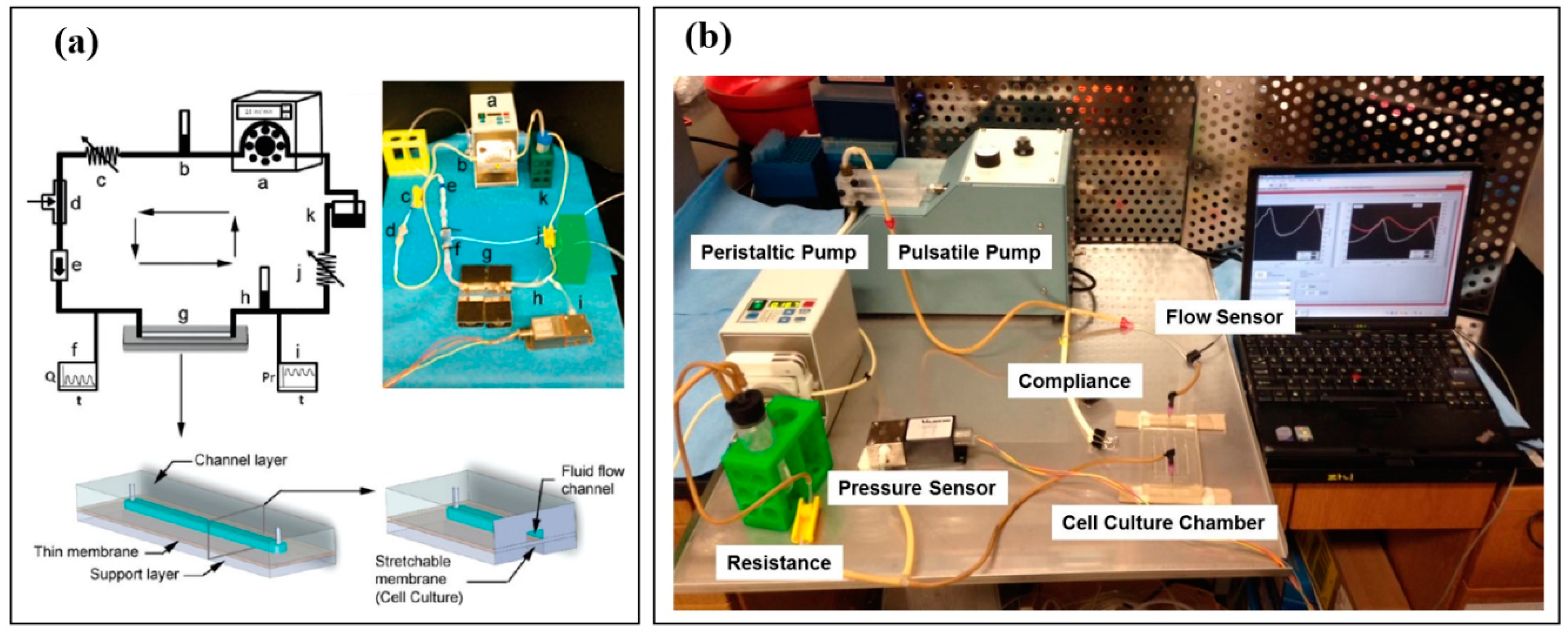
3.5. Examples of Vascular Tissue Chips
3.6. Limitations
4. Liver Tissue Chips
4.1. Function
4.2. Cell Types and Extracellular Matrix
4.3. Cellular Organization
4.4. Physical Stresses and Fluid Flow
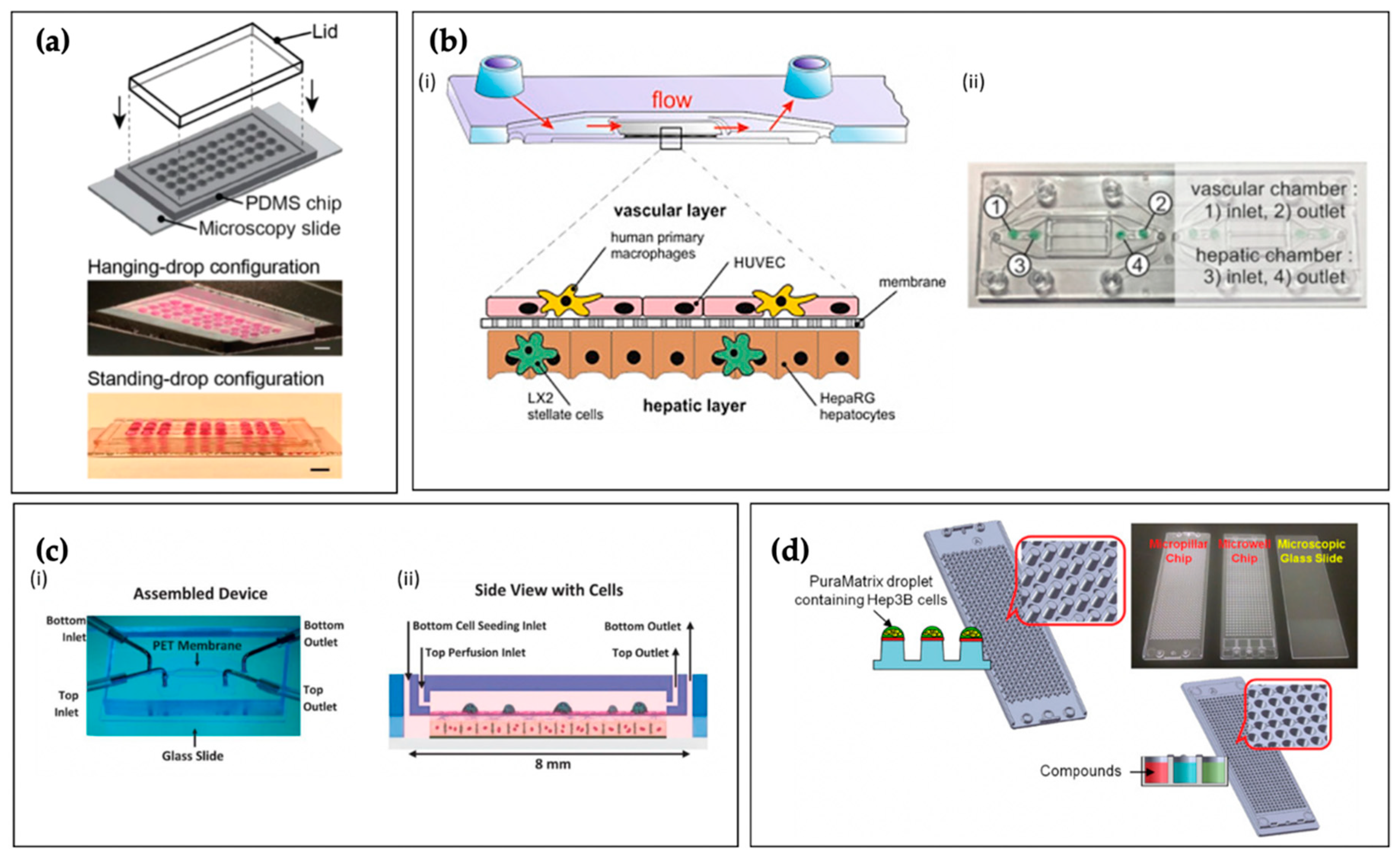
4.5. Examples of Liver Tissue Chips
4.6. Limitations
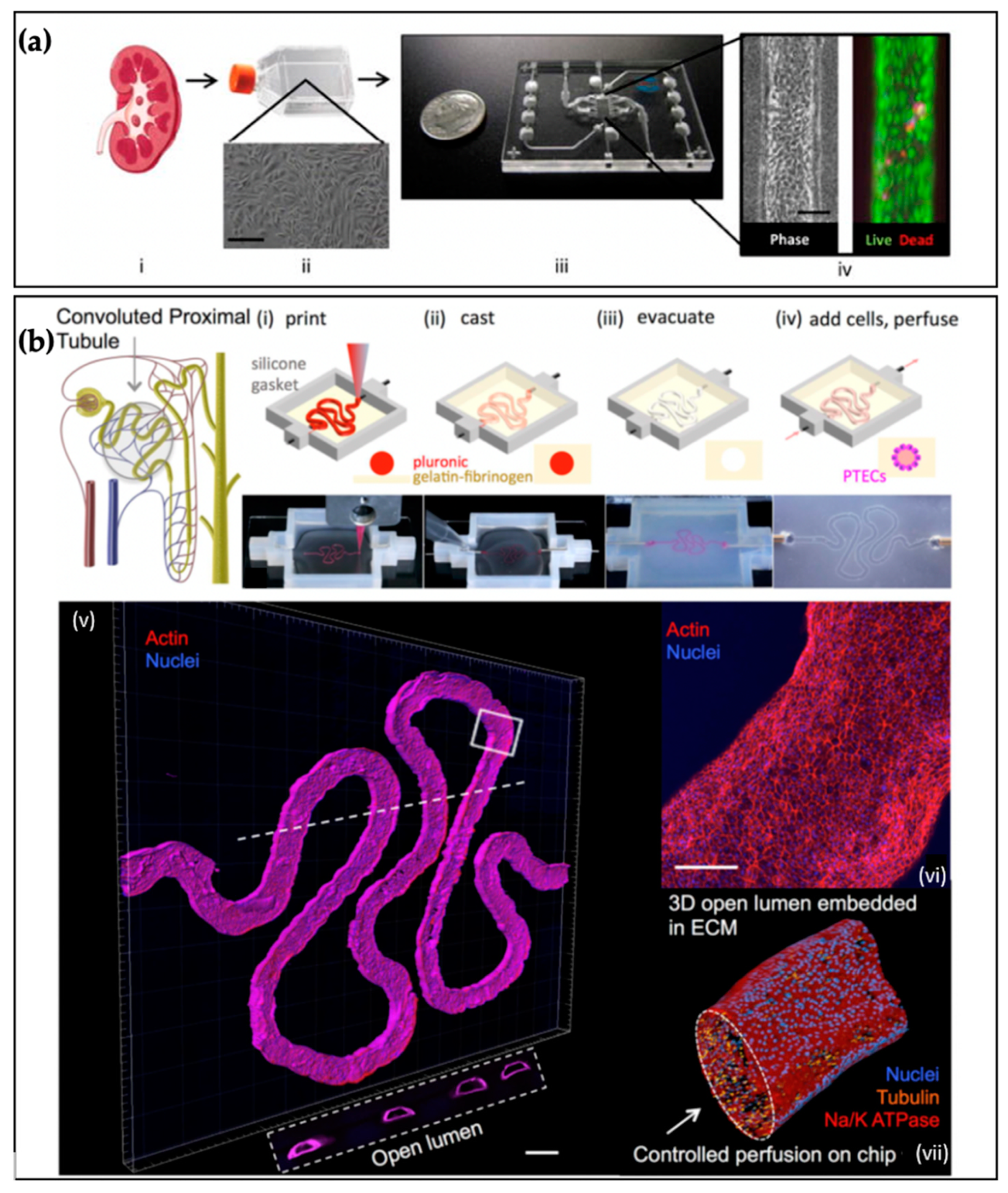
5. Kidney Tissue Chips
5.1. Function
5.2. Cell Types and Extracellular Matrix
5.3. Cellular Organization
5.4. Physical Stresses and Fluid Flow
5.5. Examples of Kidney Tissue Chips
5.6. Limitations
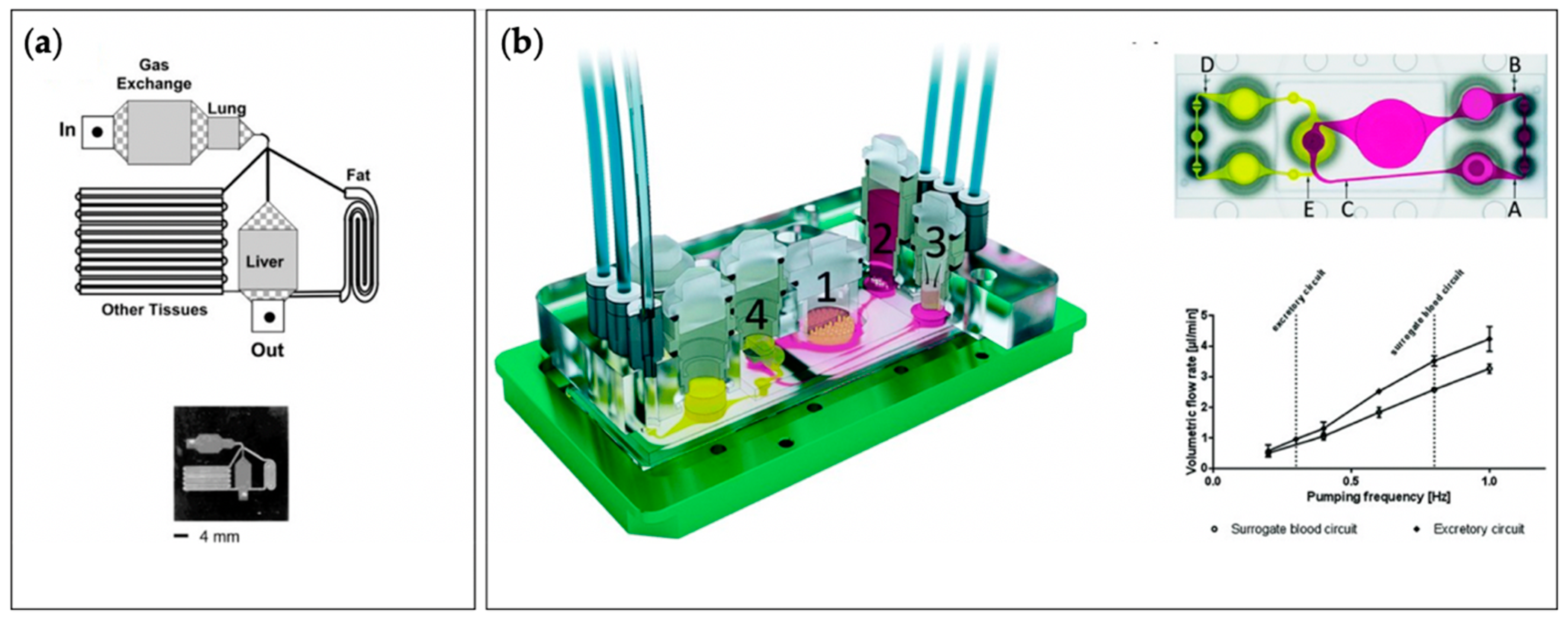
6. Microphysiological Systems
7. Challenges Associated with Design and Construction of Microphysiological Systems
7.1. Communication
7.2. Nondestructive Monitoring
7.3. Material Selection and Fabrication of TCs and MPS
8. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417–429. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Tagle, D. National Center for Advancing Translational Sciences: About Tissue Chip. Available online: https://ncats.nih.gov/tissuechip/about (accessed on 28 November 2020).
- Tagle, D. National Center for Advancing Translational Sciences: Tissue Chip Initiatives & Projects. Available online: https://ncats.nih.gov/tissuechip/projects (accessed on 28 November 2020).
- Raimondi, I.; Izzo, L.; Tunesi, M.; Comar, M.; Albani, D.; Giordano, C. Organ-On-A-Chip in vitro Models of the Brain and the Blood-Brain Barrier and Their Value to Study the Microbiota-Gut-Brain Axis in Neurodegeneration. Front. Bioeng. Biotechnol 2019, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Doryab, A.; Amoabediny, G.; Salehi-Najafabadi, A. Advances in pulmonary therapy and drug development: Lung tissue engineering to lung-on-a-chip. Biotechnol. Adv. 2016, 34, 588–596. [Google Scholar] [CrossRef]
- Bein, A.; Shin, W.; Jalili-Firoozinezhad, S.; Park, M.H.; Sontheimer-Phelps, A.; Tovaglieri, A.; Chalkiadaki, A.; Kim, H.J.; Ingber, D.E. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Abadpour, S.; Aizenshtadt, A.; Olsen, P.A.; Shoji, K.; Wilson, S.R.; Krauss, S.; Scholz, H. Pancreas-on-a-Chip Technology for Transplantation Applications. Curr. Diab. Rep. 2020, 20, 72. [Google Scholar] [CrossRef]
- Heidari-Khoei, H.; Esfandiari, F.; Hajari, M.A.; Ghorbaninejad, Z.; Piryaei, A.; Baharvand, H. Organoid technology in female reproductive biomedicine. Reprod. Biol. Endocrinol. 2020, 18, 64. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2020, 1–17. [Google Scholar] [CrossRef]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Kashaninejad, N.; Nikmaneshi, M.R.; Moghadas, H.; Kiyoumarsi Oskouei, A.; Rismanian, M.; Barisam, M.; Saidi, M.S.; Firoozabadi, B. Organ-Tumor-on-a-Chip for Chemosensitivity Assay: A Critical Review. Micromachines 2016, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Fritz, J.M.; O’Hare, J.E. The mechanical nature of the heart as a pump. Am. Heart J. 1967, 73, 92–105. [Google Scholar] [CrossRef]
- Ehler, E. Cardiac cytoarchitecture—Why the “hardware” is important for heart function! Biochim. Biophys. Acta 2016, 1863, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and brain natriuretic peptides: Hormones secreted from the heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Avolio, E.; Alvino, V.V.; Ghorbel, M.T.; Campagnolo, P. Perivascular cells and tissue engineering: Current applications and untapped potential. Pharmacol. Ther. 2017, 171, 83–92. [Google Scholar] [CrossRef]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef]
- Achanta, S.; Gorky, J.; Leung, C.; Moss, A.; Robbins, S.; Eisenman, L.; Chen, J.; Tappan, S.; Heal, M.; Farahani, N.; et al. A Comprehensive Integrated Anatomical and Molecular Atlas of Rat Intrinsic Cardiac Nervous System. iScience 2020, 23, 101140. [Google Scholar] [CrossRef]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef]
- Samarel, A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2291–H2301. [Google Scholar] [CrossRef]
- Vaidya, D.; Tamaddon, H.S.; Lo, C.W.; Taffet, S.M.; Delmar, M.; Morley, G.E.; Jalife, J. Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ. Res. 2001, 88, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Atmanli, A.; Domian, I.J. Generation of aligned functional myocardial tissue through microcontact printing. J. Vis. Exp. 2013, 73, e50288. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lipke, E.A.; Kim, P.; Cheong, R.; Thompson, S.; Delannoy, M.; Suh, K.Y.; Tung, L.; Levchenko, A. Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc. Natl. Acad. Sci. USA 2010, 107, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology (Bethesda) 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Savoji, H.; Mohammadi, M.H.; Rafatian, N.; Toroghi, M.K.; Wang, E.Y.; Zhao, Y.; Korolj, A.; Ahadian, S.; Radisic, M. Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials 2019, 198, 3–26. [Google Scholar] [CrossRef]
- Zuppinger, C. 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front. Cardiovasc. Med. 2019, 6, 87. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Fink, C.; Remmers, U.; Scholz, H.; Wattchow, J.; Weil, J.; Zimmermann, W.; Dohmen, H.H.; Schäfer, H.; Bishopric, N.; et al. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: A new heart muscle model system. FASEB J. 1997, 11, 683–694. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab. Chip 2016, 16, 599–610. [Google Scholar] [CrossRef]
- Giridharan, G.A.; Nguyen, M.D.; Estrada, R.; Parichehreh, V.; Hamid, T.; Ismahil, M.A.; Prabhu, S.D.; Sethu, P. Microfluidic cardiac cell culture model (μCCCM). Anal. Chem. 2010, 82, 7581–7587. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Tinney, J.P.; Ye, F.; Elnakib, A.A.; Yuan, F.; El-Baz, A.; Sethu, P.; Keller, B.B.; Giridharan, G.A. Effects of physiologic mechanical stimulation on embryonic chick cardiomyocytes using a microfluidic cardiac cell culture model. Anal. Chem. 2015, 87, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Fast, V.G.; Sethu, P. Biomimetic Cardiac Tissue Model Enables the Adaption of Human Induced Pluripotent Stem Cell Cardiomyocytes to Physiological Hemodynamic Loads. Anal. Chem. 2016, 88, 9862–9868. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Miller, J.M.; Kannappan, R.; Sethu, P. Cardiac Tissue Chips (CTCs) for Modeling Cardiovascular Disease. IEEE Trans. Biomed. Eng. 2019, 66, 3436–3443. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Eder, A.; Bönstrup, M.; Flato, M.; Mewe, M.; Schaaf, S.; Aksehirlioglu, B.; Schwoerer, A.P.; Uebeler, J.; Eschenhagen, T. Development of a drug screening platform based on engineered heart tissue. Circ. Res. 2010, 107, 35–44. [Google Scholar] [CrossRef]
- Mannhardt, I.; Breckwoldt, K.; Letuffe-Brenière, D.; Schaaf, S.; Schulz, H.; Neuber, C.; Benzin, A.; Werner, T.; Eder, A.; Schulze, T.; et al. Human Engineered Heart Tissue: Analysis of Contractile Force. Stem Cell Rep. 2016, 7, 29–42. [Google Scholar] [CrossRef]
- Breckwoldt, K.; Letuffe-Brenière, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef]
- Aung, A.; Bhullar, I.S.; Theprungsirikul, J.; Davey, S.K.; Lim, H.L.; Chiu, Y.J.; Ma, X.; Dewan, S.; Lo, Y.H.; McCulloch, A.; et al. 3D cardiac μtissues within a microfluidic device with real-time contractile stress readout. Lab. Chip 2016, 16, 153–162. [Google Scholar] [CrossRef]
- Van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef]
- Stephenson, R.S.; Atkinson, A.; Kottas, P.; Perde, F.; Jafarzadeh, F.; Bateman, M.; Iaizzo, P.A.; Zhao, J.; Zhang, H.; Anderson, R.H.; et al. High resolution 3-Dimensional imaging of the human cardiac conduction system from microanatomy to mathematical modeling. Sci. Rep. 2017, 7, 7188. [Google Scholar] [CrossRef]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Yang, X.J.; Li, H.X.; Han, L.H.; Jiang, W.P. Genetically-engineered mesenchymal stem cells transfected with human HCN1 gene to create cardiac pacemaker cells. J. Int. Med. Res. 2013, 41, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Aleman, J.; Arneri, A.; Bersini, S.; Piraino, F.; Shin, S.R.; Dokmeci, M.R.; Khademhosseini, A. From cardiac tissue engineering to heart-on-a-chip: Beating challenges. Biomed. Mater. 2015, 10, 034006. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, C.M.; Klimas, A.; Yu, J.; Entcheva, E. Cardiac applications of optogenetics. Prog. Biophys. Mol. Biol. 2014, 115, 294–304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Savchenko, A.; Cherkas, V.; Liu, C.; Braun, G.B.; Kleschevnikov, A.; Miller, Y.I.; Molokanova, E. Graphene biointerfaces for optical stimulation of cells. Sci. Adv. 2018, 4, eaat0351. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Marsano, A.; Maidhof, R.; Numata, K.; Montouri-Sorrentino, C.; Cannizzaro, C.; Voldman, J.; Vunjak-Novakovic, G. Surface-patterned electrode bioreactor for electrical stimulation. Lab. Chip 2010, 10, 692–700. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, Q.; Liu, H.; Yang, H.; Yun, J.X.; Eisenberg, C.; Borg, T.K.; Xu, M.; Gao, B.Z. Laser-patterned stem-cell bridges in a cardiac muscle model for on-chip electrical conductivity analyses. Lab. Chip 2012, 12, 566–573. [Google Scholar] [CrossRef]
- Qian, F.; Huang, C.; Lin, Y.D.; Ivanovskaya, A.N.; O’Hara, T.J.; Booth, R.H.; Creek, C.J.; Enright, H.A.; Soscia, D.A.; Belle, A.M.; et al. Simultaneous electrical recording of cardiac electrophysiology and contraction on chip. Lab. Chip 2017, 17, 1732–1739. [Google Scholar] [CrossRef]
- Kujala, V.J.; Pasqualini, F.S.; Goss, J.A.; Nawroth, J.C.; Parker, K.K. Laminar ventricular myocardium on a microelectrode array-based chip. J. Mater. Chem. B 2016, 4, 3534–3543. [Google Scholar] [CrossRef]
- Lin, Z.C.; Xie, C.; Osakada, Y.; Cui, Y.; Cui, B. Iridium oxide nanotube electrodes for sensitive and prolonged intracellular measurement of action potentials. Nat. Commun. 2014, 5, 3206. [Google Scholar] [CrossRef]
- Acker, C.D.; Yan, P.; Loew, L.M. Recent progress in optical voltage-sensor technology and applications to cardiac research: From single cells to whole hearts. Prog. Biophys. Mol. Biol. 2020, 154, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Fedele, L.; Brand, T. The Intrinsic Cardiac Nervous System and Its Role in Cardiac Pacemaking and Conduction. J. Cardiovasc. Dev. Dis. 2020, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Shimba, K.; Ishizuka, K.; Yang, Z.; Oiwa, K.; Takeuchi, A.; Kotani, K.; Jimbo, Y. Functional innervation of human induced pluripotent stem cell-derived cardiomyocytes by co-culture with sympathetic neurons developed using a microtunnel technique. Biochem. Biophys. Res. Commun. 2017, 494, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Cho, G.S.; Li, Z.; Hong, I.; Zhu, R.; Kim, M.J.; Kim, Y.J.; Tampakakis, E.; Tung, L.; Huganir, R.; et al. Functional Coupling with Cardiac Muscle Promotes Maturation of hPSC-Derived Sympathetic Neurons. Cell Stem Cell 2016, 19, 95–106. [Google Scholar] [CrossRef]
- Kagemoto, T.; Li, A.; Dos Remedios, C.; Ishiwata, S. Spontaneous oscillatory contraction (SPOC) in cardiomyocytes. Biophys. Rev. 2015, 7, 15–24. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Fleischmann, B.K.; Liu, Q.; Sauer, H.; Gryshchenko, O.; Ji, G.J.; Hescheler, J. Intracellular Ca2+ oscillations drive spontaneous contractions in cardiomyocytes during early development. Proc. Natl. Acad. Sci. USA 1999, 96, 8259–8264. [Google Scholar] [CrossRef]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sato, K.; Shimizu, T.; Yamato, M.; Okano, T.; Kitamori, T. A micro-spherical heart pump powered by cultured cardiomyocytes. Lab. Chip 2007, 7, 207–212. [Google Scholar] [CrossRef]
- MacQueen, L.A.; Sheehy, S.P.; Chantre, C.O.; Zimmerman, J.F.; Pasqualini, F.S.; Liu, X.; Goss, J.A.; Campbell, P.H.; Gonzalez, G.M.; Park, S.J.; et al. A tissue-engineered scale model of the heart ventricle. Nat. Biomed. Eng. 2018, 2, 930–941. [Google Scholar] [CrossRef]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, N. Mechanisms and modifiers of reflex induced cutaneous vasodilation and vasoconstriction in humans. J. Appl. Physiol. 2010, 109, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular permeability--the essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharmacol. 2017, 78, 323–350. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef]
- Van der Loop, F.T.; Gabbiani, G.; Kohnen, G.; Ramaekers, F.C.; van Eys, G.J. Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 665–671. [Google Scholar] [CrossRef]
- Bruno, R.M.; Ghiadoni, L.; Seravalle, G.; Dell’oro, R.; Taddei, S.; Grassi, G. Sympathetic regulation of vascular function in health and disease. Front. Physiol. 2012, 3, 284. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of Cells from the Arterial Vessel Wall in Atherosclerosis. Mediators Inflamm. 2017, 2017, 8135934. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: Molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, R.M.; Mohandas, N.; Blackshear, P.L., Jr. Measurement of the elastic modulus for red cell membrane using a fluid mechanical technique. Biophys. J. 1973, 13, 747–762. [Google Scholar] [CrossRef]
- Ives, C.L.; Eskin, S.G.; McIntire, L.V.; DeBakey, M.E. The importance of cell origin and substrate in the kinetics of endothelial cell alignment in response to steady flow. Trans. Am. Soc. Artif. Intern. Organs 1983, 29, 269–274. [Google Scholar] [PubMed]
- Frangos, J.A.; Eskin, S.G.; McIntire, L.V.; Ives, C.L. Flow effects on prostacyclin production by cultured human endothelial cells. Science 1985, 227, 1477–1479. [Google Scholar] [CrossRef]
- Lin, K.; Hsu, P.P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.; Li, Y.S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389. [Google Scholar] [CrossRef]
- Brown, T.D. Techniques for mechanical stimulation of cells in vitro: A review. J. Biomech. 2000, 33, 3–14. [Google Scholar] [CrossRef]
- Chiu, J.J.; Wang, D.L.; Chien, S.; Skalak, R.; Usami, S. Effects of disturbed flow on endothelial cells. J. Biomech. Eng. 1998, 120, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.R.; García-Cardeña, G.; Gimbrone, M.A., Jr. A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J. Biomech. Eng. 2002, 124, 397–407. [Google Scholar] [CrossRef]
- Estrada, R.; Giridharan, G.A.; Nguyen, M.D.; Prabhu, S.D.; Sethu, P. Microfluidic endothelial cell culture model to replicate disturbed flow conditions seen in atherosclerosis susceptible regions. Biomicrofluidics 2011, 5, 32006–3200611. [Google Scholar] [CrossRef] [PubMed]
- Estrada, R.; Giridharan, G.A.; Nguyen, M.D.; Roussel, T.J.; Shakeri, M.; Parichehreh, V.; Prabhu, S.D.; Sethu, P. Endothelial cell culture model for replication of physiological profiles of pressure, flow, stretch, and shear stress in vitro. Anal. Chem. 2011, 83, 3170–3177. [Google Scholar] [CrossRef] [PubMed]
- Patibandla, P.K.; Rajasekaran, N.S.; Shelar, S.B.; Giridharan, G.A.; Litovsky, S.H.; Sethu, P. Evaluation of the effect of diminished pulsatility as seen in continuous flow ventricular assist devices on arterial endothelial cell phenotype and function. J. Heart Lung Transplant. 2016, 35, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Haglund, T.A.; Rajasekaran, N.S.; Smood, B.; Giridharan, G.A.; Hoopes, C.W.; Holman, W.L.; Mauchley, D.C.; Prabhu, S.D.; Pamboukian, S.V.; Tallaj, J.A.; et al. Evaluation of flow-modulation approaches in ventricular assist devices using an in-vitro endothelial cell culture model. J. Heart Lung Transplant. 2019, 38, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Donoghue, L.; Giridharan, G.A.; Naber, J.P.; Vincent, D.; Fukamachi, K.; Kotru, A.; Sethu, P. Acute Response of Human Aortic Endothelial Cells (HAECs) to Loss of Pulsatility as Seen During Cardiopulmonary Bypass. Cells Tissues Organs 2021. accepted. [Google Scholar]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef]
- Bonnier, F.; Keating, M.E.; Wróbel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef]
- Moya, M.L.; Hsu, Y.H.; Lee, A.P.; Hughes, C.C.; George, S.C. In vitro perfused human capillary networks. Tissue Eng. Part C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef]
- Alonzo, L.F.; Moya, M.L.; Shirure, V.S.; George, S.C. Microfluidic device to control interstitial flow-mediated homotypic and heterotypic cellular communication. Lab. Chip 2015, 15, 3521–3529. [Google Scholar] [CrossRef]
- Shirure, V.S.; Lezia, A.; Tao, A.; Alonzo, L.F.; George, S.C. Low levels of physiological interstitial flow eliminate morphogen gradients and guide angiogenesis. Angiogenesis 2017, 20, 493–504. [Google Scholar] [CrossRef]
- Griffith, C.K.; Miller, C.; Sainson, R.C.; Calvert, J.W.; Jeon, N.L.; Hughes, C.C.; George, S.C. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Eng. 2005, 11, 257–266. [Google Scholar] [CrossRef]
- Zandonella, C. Tissue engineering: The beat goes on. Nature 2003, 421, 884–886. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Davoudi, F.; Walch, P.; Manbachi, A.; Luo, X.; Dell’Erba, V.; Miri, A.K.; Albadawi, H.; Arneri, A.; Li, X.; et al. Bioprinted thrombosis-on-a-chip. Lab. Chip 2016, 16, 4097–4105. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, J.; De Lorenzi, F.; Theek, B.; Blaeser, A.; Rommel, D.; Kuehne, A.J.C.; Kießling, F.; Fischer, H. Engineering biofunctional in vitro vessel models using a multilayer bioprinting technique. Sci. Rep. 2018, 8, 10430. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Vildhede, A.; Norén, A.; Artursson, P. In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J. Proteomics 2016, 136, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wei, W.; Chen, Z.; Lin, B.; Zhao, W.; Luo, Y.; Zhang, X. Engineered Liver-on-a-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review. Micromachines (Basel) 2019, 10, 676. [Google Scholar] [CrossRef] [PubMed]
- Klaas, M.; Kangur, T.; Viil, J.; Mäemets-Allas, K.; Minajeva, A.; Vadi, K.; Antsov, M.; Lapidus, N.; Järvekülg, M.; Jaks, V. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci. Rep. 2016, 6, 27398. [Google Scholar] [CrossRef] [PubMed]
- Usta, O.B.; McCarty, W.J.; Bale, S.; Hegde, M.; Jindal, R.; Bhushan, A.; Golberg, I.; Yarmush, M.L. Microengineered cell and tissue systems for drug screening and toxicology applications: Evolution of in-vitro liver technologies. Technology (Singapore) 2015, 3, 1–26. [Google Scholar] [CrossRef]
- Teutsch, H.F. The modular microarchitecture of human liver. Hepatology 2005, 42, 317–325. [Google Scholar] [CrossRef]
- Krishna, M. Microscopic anatomy of the liver. Clin. Liver Dis. (Hoboken) 2013, 2, S4–S7. [Google Scholar] [CrossRef]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef]
- Natarajan, V.; Harris, E.N.; Kidambi, S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed. Res. Int. 2017, 2017, 4097205. [Google Scholar] [CrossRef]
- Jansen, P.L.M. Hydrodynamics of bile flow: Lessons from computational modeling. Hepatology 2018, 67, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Sellaro, T.L.; Ranade, A.; Faulk, D.M.; McCabe, G.P.; Dorko, K.; Badylak, S.F.; Strom, S.C. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue Eng. Part. A 2010, 16, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.T.; Lin, R.Z.; Chen, R.J.; Chin, C.K.; Gong, S.E.; Chang, H.Y.; Peng, H.L.; Hsu, L.; Yew, T.R.; Chang, S.F.; et al. Liver-cell patterning lab chip: Mimicking the morphology of liver lobule tissue. Lab. Chip 2013, 13, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef]
- Khetani, S.R.; Berger, D.R.; Ballinger, K.R.; Davidson, M.D.; Lin, C.; Ware, B.R. Microengineered liver tissues for drug testing. J. Lab. Autom. 2015, 20, 216–250. [Google Scholar] [CrossRef]
- March, S.; Ramanan, V.; Trehan, K.; Ng, S.; Galstian, A.; Gural, N.; Scull, M.A.; Shlomai, A.; Mota, M.M.; Fleming, H.E.; et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat. Protoc. 2015, 10, 2027–2053. [Google Scholar] [CrossRef]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef]
- Desai, P.K.; Tseng, H.; Souza, G.R. Assembly of Hepatocyte Spheroids Using Magnetic 3D Cell Culture for CYP450 Inhibition/Induction. Int. J. Mol. Sci. 2017, 18, 1085. [Google Scholar] [CrossRef]
- Boos, J.A.; Misun, P.M.; Michlmayr, A.; Hierlemann, A.; Frey, O. Microfluidic Multitissue Platform for Advanced Embryotoxicity Testing In Vitro. Adv. Sci. (Weinheim) 2019, 6, 1900294. [Google Scholar] [CrossRef]
- Rennert, K.; Steinborn, S.; Gröger, M.; Ungerböck, B.; Jank, A.M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef]
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Lama, P.; Dunn, S.; Hong, S.; Lee, M.Y. Polymer coating on a micropillar chip for robust attachment of PuraMatrix peptide hydrogel for 3D hepatic cell culture. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 90, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab. Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Norona, L.M.; Nguyen, D.G.; Gerber, D.A.; Presnell, S.C.; LeCluyse, E.L. Editor’s Highlight: Modeling Compound-Induced Fibrogenesis In Vitro Using Three-Dimensional Bioprinted Human Liver Tissues. Toxicol. Sci. 2016, 154, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Yoshida, T.; Okabe, M.; Goto, M.; Mir, T.A.; Soko, C.; Tsukamoto, Y.; Akaike, T.; Nikaido, T.; Zhou, K.; et al. Fabrication of 3D-culture platform with sandwich architecture for preserving liver-specific functions of hepatocytes using 3D bioprinter. J. Biomed. Mater. Res. A 2017, 105, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Shrike Zhang, Y.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes (Basel) 2018, 9, 176. [Google Scholar] [CrossRef]
- McMahon, A.P. Development of the Mammalian Kidney. Curr. Top. Dev. Biol. 2016, 117, 31–64. [Google Scholar] [CrossRef]
- Koeppen, B.M.; Stanton, B.A. Structure and Function of the Kidneys. In Renal Physiology, 6th ed.; Elsevier: Philadelphia, PA, USA, 2019. [Google Scholar]
- Verschuren, E.H.J.; Castenmiller, C.; Peters, D.J.M.; Arjona, F.J.; Bindels, R.J.M.; Hoenderop, J.G.J. Sensing of tubular flow and renal electrolyte transport. Nat. Rev. Nephrol. 2020, 16, 337–351. [Google Scholar] [CrossRef]
- Gilmer, G.G.; Deshpande, V.G.; Chou, C.L.; Knepper, M. Flow resistance along the rat renal tubule. Am. J. Physiol. Renal. Physiol. 2018, 315, F1398–F1405. [Google Scholar] [CrossRef]
- Holstein-Rathlou, N.H.; Marsh, D.J. Oscillations of tubular pressure, flow, and distal chloride concentration in rats. Am. J. Physiol. 1989, 256, F1007–F1014. [Google Scholar] [CrossRef] [PubMed]
- Reinking, L.N.; Schmidt-Nielsen, B. Peristaltic flow of urine in the renal capillary collecting ducts of hamsters. Kidney Int. 1981, 20, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Craig, D.A.; Wexler, A.S.; Marsh, D.J. Fluid waves in renal tubules. Biophys. J. 1986, 50, 805–813. [Google Scholar] [CrossRef]
- Schnermann, J.; Wright, F.S.; Davis, J.M.; von Stackelberg, W.; Grill, G. Regulation of superficial nephron filtration rate by tubulo-glomerular feedback. Pflugers Arch. 1970, 318, 147–175. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Tubuloglomerular feedback and the control of glomerular filtration rate. News Physiol. Sci. 2003, 18, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Duan, Y.; Satlin, L.M.; Wang, T.; Weinstein, A.M. Mechanotransduction in the renal tubule. Am. J. Physiol. Renal. Physiol. 2010, 299, F1220–F1236. [Google Scholar] [CrossRef]
- Sgouralis, I.; Layton, A.T. Control and modulation of fluid flow in the rat kidney. Bull. Math. Biol. 2013, 75, 2551–2574. [Google Scholar] [CrossRef]
- Cabral, P.D.; Garvin, J.L. Luminal flow regulates NO and O2(-) along the nephron. Am. J. Physiol. Renal. Physiol. 2011, 300, F1047–F1053. [Google Scholar] [CrossRef]
- Loichot, C.; Krieger, J.P.; De Jong, W.; Helwig, J.J.; Nisato, D.; Imbs, J.L.; Barthelmebs, M. Shear stress modulates vasopressin-induced renal vasoconstriction in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2002, 366, 555–561. [Google Scholar] [CrossRef]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. (Cambridge) 2013, 5, 1119–1129. [Google Scholar] [CrossRef]
- Astashkina, A.I.; Mann, B.K.; Prestwich, G.D.; Grainger, D.W. A 3-D organoid kidney culture model engineered for high-throughput nephrotoxicity assays. Biomaterials 2012, 33, 4700–4711. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Rudym, D.; Cannizzaro, C.; Perrone, R.; Zhou, J.; Kaplan, D.L. Tissue-engineered three-dimensional in vitro models for normal and diseased kidney. Tissue Eng. Part A 2010, 16, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.J.; Chapron, A.; Chapron, B.D.; Voellinger, J.L.; Lidberg, K.A.; Yeung, C.K.; Wang, Z.; Yamaura, Y.; Hailey, D.W.; Neumann, T.; et al. Development of a microphysiological model of human kidney proximal tubule function. Kidney Int. 2016, 90, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Homan, K.A.; Kolesky, D.B.; Skylar-Scott, M.A.; Herrmann, J.; Obuobi, H.; Moisan, A.; Lewis, J.A. Bioprinting of 3D Convoluted Renal Proximal Tubules on Perfusable Chips. Sci. Rep. 2016, 6, 34845. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.Y.C.; Homan, K.A.; Robinson, S.S.; Kolesky, D.B.; Duarte, N.; Moisan, A.; Lewis, J.A. Renal reabsorption in 3D vascularized proximal tubule models. Proc. Natl. Acad. Sci. USA 2019, 116, 5399–5404. [Google Scholar] [CrossRef]
- Rein, J.L.; Heja, S.; Flores, D.; Carrisoza-Gaytán, R.; Lin, N.Y.C.; Homan, K.A.; Lewis, J.A.; Satlin, L.M. Effect of luminal flow on doming of mpkCCD cells in a 3D perfusable kidney cortical collecting duct model. Am. J. Physiol. Cell Physiol. 2020, 319, C136–C147. [Google Scholar] [CrossRef]
- Carrisoza-Gaytan, R.; Carattino, M.D.; Kleyman, T.R.; Satlin, L.M. An unexpected journey: Conceptual evolution of mechanoregulated potassium transport in the distal nephron. Am. J. Physiol. Cell Physiol. 2016, 310, C243–C259. [Google Scholar] [CrossRef]
- Palmer, L.G.; Schnermann, J. Integrated control of Na transport along the nephron. Clin. J. Am. Soc. Nephrol. 2015, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef]
- Roy, A.; Al-bataineh, M.M.; Pastor-Soler, N.M. Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 305–324. [Google Scholar] [CrossRef]
- Musah, S.; Mammoto, A.; Ferrante, T.C.; Jeanty, S.S.F.; Hirano-Kobayashi, M.; Mammoto, T.; Roberts, K.; Chung, S.; Novak, R.; Ingram, M.; et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Al-Awqati, Q.; Oliver, J.A. Stem cells in the kidney. Kidney Int. 2002, 61, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Saleme, B.; Sutendra, G. A Similar Metabolic Profile Between the Failing Myocardium and Tumor Could Provide Alternative Therapeutic Targets in Chemotherapy-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- Morsy, M.S.; Dishmon, D.A.; Garg, N.; Weber, K.T. Secondary Hyperparathyroidism in Heart Failure. Am. J. Med. Sci. 2017, 354, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Viravaidya, K.; Sin, A.; Shuler, M.L. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol. Prog. 2004, 20, 316–323. [Google Scholar] [CrossRef]
- McAleer, C.W.; Pointon, A.; Long, C.J.; Brighton, R.L.; Wilkin, B.D.; Bridges, L.R.; Narasimhan Sriram, N.; Fabre, K.; McDougall, R.; Muse, V.P.; et al. On the potential of in vitro organ-chip models to define temporal pharmacokinetic-pharmacodynamic relationships. Sci. Rep. 2019, 9, 9619. [Google Scholar] [CrossRef]
- Oleaga, C.; Riu, A.; Rothemund, S.; Lavado, A.; McAleer, C.W.; Long, C.J.; Persaud, K.; Narasimhan, N.S.; Tran, M.; Roles, J.; et al. Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 2018, 182, 176–190. [Google Scholar] [CrossRef]
- Vunjak-Novakovic, G.; Bhatia, S.; Chen, C.; Hirschi, K. HeLiVa platform: Integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther. 2013, 4 (Suppl. S1), S8. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Shrike Zhang, Y.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 8837. [Google Scholar] [CrossRef]
- Schimek, K.; Frentzel, S.; Luettich, K.; Bovard, D.; Rütschle, I.; Boden, L.; Rambo, F.; Erfurth, H.; Dehne, E.M.; Winter, A.; et al. Human multi-organ chip co-culture of bronchial lung culture and liver spheroids for substance exposure studies. Sci. Rep. 2020, 10, 7865. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab. Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Wagner, I.; Materne, E.M.; Brincker, S.; Süssbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab. Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lozito, T.P.; Alexander, P.G.; Gottardi, R.; Tuan, R.S. Stem cell-based microphysiological osteochondral system to model tissue response to interleukin-1β. Mol. Pharm. 2014, 11, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Wheeler, S.E.; Taylor, D.P.; Pillai, V.C.; Young, C.L.; Prantil-Baun, R.; Nguyen, T.; Stolz, D.B.; Borenstein, J.T.; Lauffenburger, D.A.; et al. A microphysiological system model of therapy for liver micrometastases. Exp. Biol. Med. (Maywood) 2014, 239, 1170–1179. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver-intestine and liver-skin co-cultures--A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef]
- Esch, M.B.; Ueno, H.; Applegate, D.R.; Shuler, M.L. Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab. Chip 2016, 16, 2719–2729. [Google Scholar] [CrossRef]
- Moura Rosa, P.; Gopalakrishnan, N.; Ibrahim, H.; Haug, M.; Halaas, Ø. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab. Chip 2016, 16, 3728–3740. [Google Scholar] [CrossRef]
- Loskill, P.; Sezhian, T.; Tharp, K.M.; Lee-Montiel, F.T.; Jeeawoody, S.; Reese, W.M.; Zushin, P.H.; Stahl, A.; Healy, K.E. WAT-on-a-chip: A physiologically relevant microfluidic system incorporating white adipose tissue. Lab. Chip 2017, 17, 1645–1654. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Chen, W.L.K.; Edington, C.D.; Stokes, C.L.; Griffith, L.G.; Cirit, M. Integrated Gut and Liver Microphysiological Systems for Quantitative In Vitro Pharmacokinetic Studies. AAPS J. 2017, 19, 1499–1512. [Google Scholar] [CrossRef]
- Yin, F.; Zhang, X.; Wang, L.; Wang, Y.; Zhu, Y.; Li, Z.; Tao, T.; Chen, W.; Yu, H.; Qin, J. HiPSC-derived multi-organoids-on-chip system for safety assessment of antidepressant drugs. Lab. Chip 2020. [Google Scholar] [CrossRef]
- Baert, Y.; Ruetschle, I.; Cools, W.; Oehme, A.; Lorenz, A.; Marx, U.; Goossens, E.; Maschmeyer, I. A multi-organ-chip co-culture of liver and testis equivalents: A first step toward a systemic male reprotoxicity model. Hum. Reprod. 2020, 35, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Pires de Mello, C.P.; Carmona-Moran, C.; McAleer, C.W.; Perez, J.; Coln, E.A.; Long, C.J.; Oleaga, C.; Riu, A.; Note, R.; Teissier, S.; et al. Microphysiological heart-liver body-on-a-chip system with a skin mimic for evaluating topical drug delivery. Lab. Chip 2020, 20, 749–759. [Google Scholar] [CrossRef]
- Kwak, B.S.; Jin, S.P.; Kim, S.J.; Kim, E.J.; Chung, J.H.; Sung, J.H. Microfluidic skin chip with vasculature for recapitulating the immune response of the skin tissue. Biotechnol. Bioeng. 2020, 117, 1853–1863. [Google Scholar] [CrossRef]
- Sung, J.H. A body-on-a-chip (BOC) system for studying gut-liver interaction. Methods Cell Biol. 2020, 158, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Allbritton, N.L.; Wells, A. Integrative microphysiological tissue systems of cancer metastasis to the liver. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.W.; Choi, N.; Lee, S.H.; Sung, J.H. Three-tissue microphysiological system for studying inflammatory responses in gut-liver Axis. Biomed. Microdevices 2020, 22, 65. [Google Scholar] [CrossRef]
- Marin, T.M.; Indolfo, N.C.; Rocco, S.A.; de Carvalho, M.; Dias, M.M.; Vasconcelos Bento, G.I.; Bortot, L.O.; Schuck, D.C.; Lorencini, M.; Pagani, E. An Intestine/Liver Microphysiological System for Drug Pharmacokinetic and Toxicological Assessment. J. Vis. Exp. 2020, 166, e60184. [Google Scholar] [CrossRef]
- Giordano, L.; Mihaila, S.M.; Eslami Amirabadi, H.; Masereeuw, R. Microphysiological Systems to Recapitulate the Gut-Kidney Axis. Trends Biotechnol. 2021. [Google Scholar] [CrossRef]
- Benedetto, A.; Accetta, G.; Fujita, Y.; Charras, G. Spatiotemporal control of gene expression using microfluidics. Lab. Chip 2014, 14, 1336–1347. [Google Scholar] [CrossRef]
- O’Grady, B.; Balikov, D.A.; Wang, J.X.; Neal, E.K.; Ou, Y.C.; Bardhan, R.; Lippmann, E.S.; Bellan, L.M. Spatiotemporal control and modeling of morphogen delivery to induce gradient patterning of stem cell differentiation using fluidic channels. Biomater. Sci. 2019, 7, 1358–1371. [Google Scholar] [CrossRef]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-chip platforms for studying drug delivery systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Chiriacò, M.S.; Bianco, M.; Nigro, A.; Primiceri, E.; Ferrara, F.; Romano, A.; Quattrini, A.; Furlan, R.; Arima, V.; Maruccio, G. Lab-on-Chip for Exosomes and Microvesicles Detection and Characterization. Sensors (Basel) 2018, 18, 3175. [Google Scholar] [CrossRef] [PubMed]
- Ellis, B.W.; Acun, A.; Can, U.I.; Zorlutuna, P. Human iPSC-derived myocardium-on-chip with capillary-like flow for personalized medicine. Biomicrofluidics 2017, 11, 024105. [Google Scholar] [CrossRef] [PubMed]
- Menon, N.V.; Tay, H.M.; Wee, S.N.; Li, K.H.H.; Hou, H.W. Micro-engineered perfusable 3D vasculatures for cardiovascular diseases. Lab. Chip 2017, 17, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.S.; Chiu, D.T. Controlling mass transport in microfluidic devices. Annu. Rev. Anal. Chem. (Palo Alto) 2011, 4, 275–296. [Google Scholar] [CrossRef]
- Velve-Casquillas, G.; Le Berre, M.; Piel, M.; Tran, P.T. Microfluidic tools for cell biological research. Nano Today 2010, 5, 28–47. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z. Microfluidic Passive Flow Regulatory Device with an Integrated Check Valve for Enhanced Flow Control. Micromachines (Basel) 2019, 10, 653. [Google Scholar] [CrossRef]
- Rogers, A.J.; Kannappan, R.; Abukhalifeh, H.; Ghazal, M.; Miller, J.M.; El-Baz, A.; Fast, V.G.; Sethu, P. Hemodynamic Stimulation Using the Biomimetic Cardiac Tissue Model (BCTM) Enhances Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cells Tissues Organs 2018, 206, 82–94. [Google Scholar] [CrossRef]
- Kratz, S.R.A.; Höll, G.; Schuller, P.; Ertl, P.; Rothbauer, M. Latest Trends in Biosensing for Microphysiological Organs-on-a-Chip and Body-on-a-Chip Systems. Biosensors (Basel) 2019, 9, 110. [Google Scholar] [CrossRef]
- Ballini, M.; Müller, J.; Livi, P.; Chen, Y.; Frey, U.; Stettler, A.; Shadmani, A.; Viswam, V.; Jones, I.L.; Jäckel, D.; et al. A 1024-Channel CMOS Microelectrode Array With 26,400 Electrodes for Recording and Stimulation of Electrogenic Cells In Vitro. IEEE J. Solid State Circuits 2014, 49, 2705–2719. [Google Scholar] [CrossRef]
- Grosberg, A.; Alford, P.W.; McCain, M.L.; Parker, K.K. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab. Chip 2011, 11, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bustamante, C.D.; Frey, U.; Kelm, J.M.; Hierlemann, A.; Fussenegger, M. Modulation of cardiomyocyte electrical properties using regulated bone morphogenetic protein-2 expression. Tissue Eng. Part A 2008, 14, 1969–1988. [Google Scholar] [CrossRef] [PubMed]
- Henry, O.Y.F.; Villenave, R.; Cronce, M.J.; Leineweber, W.D.; Benz, M.A.; Ingber, D.E. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (TEER) measurements of human epithelial barrier function. Lab. Chip 2017, 17, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.G.; Chen, C. An arduino-based sensor to measure transendothelial electrical resistance. Sens. Actuators A 2020, 314, 112216. [Google Scholar] [CrossRef]
- Gaio, N.; Waafi, A.; Vlaming, M.; Boschman, E.; Dijkstra, P.; Nacken, P.; Braam, S.; Boucsein, C.; Sarro, P.; Dekker, R. A multiwell plate Organ-on-Chip (OOC) device for in-vitro cell culture stimulation and monitoring. In Proceedings of the 2018 IEEE Micro Electro Mechanical Systems (MEMS), Belfast, UK, 21–25 January 2018; pp. 314–317. [Google Scholar]
- Shirure, V.S.; George, S.C. Design considerations to minimize the impact of drug absorption in polymer-based organ-on-a-chip platforms. Lab. Chip 2017, 17, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Torino, S.; Corrado, B.; Iodice, M.; Coppola, G. PDMS-Based Microfluidic Devices for Cell Culture. Inventions 2018, 3, 65. [Google Scholar] [CrossRef]
- Liao, Z.; Wang, J.; Zhang, P.; Zhang, Y.; Miao, Y.; Gao, S.; Deng, Y.; Geng, L. Recent advances in microfluidic chip integrated electronic biosensors for multiplexed detection. Biosens. Bioelectron. 2018, 121, 272–280. [Google Scholar] [CrossRef]
- Bernard, M.; Jubeli, E.; Pungente, M.D.; Yagoubi, N. Biocompatibility of polymer-based biomaterials and medical devices—Regulations, in vitro screening and risk-management. Biomater. Sci. 2018, 6, 2025–2053. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Author | Organs/Tissues | Application |
|---|---|---|---|
| 2004 | Viravaidya et al. [148] | Lung–Liver–Fat | Napthalene toxicity |
| 2013 | Wagner et al. [155] | Liver–Skin | Drug Testing |
| 2013 | Vunjak-Novakovic et al. [151] | Heart–Liver–Vascular | Drug Testing |
| 2014 | Lin et al. [156] | Bone–Cartilage | Drug Testing |
| 2014 | Clark et al. [157] | Liver–Tumor | Testing Therapeutic Strategies |
| 2015 | Maschmayer et al. [158] | Liver–Skin/Intestine | Drug Testing |
| 2015 | Maschmayer et al. [154] | Intestine–Liver–Skin–Kidney | Drug Pharmacodynamics and Toxicity |
| 2016 | Esch et al. [159] | GI Tract–Liver | Disease Modeling |
| 2016 | Moura Rosa et al. [160] | Lymph Node–Immune Cell | Disease Modeling and Drug Testing |
| 2017 | Loskill et al. [161] | Adipose–Vascular | Disease Modeling |
| 2017 | Skardal et al. [152] | Liver–Heart–Lung | Drug Efficacy and Toxicity |
| 2017 | Tsamandouras et al. [162] | Gut–Liver | Drug Pharmacokinetics |
| 2018 | Oleaga et al. [150] | Heart–Liver | Cardiotoxicity |
| 2019 | McAleer et al. [149] | Heart–Liver | Terfenadine Pharmacokinetics |
| 2020 | Yin et al. [163] | Heart–Liver | Testing Anti-depressant Drugs |
| 2020 | Schimek et al. [153] | Lung–Liver | Toxicity of inhaled substances |
| 2020 | Baert et al. [164] | Liver–Testis | Reproductive toxicity |
| 2020 | de Mello et al. [165] | Heart–Liver–Skin | Topical Drug Delivery |
| 2020 | Kwak et al. [166] | Skin–Vasculature | Immune responses |
| 2020 | Sung et al. [167] | Gut–Liver | Drug Testing |
| 2020 | Clark et al. [168] | Liver–Tumor | Tumor Metastasis |
| 2020 | Jeon et al. [169] | Gut–Liver–Immune Cell | Modeling inflammatory responses |
| 2020 | Marin et al. [170] | Liver–Intestine | Drug pharmacological and toxicological assessment |
| 2021 | Giordano et al. [171] | Gut–Kidney | Chronic Kidney Disease Modeling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donoghue, L.; Nguyen, K.T.; Graham, C.; Sethu, P. Tissue Chips and Microphysiological Systems for Disease Modeling and Drug Testing. Micromachines 2021, 12, 139. https://doi.org/10.3390/mi12020139
Donoghue L, Nguyen KT, Graham C, Sethu P. Tissue Chips and Microphysiological Systems for Disease Modeling and Drug Testing. Micromachines. 2021; 12(2):139. https://doi.org/10.3390/mi12020139
Chicago/Turabian StyleDonoghue, Leslie, Khanh T. Nguyen, Caleb Graham, and Palaniappan Sethu. 2021. "Tissue Chips and Microphysiological Systems for Disease Modeling and Drug Testing" Micromachines 12, no. 2: 139. https://doi.org/10.3390/mi12020139
APA StyleDonoghue, L., Nguyen, K. T., Graham, C., & Sethu, P. (2021). Tissue Chips and Microphysiological Systems for Disease Modeling and Drug Testing. Micromachines, 12(2), 139. https://doi.org/10.3390/mi12020139