Characterization and Optimization of Isotachophoresis Parameters for Pacific Blue Succinimidyl Ester Dye on a PDMS Microfluidic Chip

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
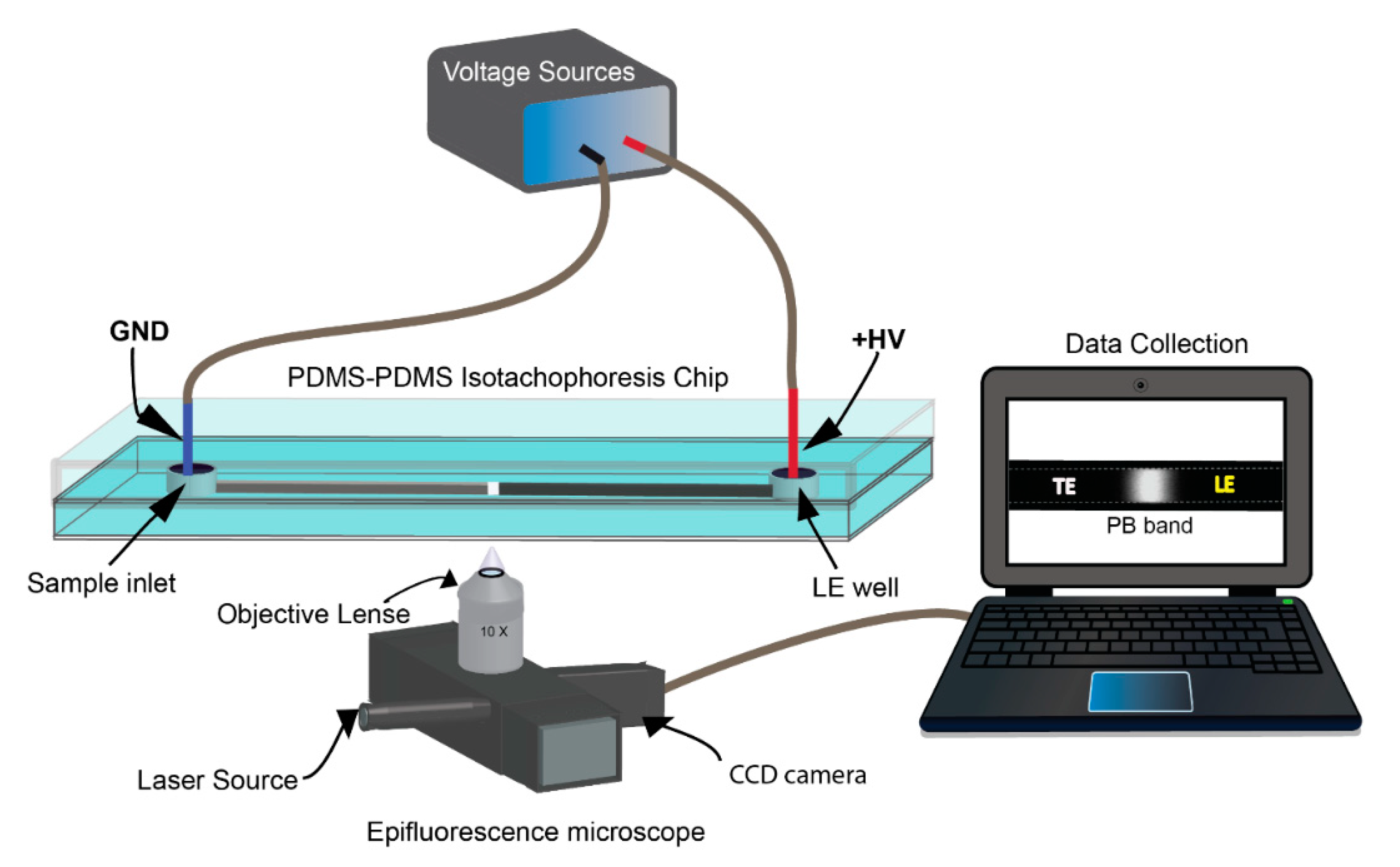
2.1. Fabrication and Preparation of PDMS Microfluidic Chip
2.2. Reagent Preparation for ITP
2.3. ITP Protocol
2.4. Determination of the TE and the LE Electrolytes
2.5. Optimization of Electric Field (EF) and Measurement Point
2.6. Optimization of LE and TE Concentrations
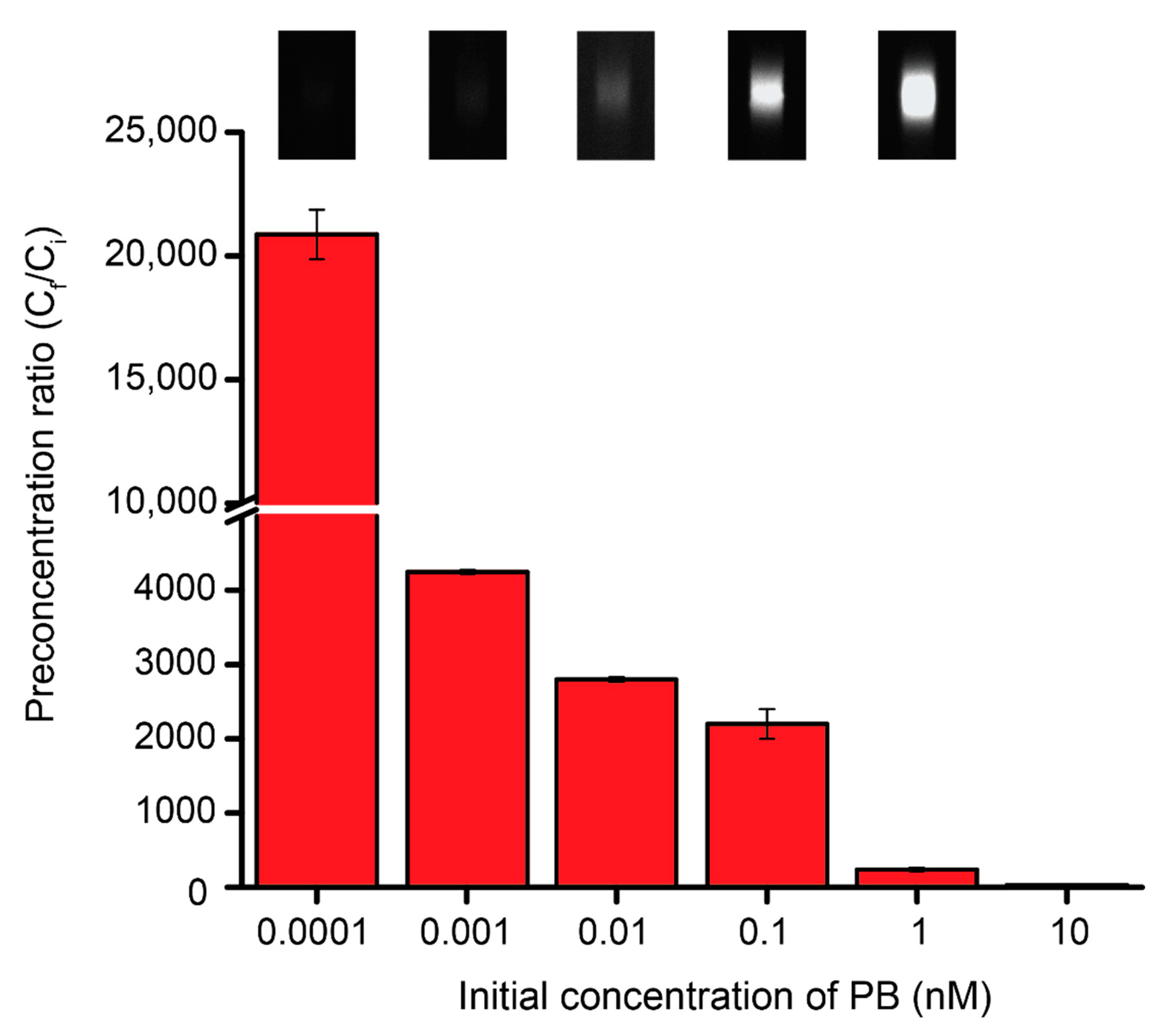
2.7. The Preconcentration Ratio of PB
2.8. LDH Measurement with PB
3. Results and Discussion
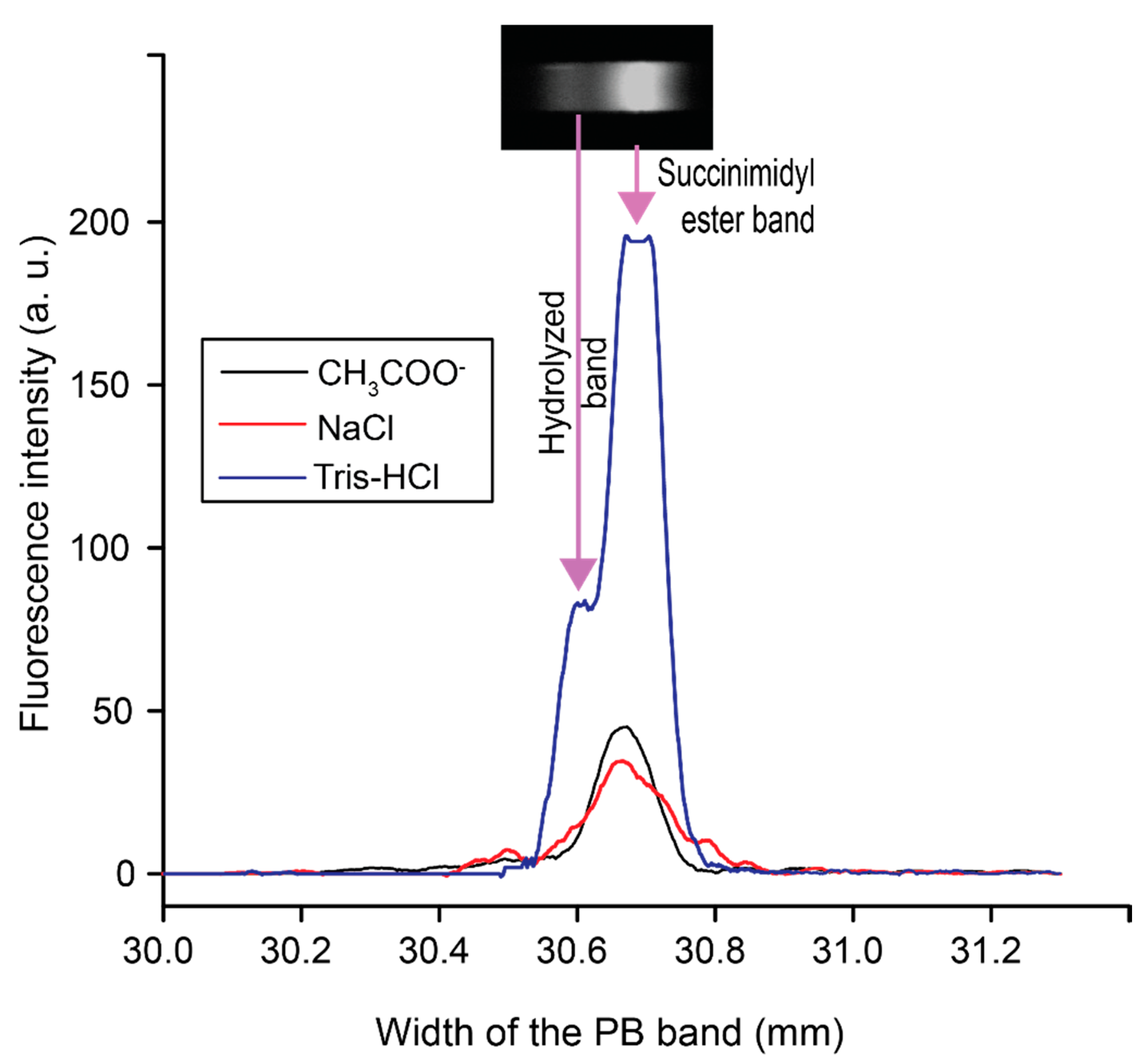
3.1. Selecting Electrolytes to Preconcentrate PB on a PDMS Microfluidic Chip
3.2. The Preconcentration Ratio of PB
3.3. Preconcentrating LDH Using PB Conjugation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleparnik, K.; Bocek, P. DNA diagnostics by capillary electrophoresis. Chem. Rev. 2007, 107, 5279–5317. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, J.; Verpoorte, E.; de Rooij, N.F. Sample preconcentration by field amplification stacking for microchip-based capillary electrophoresis. Electrophoresis 2001, 22, 258–271. [Google Scholar] [CrossRef]
- Britz-McKibbin, P.; Chen, D.D.Y. Selective focusing of catecholamines and weakly acidic compounds by capillary electrophoresis using a dynamic pH junction. Anal. Chem. 2000, 72, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.A., 3rd; Morris, M.D.; Burns, M.A.; Johnson, B.N. Isotachophoretic separations on a microchip. Normal Raman spectroscopy detection. Anal. Chem. 1998, 70, 3766–3769. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Bharadwaj, R.; Santiago, J.G. On-chip millionfold sample stacking using transient isotachophoresis. Anal. Chem. 2006, 78, 2319–2327. [Google Scholar] [CrossRef]
- Liu, D.; Shi, M.; Huang, H.; Long, Z.; Zhou, X.; Qin, J.; Lin, B. Isotachophoresis preconcentration integrated microfluidic chip for highly sensitive genotyping of the hepatitis B virus. J. Chromatogr. B 2006, 844, 32–38. [Google Scholar] [CrossRef]
- Eid, C.; Santiago, J.G. Isotachophoresis applied to biomolecular reactions. Lab Chip 2017, 18, 11–26. [Google Scholar] [CrossRef]
- Bahga, S.S.; Santiago, J.G. Coupling isotachophoresis and capillary electrophoresis: A review and comparison of methods. Analyst 2013, 138, 735–754. [Google Scholar] [CrossRef]
- Stockton, A.M.; Mora, M.F.; Cable, M.L.; Davenport, T.C.; Tilley, T.D.; Willis, P.A. Hydrolysis of 3-carboxy-6,8-difluoro-7-hydroxycoumarin (Pacific Blue (TM)) succinimidyl ester under acidic and basic conditions. Dyes Pigm. 2013, 96, 148–151. [Google Scholar] [CrossRef]
- Chiesl, T.N.; Chu, W.K.; Stockton, A.M.; Amashukeli, X.; Grunthaner, F.; Mathies, R.A. Enhanced Amine and Amino Acid Analysis Using Pacific Blue and the Mars Organic Analyzer Microchip Capillary Electrophoresis System. Anal. Chem. 2009, 81, 2537–2544. [Google Scholar] [CrossRef]
- Stockton, A.M.; Chiesl, T.N.; Lowenstein, T.K.; Amashukeli, X.; Grunthaner, F.; Mathies, R.A. Capillary Electrophoresis Analysis of Organic Amines and Amino Acids in Saline and Acidic Samples Using the Mars Organic Analyzer. Astrobiology 2009, 9, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Jacroux, T.; Bottenus, D.; Rieck, B.; Ivory, C.F.; Dong, W.J. Cationic isotachophoresis separation of the biomarker cardiac troponin I from a high-abundance contaminant, serum albumin. Electrophoresis 2014, 35, 2029–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, T.K.; Santiago, J.G. Preconcentration, separation, and indirect detection of nonfluorescent analytes using fluorescent mobility markers. Anal. Chem. 2008, 80, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Dolník, V.; Liu, S.; Jovanovich, S. Capillary electrophoresis on microchip. Electrophoresis 2000, 21, 41–54. [Google Scholar] [CrossRef]
- Guckenberger, D.J.; de Groot, T.E.; Wan, A.M.; Beebe, D.J.; Young, E.W. Micromilling: A method for ultra-rapid prototyping of plastic microfluidic devices. Lab Chip 2015, 15, 2364–2378. [Google Scholar] [CrossRef] [Green Version]
- Okagbare, P.I.; Emory, J.M.; Datta, P.; Goettert, J.; Soper, S.A. Fabrication of a cyclic olefin copolymer planar waveguide embedded in a multi-channel poly(methyl methacrylate) fluidic chip for evanescence excitation. Lab Chip 2010, 10, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Xiang, J.Z.; Ai, Z.; Zhang, S.K.; Fang, Y.; Chen, T.; Zhou, Q.W.; Li, S.Z.; Wang, S.X.; Zhang, N.G. PMMA microfluidic chip fabrication using laser ablation and low temperature bonding with OCA film and LOCA. Microsyst. Technol. 2017, 23, 1937–1942. [Google Scholar] [CrossRef]
- Chou, S.Y.; Krauss, P.R.; Renstrom, P.J. Imprint lithography with 25-nanometer resolution. Science 1996, 272, 85–87. [Google Scholar] [CrossRef]
- Li, M.W.; Huynh, B.H.; Hulvey, M.K.; Lunte, S.M.; Martin, R.S. Design and characterization of poly(dimethylsiloxane)-based valves for interfacing continuous-flow sampling to microchip electrophoresis. Anal. Chem. 2006, 78, 1042–1051. [Google Scholar] [CrossRef]
- McDonald, J.C.; Whitesides, G.M. Poly(dimethylsiloxane) as a material for fabricating microfluidic devices. Acc. Chem. Res. 2002, 35, 491–499. [Google Scholar] [CrossRef]
- Jeong, Y.W.; Choi, K.W.; Kang, M.K.; Chun, K.J.; Chung, D.S. Transient isotachophoresis of highly saline samples using a microchip. Sens. Actuat. B Chem. 2005, 104, 269–275. [Google Scholar] [CrossRef]
- Cui, H.; Dutta, P.; Ivory, C.F. Isotachophoresis of proteins in a networked microfluidic chip: Experiment and 2-D simulation. Electrophoresis 2007, 28, 1138–1145. [Google Scholar] [CrossRef]
- Han, C.M.; Catoe, D.; Munro, S.A.; Khnouf, R.; Snyder, M.P.; Santiago, J.G.; Salit, M.L.; Cenik, C. Simultaneous RNA purification and size selection using on-chip isotachophoresis with an ionic spacer. Lab Chip 2019, 19, 2741–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.M.; Katilius, E.; Santiago, J.G. Increasing hybridization rate and sensitivity of DNA microarrays using isotachophoresis. Lab Chip 2014, 14, 2958–2967. [Google Scholar] [CrossRef]
- Janasek, D.; Schilling, M.; Franzke, J.; Manz, A. Isotachophoresis in free-flow using a miniaturized device. Anal. Chem. 2006, 78, 3815–3819. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, M.; Jensen, E.C.; Mathies, R.A. Lifting gate polydimethylsiloxane microvalves and pumps for microfluidic control. Anal. Chem. 2012, 84, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Stockton, A.M.; Jensen, E.C.; Mathies, R.A. Pneumatically actuated microvalve circuits for programmable automation of chemical and biochemical analysis. Lab Chip 2016, 16, 812–819. [Google Scholar] [CrossRef]
- Jang, L.W.; Lee, J.; Razu, M.E.; Jensen, E.C.; Kim, J. Fabrication of PDMS Nanocomposite Materials and Nanostructures for Biomedical Nanosystems. IEEE Trans. NanoBiosci. 2015, 14, 841–849. [Google Scholar] [CrossRef]
- Khurana, T.K.; Santiago, J.G. Sample zone dynamics in peak mode isotachophoresis. Anal. Chem. 2008, 80, 6300–6307. [Google Scholar] [CrossRef]
- Lim, A.E.; Lim, C.Y.; Lam, Y.C. Electroosmotic flow hysteresis for dissimilar ionic solutions. Biomicrofluidics 2015, 9, 024113. [Google Scholar] [CrossRef] [Green Version]
- Routs, R.J. Electrolyte Systems in Isotachphoresis and Their Applications to Some Protein Seaparations; Solna Skriv- & Stenograftjiinst AB: Solna, Sweden, 1971. [Google Scholar]
- Jung, B.G.; Zhu, Y.G.; Santiago, J.G. Detection of 100 aM fluorophores using a high-sensitivity on-chip CE system and transient isotachophoresis. Anal. Chem. 2007, 79, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Bahga, S.S.; Bercovici, M.; Santiago, J.G. Ionic strength effects on electrophoretic focusing and separations. Electrophoresis 2010, 31, 910–919. [Google Scholar] [CrossRef]
- Krivankova, L.; Brezkova, M.; Gebauer, P.; Bocek, P. Importance of the counterion in optimization of a borate electrolyte system for analyses of anions in samples with complex matrices performed by capillary zone electrophoresis. Electrophoresis 2004, 25, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Schwarz, G.; Rogacs, A.; Bahga, S.S.; Santiago, J.G. On-chip isotachophoresis for separation of ions and purification of nucleic acids. J. Vis. Exp. 2012, e3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, E.C.; He, J.G.; Liu, Z.H.; Ni, X.H.; Zheng, Y.G.; Gu, Q.; Zhao, Z.H.; Xiong, C.M. High levels of serum lactate dehydrogenase correlate with the severity and mortality of idiopathic pulmonary arterial hypertension. Exp. Ther. Med. 2015, 9, 2109–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.J.; Gong, X.Y.; Yeung, E.S. Combinatorial screening of enzyme activity by using multiplexed capillary electrophoresis. Anal. Chem. 2000, 72, 3383–3387. [Google Scholar] [CrossRef]
- Markert, C.L. Lactate dehydrogenase. Biochemistry and function of lactate dehydrogenase. Cell Biochem. Funct. 1984, 2, 131–134. [Google Scholar] [CrossRef]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ahsan, S.S.; Santiago-Berrios, M.B.; Abruna, H.D.; Webb, W.W. Mechanisms of quenching of Alexa fluorophores by natural amino acids. J. Am. Chem. Soc. 2010, 132, 7244–7245. [Google Scholar] [CrossRef] [Green Version]
- Cable, M.L.; Stockton, A.M.; Mora, M.F.; Willis, P.A. Low-temperature microchip nonaqueous capillary electrophoresis of aliphatic primary amines: Applications to Titan chemistry. Anal. Chem. 2013, 85, 1124–1131. [Google Scholar] [CrossRef]
- Kim, J.; Jensen, E.C.; Stockton, A.M.; Mathies, R.A. Universal microfluidic automaton for autonomous sample processing: Application to the Mars Organic Analyzer. Anal. Chem. 2013, 85, 7682–7688. [Google Scholar] [CrossRef] [PubMed]
- Jang, L.W.; Razu, M.E.; Jensen, E.C.; Jiao, H.; Kim, J. A fully automated microfluidic micellar electrokinetic chromatography analyzer for organic compound detection. Lab Chip 2016, 16, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Estlack, Z.; Somaweera, H.; Wang, X.; Lacerda, C.M.R.; Kim, J. A microfluidic cardiac flow profile generator for studying the effect of shear stress on valvular endothelial cells. Lab Chip 2018, 18, 2946–2954. [Google Scholar] [CrossRef]
- Linshiz, G.; Jensen, E.; Stawski, N.; Bi, C.; Elsbree, N.; Jiao, H.; Kim, J.; Mathies, R.; Keasling, J.D.; Hillson, N.J. End-to-end automated microfluidic platform for synthetic biology: From design to functional analysis. J. Biol. Eng. 2016, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaigala, G.V.; Bercovici, M.; Behnam, M.; Elliott, D.; Santiago, J.G.; Backhouse, C.J. Miniaturized system for isotachophoresis assays. Lab Chip 2010, 10, 2242–2250. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somaweera, H.; Estlack, Z.; Devadhasan, J.P.; Kim, J.; Kim, J. Characterization and Optimization of Isotachophoresis Parameters for Pacific Blue Succinimidyl Ester Dye on a PDMS Microfluidic Chip. Micromachines 2020, 11, 951. https://doi.org/10.3390/mi11110951
Somaweera H, Estlack Z, Devadhasan JP, Kim J, Kim J. Characterization and Optimization of Isotachophoresis Parameters for Pacific Blue Succinimidyl Ester Dye on a PDMS Microfluidic Chip. Micromachines. 2020; 11(11):951. https://doi.org/10.3390/mi11110951
Chicago/Turabian StyleSomaweera, Himali, Zachary Estlack, Jasmine Pramila Devadhasan, Jungtae Kim, and Jungkyu Kim. 2020. "Characterization and Optimization of Isotachophoresis Parameters for Pacific Blue Succinimidyl Ester Dye on a PDMS Microfluidic Chip" Micromachines 11, no. 11: 951. https://doi.org/10.3390/mi11110951
APA StyleSomaweera, H., Estlack, Z., Devadhasan, J. P., Kim, J., & Kim, J. (2020). Characterization and Optimization of Isotachophoresis Parameters for Pacific Blue Succinimidyl Ester Dye on a PDMS Microfluidic Chip. Micromachines, 11(11), 951. https://doi.org/10.3390/mi11110951