
Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism



Abstract
:
1. Introduction
2. Results
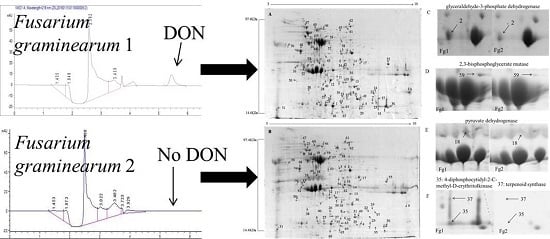
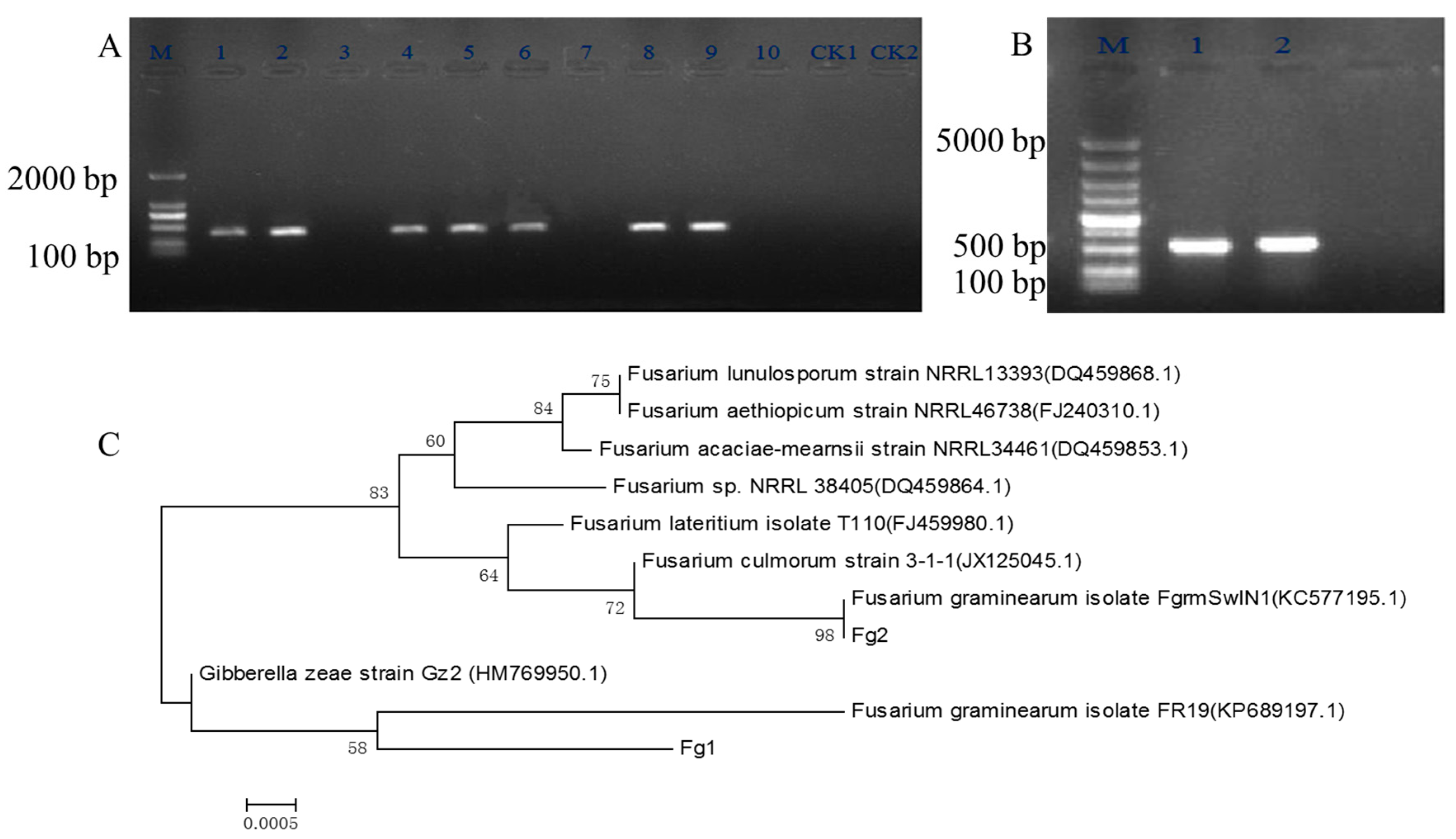
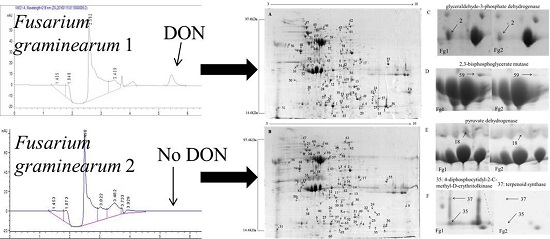
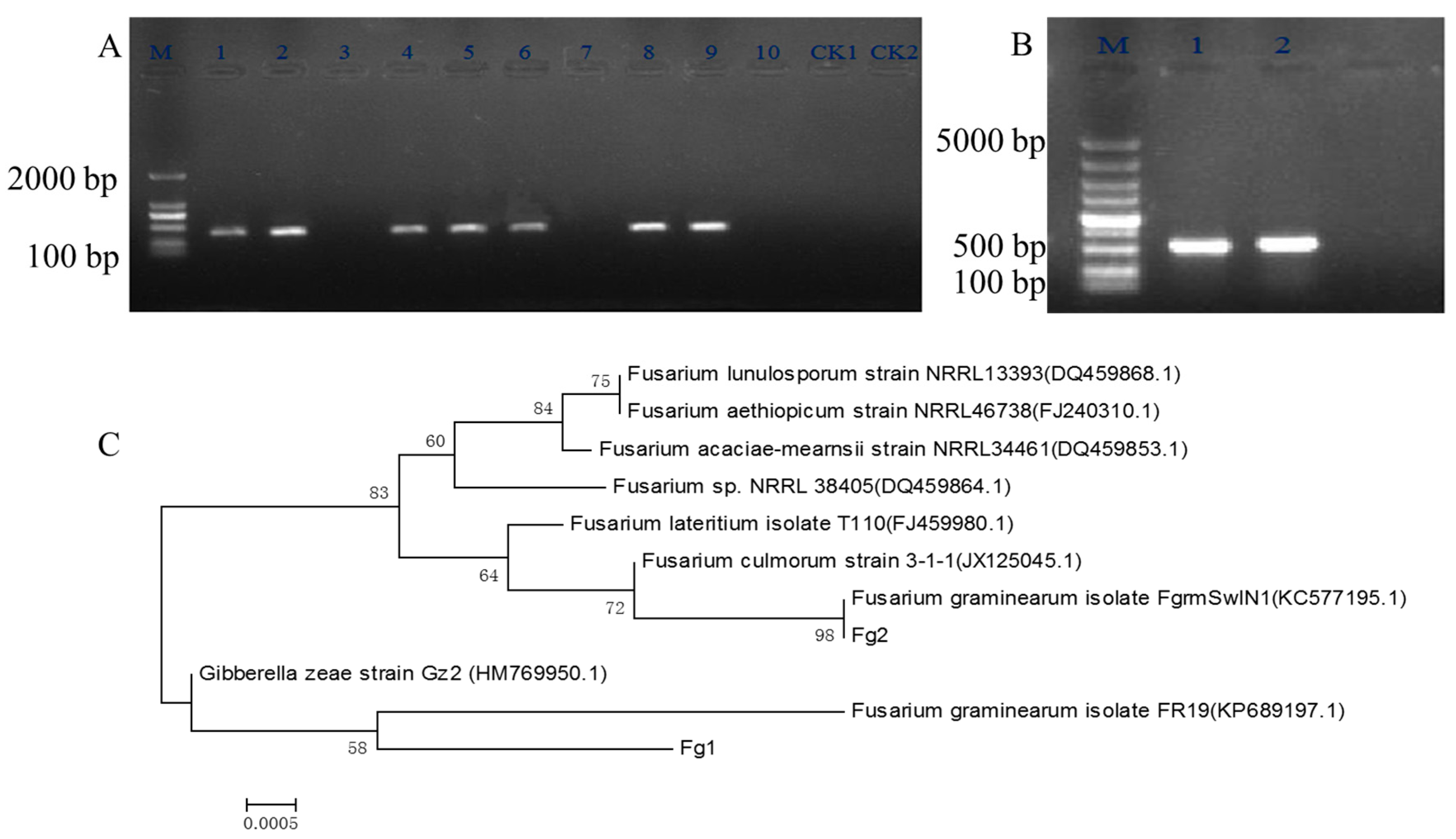
2.1. Screening and Identification of F. graminearum
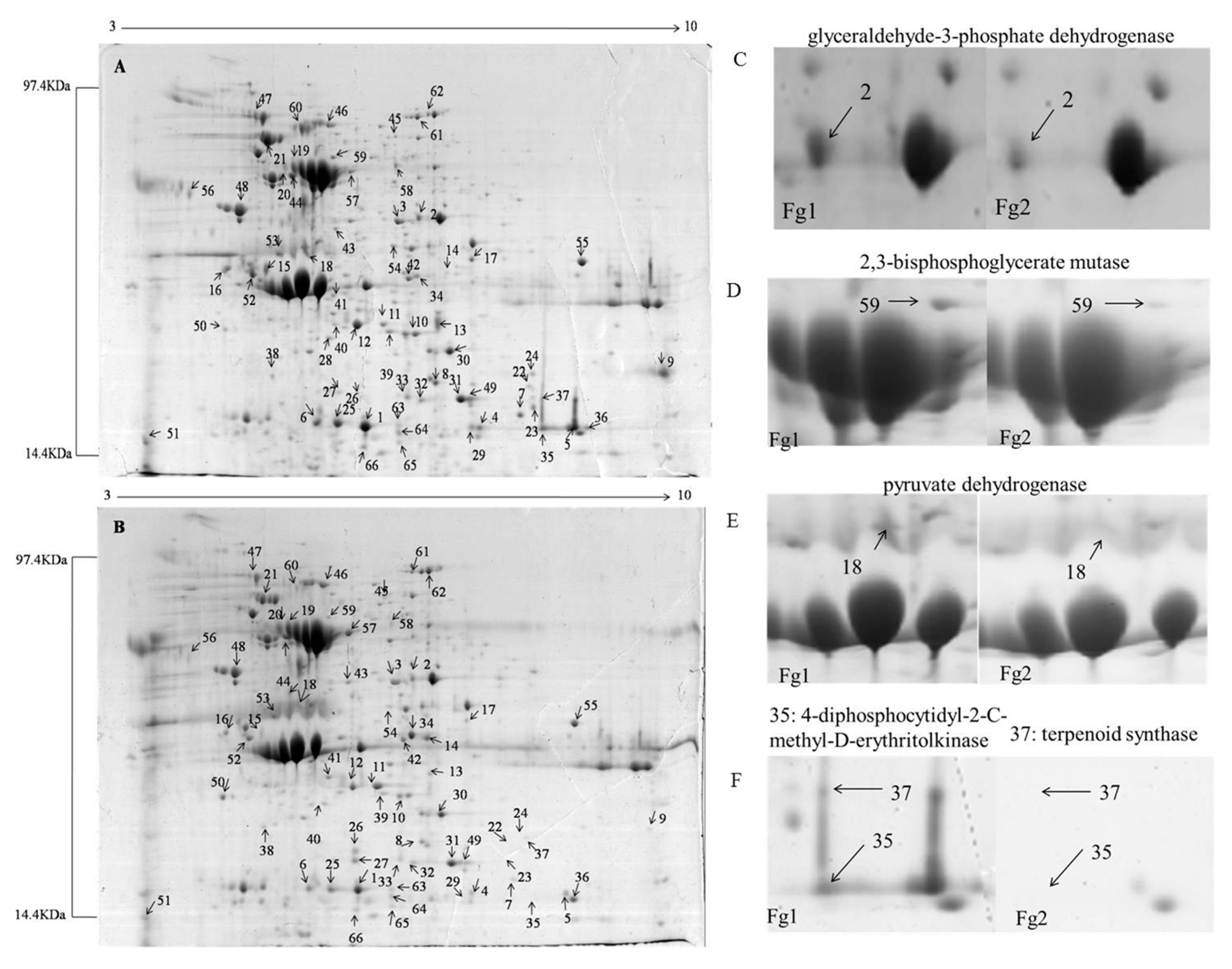
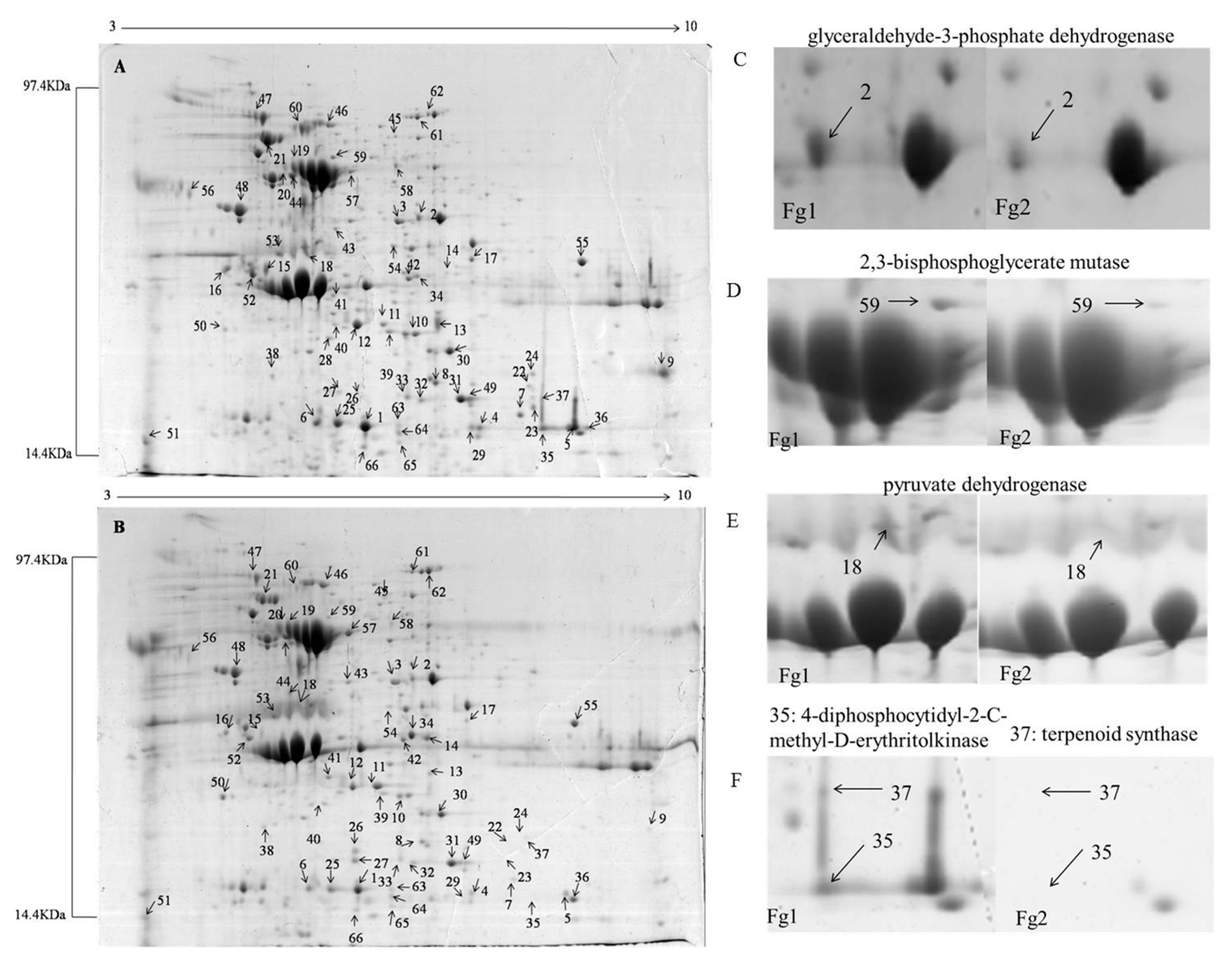
2.2. Identification of Differentially Expressed Proteins in Fg1 and Fg2 Strains
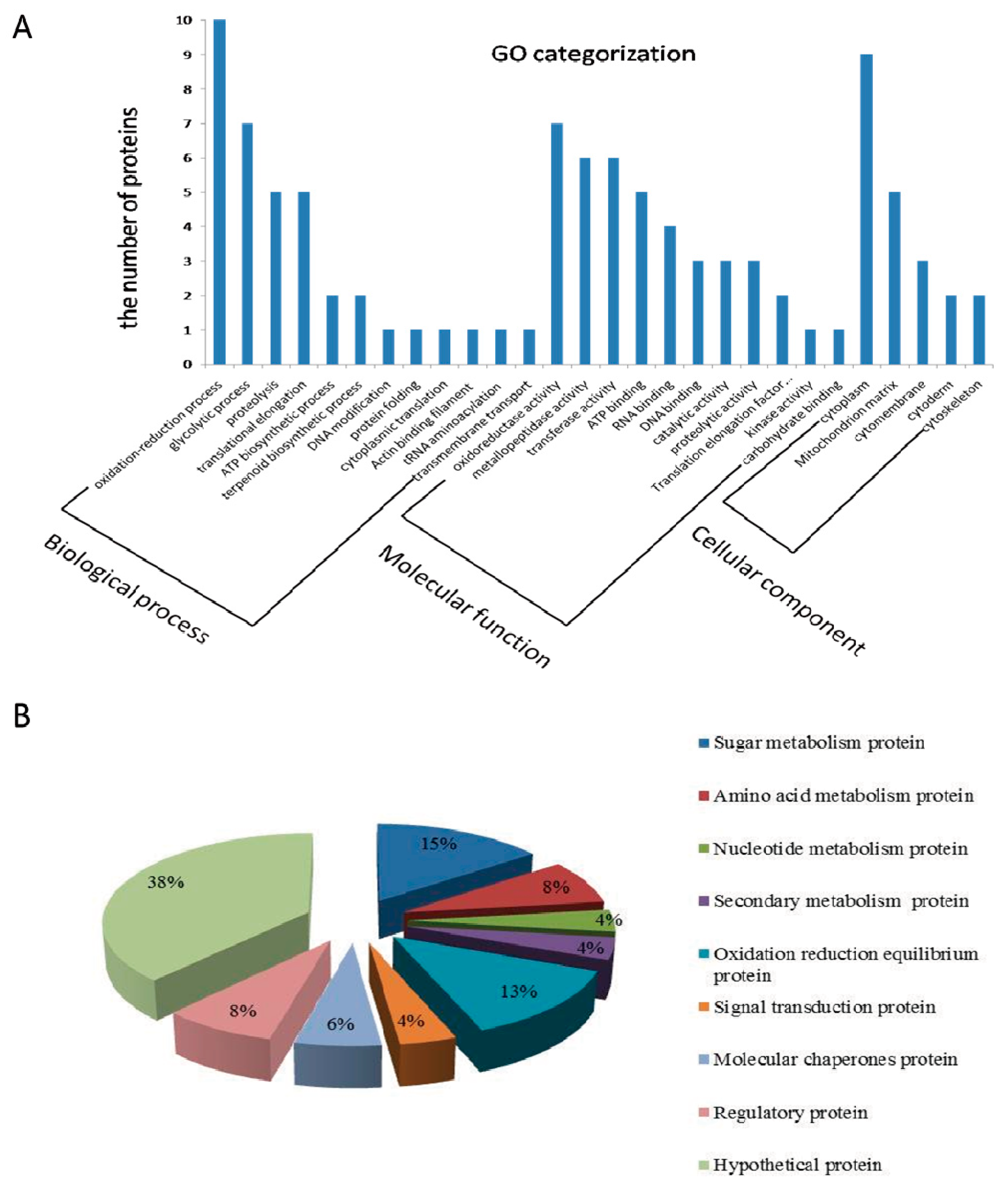
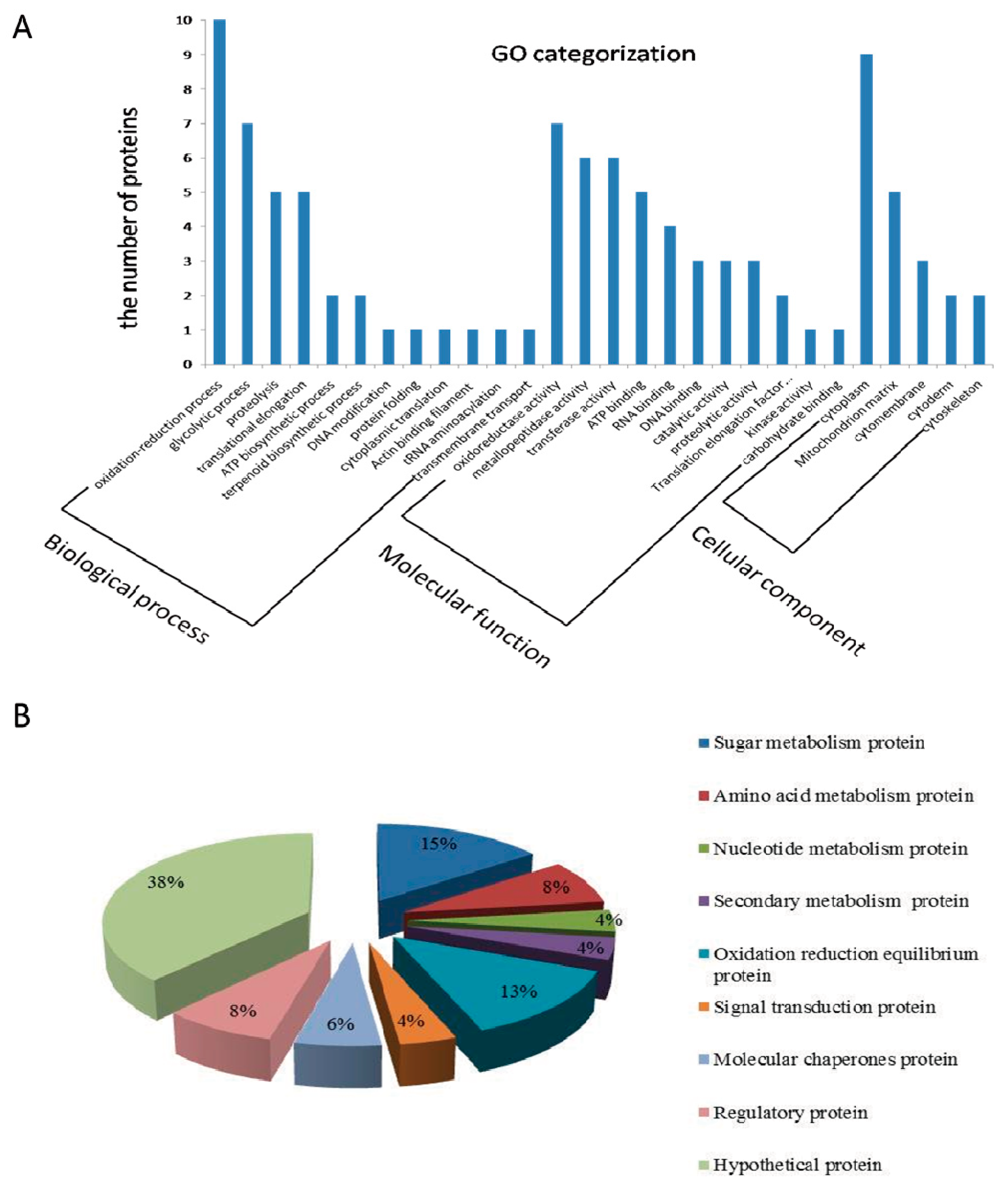
2.3. Gene Ontology (GO) Analysis and Function Classification of Differentially Expressed Proteins in Fg1 and Fg2 Strains
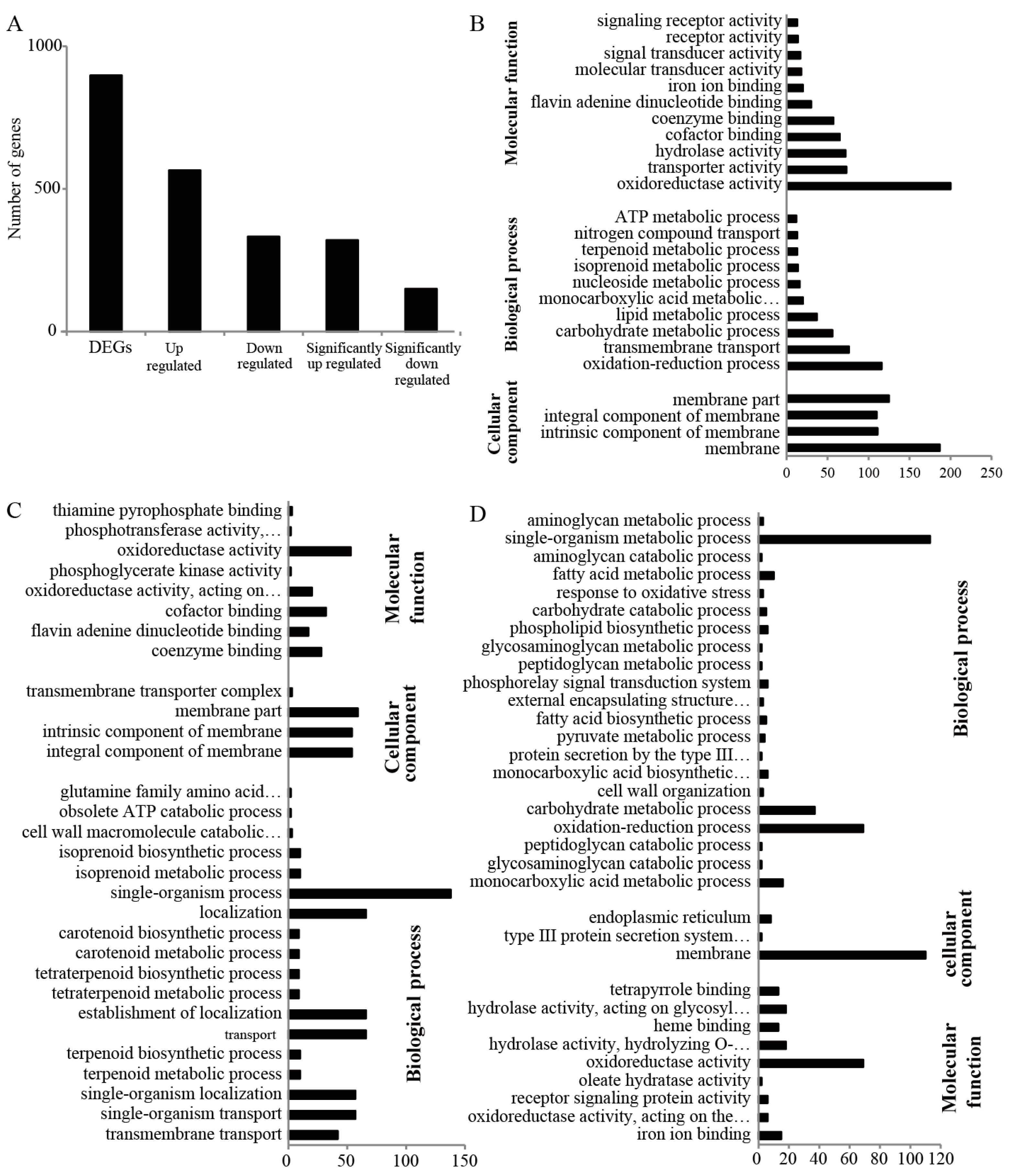
2.4. mRNA-Seq and GO Analysis of the Different Expressed Genes of Fg1 and Fg2
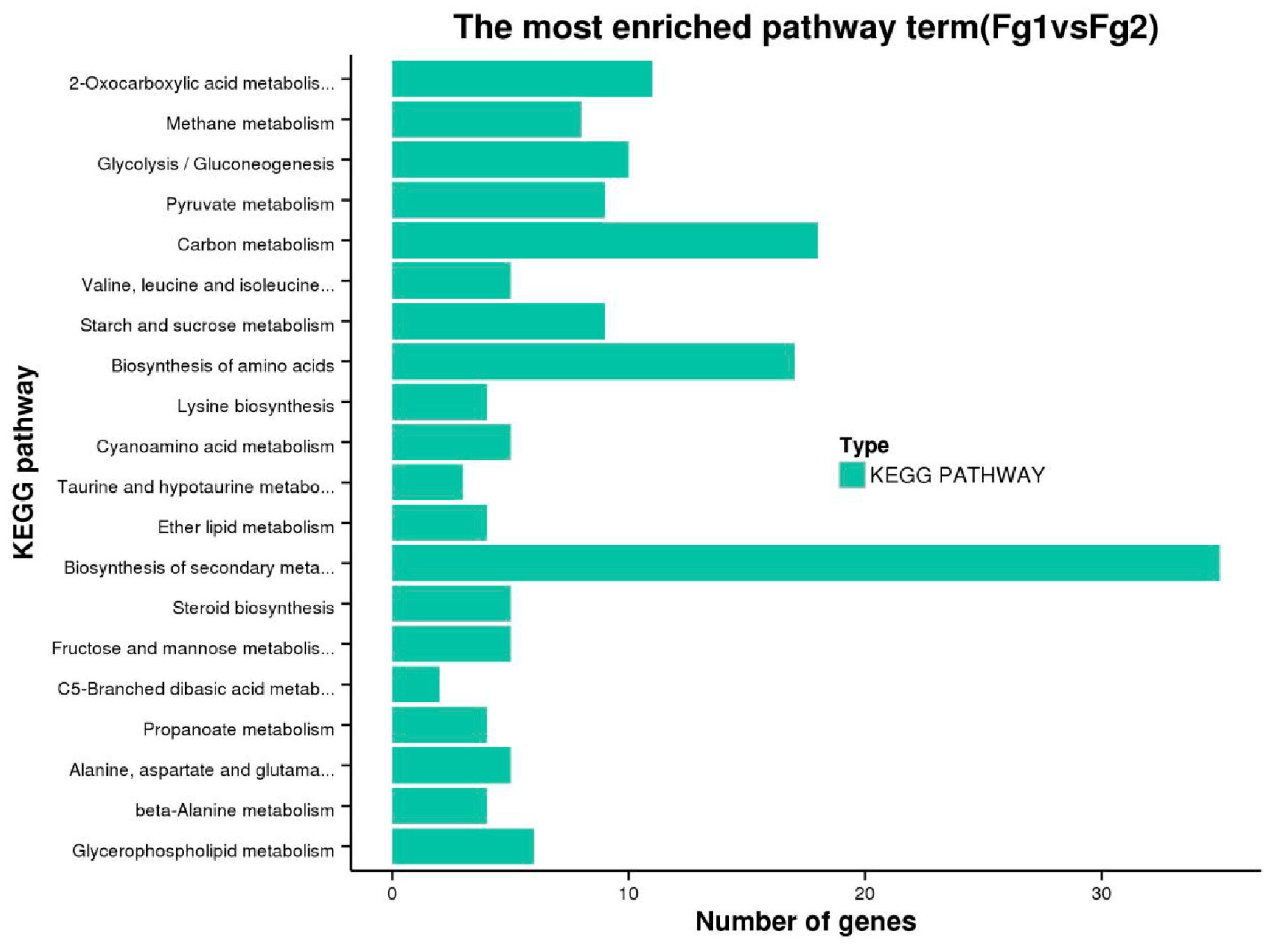
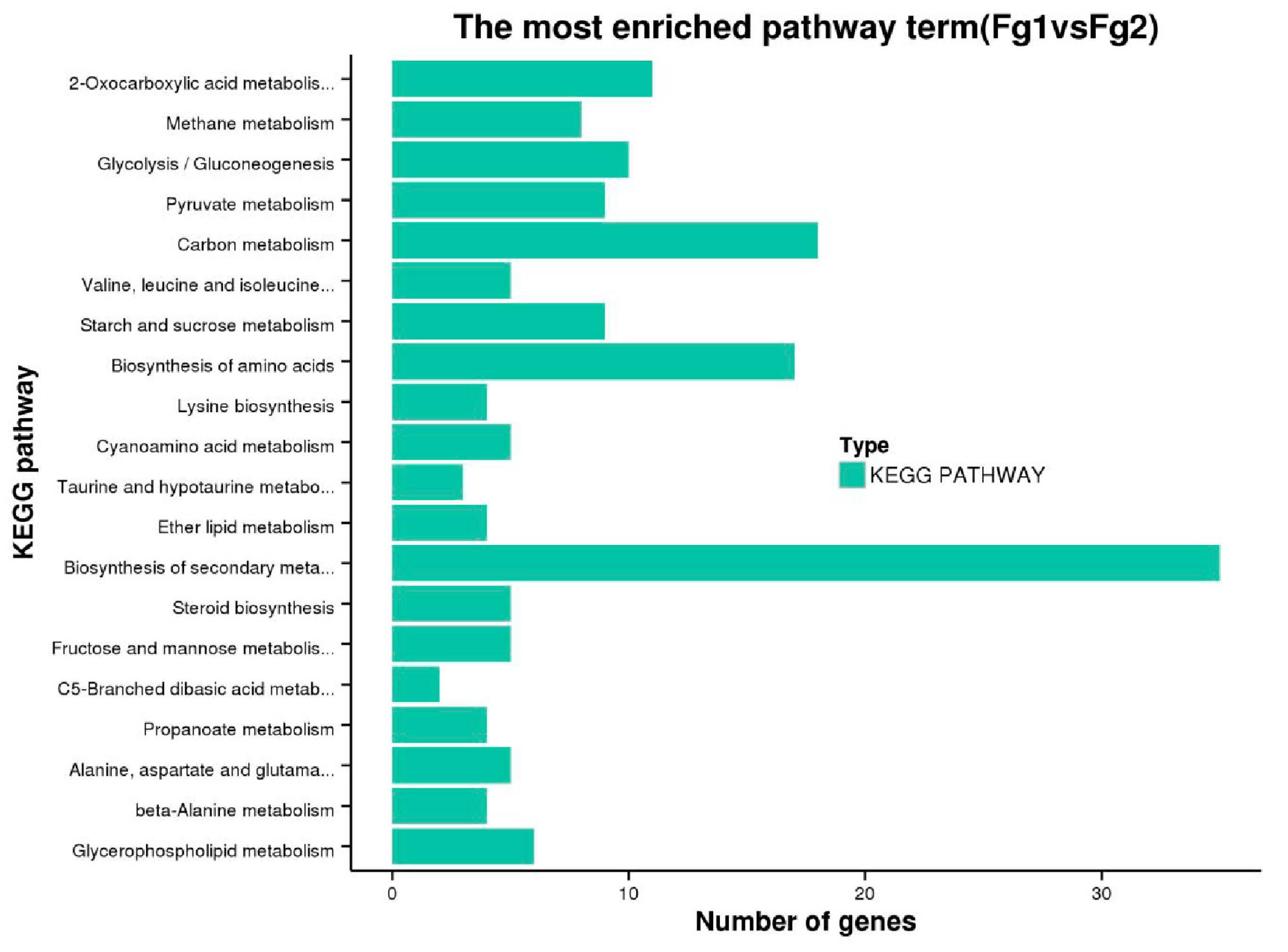
2.5. Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of DEGs of Fg1 and Fg2 Strains
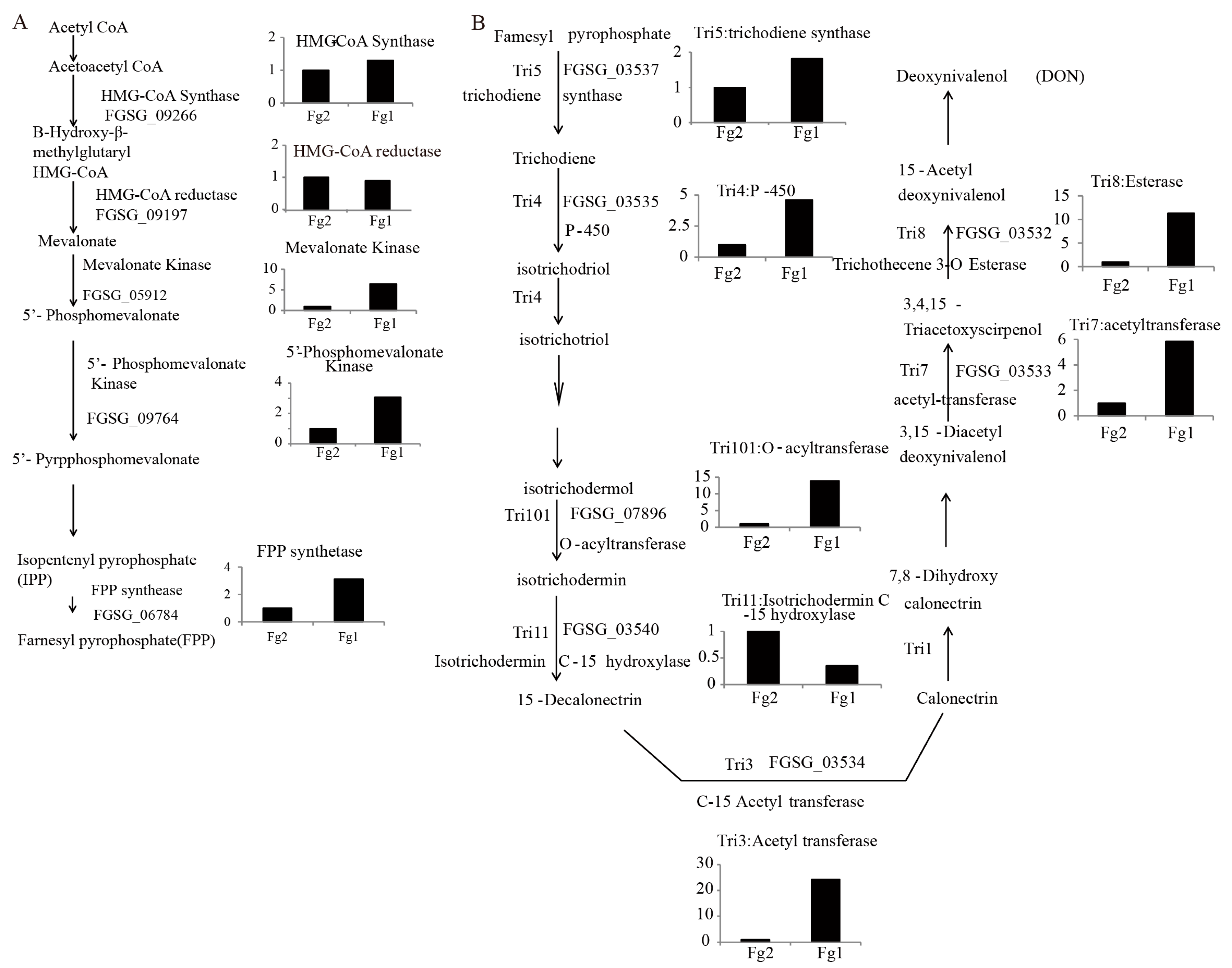
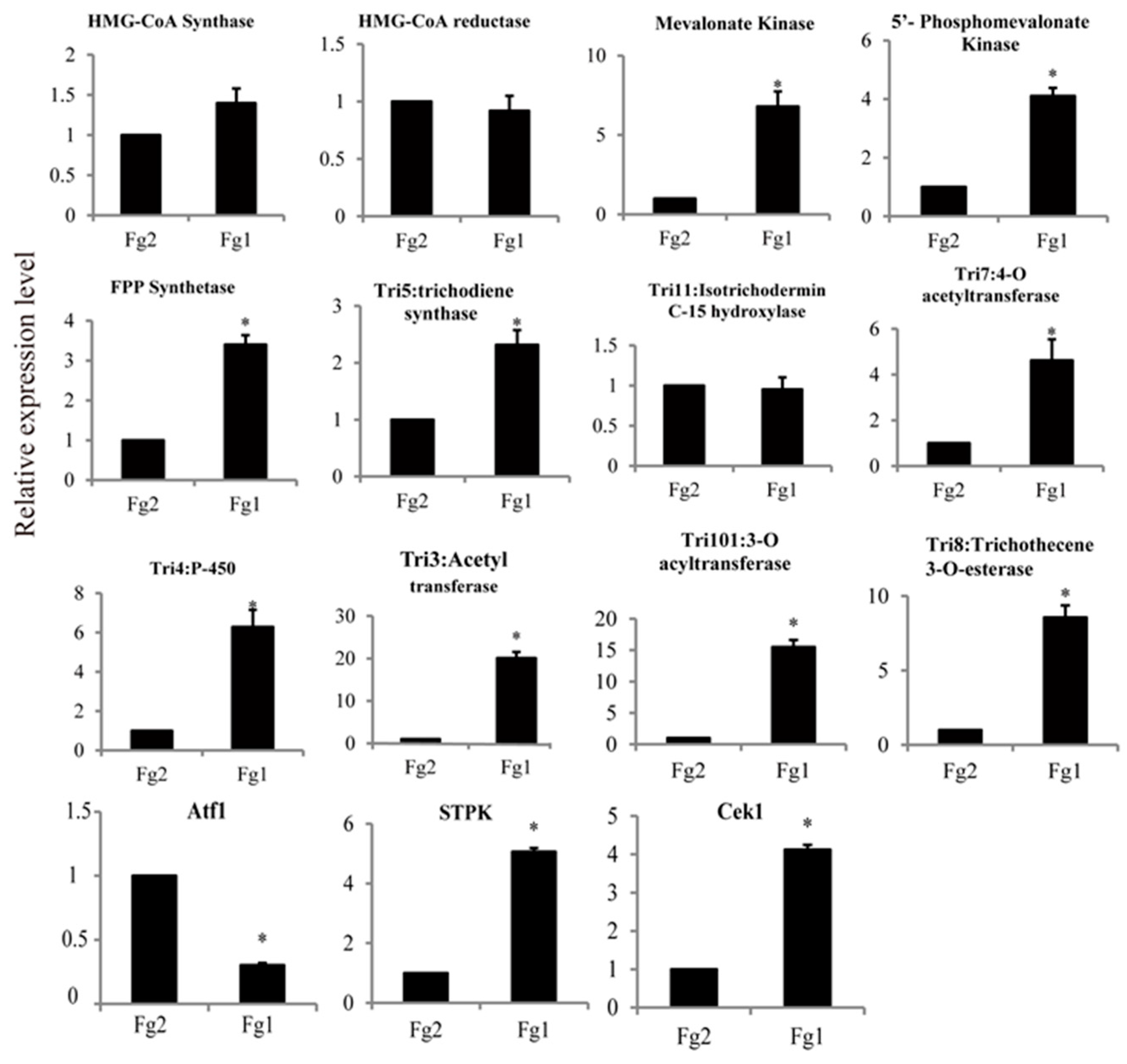
2.6. The Expression Level of Genes Involved in DON Biosynthesis in Fg1 and Fg2
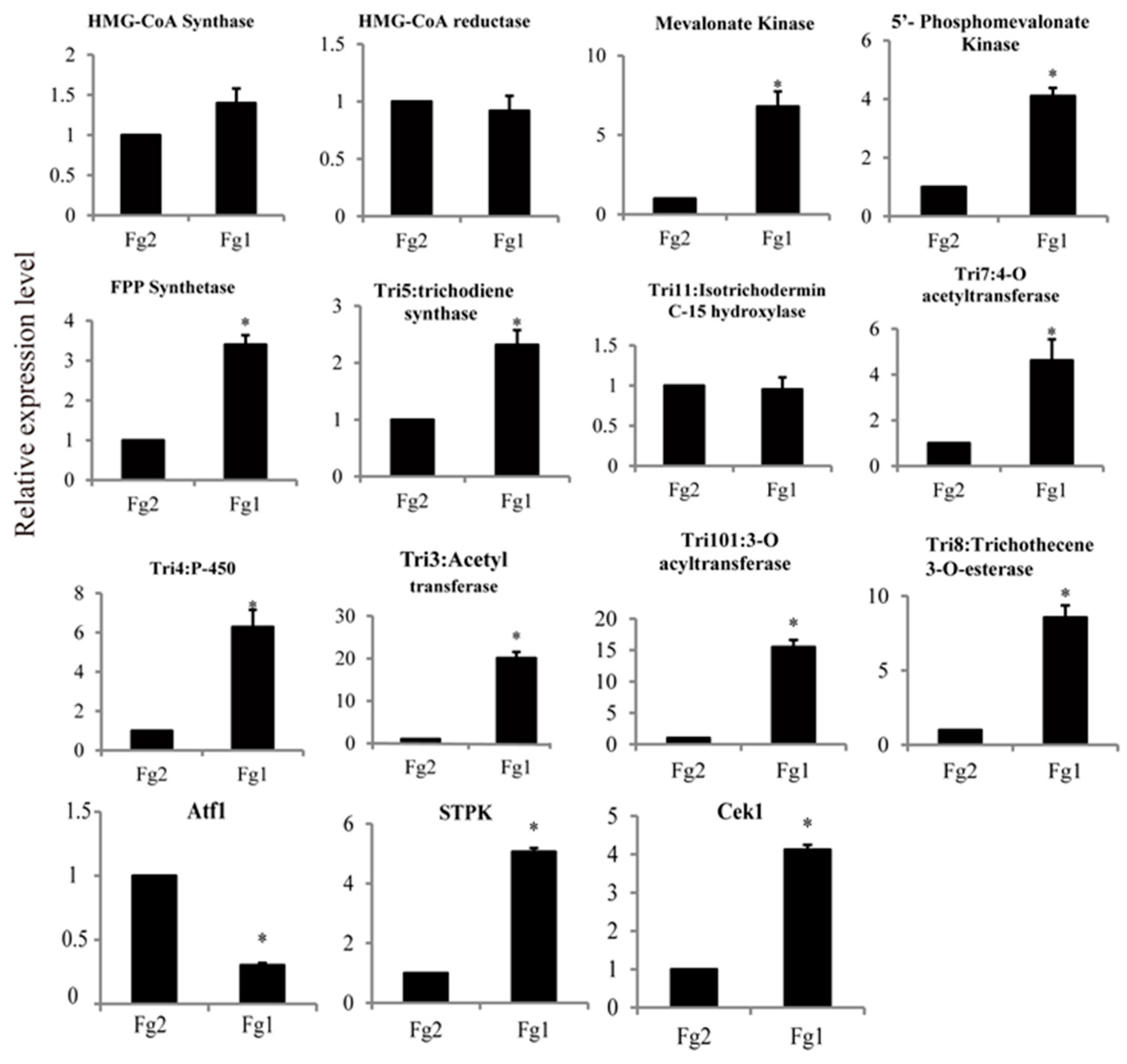
2.7. Verification of the Gene Expression Level by Real Time PCR
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strains, Media and Culture Conditions
5.2. Screening of F. graminearum with Specific PCR
5.3. DON Extraction and HPLC-UV Analysis
5.4. Proteome Analysis
5.4.1. Protein Sample Preparation
5.4.2. Two-Dimensional Gel Electrophoresis and Image Analysis
5.4.3. In-Gel Digestion
5.4.4. MS Analysis and Database Query
5.5. Transcriptome Analysis
5.5.1. RNA Extraction and Quality Test
5.5.2. RNA-Seq Library Construction and Sequencing
5.5.3. Bioinformatics Analysis of RNA-Seq Data
5.5.4. GO and KEGG Pathway Enrichment Analysis
5.6. qPCR
5.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodrigues, I.; Naehrer, K. A Three-Year Survey on the Worldwide Occurrence of Mycotoxins in Feedstuffs and Feed. Toxins 2012, 4, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Savi, G.D.; Piacentini, K.C.; Tibola, C.S.; Scussel, V.M. Mycoflora and deoxynivalenol in whole wheat grains (L.) from Southern Brazil. Food Addit. Contam. B 2014, 7, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Yu, F.; Jia, Y.; Lei, S.; Ma, S.; Cheng, J. Occurrence of mycotoxins in feed ingredients and complete feeds obtained from the Beijing region of China. J. Anim. Sci. Biotechnol. 2014, 5, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.W.; Richardson, A.J.; Duncan, G.; Holtrop, G. Annual variation of dietary deoxynivalenol exposure during years of different Fusarium prevalence: A pilot biomonitoring study. Food Addit. Contam. A 2014, 31, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Barros, G.; Chulze, S.; Ramos, A.J.; Sanchis, V.; Marín, S. Impact of cycling temperatures on Fusarium verticillioides and Fusarium graminearum growth and mycotoxins production in soybean. J. Sci. Food Agric. 2012, 92, 2952–3959. [Google Scholar] [CrossRef] [PubMed]
- Sobrova, P.; Adam, V.; Vasatkova, A.; Beklova, M.; Zeman, L.; Kizek, R. Deoxynivalenol and its toxicity. Interdiscip. Toxicol. 2010, 3, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.R.; Lima, N. Toxicology of mycotoxins. In Experientia Supplementum; Birkhäuser: Basel, Switzerlands; Boston, MA, USA; Berlin, Germany, 2010. [Google Scholar]
- Mclaughlin, J.E.; Bin-Umer, M.A.; Tortora, A.; Mendez, N.; Mccormick, S.; Tumer, N.E.; Wennstrom, J. A genome-wide screen in Saccharomyces cerevisiae reveals a critical role for the mitochondria in the toxicity of a trichothecene mycotoxin. Proc. Natl. Acad. Sci. USA 2009, 106, 21883–21888. [Google Scholar] [CrossRef] [PubMed]
- Mccormick, S.P.; Stanley, A.M.; Stover, N.A.; Alexander, N.J. Trichothecenes: From Simple to Complex Mycotoxins. Toxins 2011, 3, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tokai, T.; Takahashiando, N.; Ohsato, S.; Fujimura, M. Molecular and Genetic Studies of Fusarium Trichothecene Biosynthesis: Pathways, Genes, and Evolution. Biosci. Biotechnol. Biochem. 2007, 71, 2105–2123. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hohn, T.M. Altered Regulation of 15-Acetyldeoxynivalenol Production in Fusarium graminearum. Appl. Environ. Microbiol. 2000, 66, 2062–2065. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Han, Y.K.; Kim, K.H.; Yun, S.H.; Lee, Y.W. Tri13 and Tri7 Determine Deoxynivalenol- and Nivalenol-Producing Chemotypes of Gibberella zeae. Appl. Environ. Microbiol. 2002, 68, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.; Pasquali, M.; Song, X.J.; Hilburn, K.; Mccormick, S.; Dong, Y.; Xu, J.; Kistler, H. Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 2009, 72, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Garvey, G.S.; Mccormick, S.P.; Alexander, N.J.; Rayment, I. Structural and functional characterization of TRI3 trichothecene 15-O-acetyltransferase from Fusarium sporotrichioides. Prot. Sci. 2009, 18, 747–761. [Google Scholar] [CrossRef]
- Nasmith, C.G.; Walkowiak, S.; Wang, L.; Leung, W.W.Y.; Gong, Y.; Johnston, A.; Harris, L.J.; Guttman, D.S.; Subramaniam, R. Tri6 is a global transcription regulator in the phytopathogen Fusarium graminearum. PLoS Pathog. 2011, 7, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Dong, Y.; Kistler, H.C. Fusarium graminearum Tri12p influences virulence to wheat and trichothecene accumulation. Mol. Plant Microbe Interact. 2012, 25, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Hohn, T.M.; Beremand, M.N. Regulation of Trichodiene Synthase in Fusarium sporotrichioides and Gibberella pulicaris (Fusarium sambucinum). Appl. Environ. Microbiol. 1989, 55, 1500–1503. [Google Scholar] [PubMed]
- Hohn, T.M.; Beremand, P.D. Isolation and nucleotide sequence of a sesquiterpene cyclase gene from the trichothecene-producing fungus Fusarium sporotrichioides. Gene 1989, 79, 1381–1388. [Google Scholar] [CrossRef]
- Mccormick, S.P.; Harris, L.J.; Alexander, N.J.; Ouellet, T.; Saparno, A.; Allard, S.; Desjardins, A.E. Tri1 in Fusarium graminearum encodes a P450 oxygenase. Appl. Environ. Microbiol. 2004, 70, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Mccormick, S.P.; Alexander, N.J.; Proctor, R.H. Heterologous expression of two trichothecene P450 genes in Fusarium verticillioides. Can. J. Microbiol. 2006, 52, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Peplow, A.W.; Meek, I.B.; Wiles, M.C.; Phillips, T.D.; Beremand, M.N. Tri16 is required for esterification of position C-8 during trichothecene mycotoxin production by Fusarium sporotrichioides. Appl. Environ. Microbiol. 2003, 69, 5935–5940. [Google Scholar] [CrossRef] [PubMed]
- Alexander, N. The TRI101 story: Engineering wheat and barley to resist Fusarium head blight. World Mycotoxin J. 2008, 1, 31–37. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, H.; Qi, L.; Zhang, S.; Zhou, X.; Zhang, Y.; Xu, J.R. FgKin1 kinase localizes to the septal pore and plays a role in hyphal growth, ascospore germination, pathogenesis, and localization of Tub1 beta-tubulins in Fusarium graminearum. New Phytol. 2014, 204, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, N.; Tokai, T.; Nishiuchi, T.; Takahashi-Ando, N.; Fujimura, M.; Kimura, M. Involvement of the osmosensor histidine kinase and osmotic stress-activated protein kinases in the regulation of secondary metabolism in Fusarium graminearum. Biochem. Biophs. Res. C 2007, 363, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wang, G.; Jiang, C.; Xu, J.R.; Wang, C. Fgk3 glycogen synthase kinase is important for development, pathogenesis, and stress responses in Fusarium graminearum. Sci. Rep. UK 2015, 5, 8504. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Gu, Q.; Yun, Y.; Yin, Y.; Xu, J.R.; Shim, W.B.; Ma, Z. The TOR signaling pathway regulates vegetative development and virulence in Fusarium graminearum. New Phytol. 2014, 203, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Kröger, C.; Bönnighausen, J.; Schäfer, W.; Bormann, J. The ATF/CREB transcription factor Atf1 is essential for full virulence, deoxynivalenol production, and stress tolerance in the cereal pathogen Fusarium graminearum. Mol. Plant Microbe Interact. 2013, 26, 1378–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.; Wang, J.; Zhai, Z.; Zhang, L.; Zheng, W.; Zheng, W.; Yu, W.; Zhou, J.; Lu, G. Functional characterization of Rho family small GTPases in Fusarium graminearum. Fungal Genet. Biol. 2013, 61, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Hestbjerg, H.; Felding, G.; Elmholt, S. Fusarium culmorum, Infection of Barley Seedlings: Correlation between Aggressiveness and Deoxynivalenol Content. J. Phytopathol. 2002, 150, 308–312. [Google Scholar] [CrossRef]
- Maier, F.J.; Miedaner, T.; Hadeler, B.; Felk, A.; Salomon, S.; Lemmens, M. Involvement of trichothecenes in fusarioses of wheat, barley and maize evaluated by gene disruption of the trichodiene synthase (Tri5) gene in three field isolates of different chemotype and virulence. Mol. Plant Pathol. 2006, 7, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Takagi, M.; Kaneda, K.; Watanabe, H.; Dairi, T.; Seto, H. Studies on the nonmevalonate pathway: Conversion of 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol to its 2-phospho derivative by 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol kinase. Tetrahedron Lett. 2000, 41, 2925–2928. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Poulter, C.D. Creating isoprenoid diversity. Science 1997, 277, 1788–1789. [Google Scholar] [CrossRef] [PubMed]
- Camp, P.J.; Randall, D.D. Purification and Characterization of the Pea Chloroplast Pyruvate Dehydrogenase Complex: A Source of Acetyl-CoA and NADH for Fatty Acid Biosynthesis. Plant Physiol. 1985, 77, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Herz, S.; Wungsintaweekul, J.; Schuhr, C.A.; Hecht, S.; Luttgen, H.; Sagner, S.; Fellermeier, M.; Eisenreich, W.; Zenk, M.H.; Bacher, A.; et al. Biosynthesis of terpenoids: YgbB protein converts 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate to 2C-methyl-D-erythritol 2,4-cyclodiphosphate. Proc. Natl. Acad. Sci. USA 2000, 97, 2486–2490. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, P.; Simpson, D.R.; Weston, G.; Rezanoor, H.N.; Lees, A.K.; Parry, D.W.; Joyce, D. Detection and quantification of Fusarium culmorum and Fusarium graminearum in cereals using PCR assays. Physiol. Mol. Plant Pathol. 1998, 53, 17–37. [Google Scholar] [CrossRef]
- Manter, D.K.; Vivanco, J.M. Use of the ITS primers, ITS1F and ITS4, to characterize fungal abundance and diversity in mixed-template samples by qPCR and length heterogeneity analysis. J. Microbiol. Methods 2007, 71, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bradford, M.M.A. A Rapid and Sensitive Method for Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analyt. Biochem. 1976, 25, 248–256. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.H.; Li, Q.; Ma, Z.; Chu, C. Differential proteomic analysis of proteins in wheat spikes induced by Fusarium graminearum. Proteomics 2005, 5, 4496–4503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, Z.; Jiang, L.; Jiang, J.; Luo, H.; Fu, L. Effect of post-harvest heat treatment on proteome change of peach fruit during ripening. J. Proteom. 2011, 74, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, Q.; Zhang, H.; Cao, J.; Zhang, X.; Apaliya, M.T. The possible mechanisms involved in degradation of patulin by pichia caribbica. Toxins 2016, 8, 289. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Yun, S.H. Evaluation of Potential Reference Genes for Quantitative RT-PCR Analysis in Fusarium graminearum under Different Culture Conditions. Plant Pathol. J. 2011, 27, 301–309. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DON (μg/g) |
|---|---|
| Fg1 | 1.02 ± 0.015 |
| Fg2 | 0.00 ± 0 |
| Fg3 | 0.86 ± 0.012 |
| Fg4 | 0.24 ± 0.012 |
| Fg5 | 0.43 ± 0.013 |
| Fg6 | 0.59 ± 0.015 |
| Fg7 | 0.76 ± 0.013 |
| Protein Spot | Protein Name | NCBI Accession | Mass | PI | Species | Score | Peptide Matches |
|---|---|---|---|---|---|---|---|
| 1 | lithostathine precursor | gi|45430003 | 19720 | 5.75 | Bos taurus | 446 | 6(4) |
| 2 | glyceraldehyde-3-phosph ate dehydrogenase | gi|475671174 | 36199 | 6.10 | Fusarium oxysporum | 297 | 4(2) |
| 3 | aldehyde reductase 1 | gi|758198063 | 37477 | 5.83 | F. graminearum | 501 | 7(4) |
| 4 | glyceraldehyde-3-phosphate dehydrogenase | gi|758205105 | 32320 | 5.87 | F. graminearum | 147 | 3(0) |
| 5 | type i restriction endonuclease subunit m | gi|503927174 | 63891 | 5.79 | Pseudoxanthomonas spadix | 72 | 1(1) |
| 6 | regenerating islet-derived 3 alpha | gi|134024814 | 19560 | 5.25 | Bos taurus | 168 | 2(1) |
| 7 | hypothetical protein fpse_07721 | gi|685864163 | 59116 | 6.21 | Fusarium pseudograminearum | 420 | 4(4) |
| 8 | glyceraldehyde-3-phosphate dehydrogenase | gi|758205105 | 32320 | 6.10 | F. graminearum | 163 | 3(1) |
| 9 | hypothetical protein fpse_10416 | gi|685869551 | 18507 | 8.86 | F. pseudograminearum | 333 | 5(2) |
| 10 | superoxide dismutase | gi|758197546 | 24948 | 7.14 | F. graminearum | 556 | 4(3) |
| 12 | predicted protein | gi|302897541 | 23291 | 5.94 | Nectria haematococca mpVI 77-13-4 | 202 | 2(2) |
| 13 | hypothetical protein fpse_03954 | gi|758198681 | 33014 | 5.32 | F. graminearum PH-1 | 114 | 1(1) |
| 14 | hypothetical protein fpse_00105 | gi|758186649 | 33716 | 6.12 | F. graminearum PH-1 | 372 | 4(3) |
| 15 | hypothetical protein fpse_05320 | gi|685859361 | 32998 | 5.32 | F. pseudograminearum CS3096 | 271 | 3(3) |
| 16 | 78 kda glucose-regulated protein like protein | gi|477521896 | 70770 | 4.94 | F. oxysporum | 715 | 6(5) |
| 17 | elongation factor 2 | gi|584132859 | 93373 | 6.34 | F. verticillioides 7600 | 540 | 7(3) |
| 18 | pyruvate dehydrogenase e1 component subunit beta | gi|758201343 | 41784 | 6.07 | F. graminearum PH-1 | 377 | 6(3) |
| 19 | hypothetical protein td95_000288 | gi|802102353 | 29901 | 4.87 | Thielaviopsis punctulata | 137 | 1(1) |
| 20 | hypothetical protein fpse_01472 | gi|685851669 | 53851 | 5.39 | F. pseudograminearum CS3096 | 829 | 10(7) |
| 21 | hypothetical protein fg05_03462 | gi|596545344 | 108299 | 5.69 | F. graminearum | 454 | 5(5) |
| 23 | hypothetical protein fpse_06578 | gi|685861877 | 20437 | 7.02 | F. pseudograminearum CS3096 | 611 | 6(6) |
| 25 | ornithine carbamoyltransferase | gi|749661859 | 34612 | 5.75 | Pseudomonas sp. CB1 | 64 | 1(1) |
| 26 | islet-derived protein 3-beta-like isoform x1 | gi|512910064 | 19965 | 6.70 | HeterocepHalus glaber | 88 | 2(0) |
| 27 | hypothetical protein fpsg_08677 | gi|758195374 | 17923 | 5.46 | F. graminearum PH-1 | 413 | 5(4) |
| 29 | hypothetical protein fpse_10576 | gi|685869871 | 16463 | 7.78 | F. pseudograminearum CS3096 | 255 | 3(3) |
| 30 | hypothetical protein fpse_07721 | gi|685864163 | 59116 | 6.21 | F. pseudograminearum CS3096 | 371 | 4(2) |
| 31 | peptidylprolyl isomerase (ec 5.2.1.8) a precursor | gi|2118328 | 24688 | 9.23 | Tolypocladium inflate | 235 | 3(2) |
| 32 | cyclophilin, cytosolic form | gi|642360 | 19495 | 9.23 | Tolypocladium inflatum | 227 | 2(2) |
| 33 | hypothetical protein fpsg_00105 | gi|699040963 | 33716 | 8.64 | F. graminearum PH-1 | 442 | 4(3) |
| 34 | prolyl-tRNA synthetase | gi|602541635 | 65726 | 5.11 | Mycobacterium mageritense DSM 44476 | 64 | 1(1) |
| 35 | 4-diphosphocytidyl-2-C-methyl-D-erythritolkinase | gi|329748844 | 34575 | 7.74 | SpHaerochaeta coccoides DSM 17374 | 62 | 1(1) |
| 37 | terpenoid synthase 19 | gi|15231879 | 69187 | 5.68 | Arabidopsis thaliana | 137 | 1(1) |
| 39 | putative ATP synthase beta mitochondrial precursor protein | gi|629659977 | 54945 | 5.46 | Eutypa lata UCREL1 | 454 | 5(3) |
| 44 | hypothetical protein fg05_11228 | gi|596542472 | 70636 | 5.11 | F. graminearum | 200 | 3(1) |
| 46 | hypothetical protein fgsg_05797 | gi|758204059 | 63567 | 5.09 | F. graminearum PH-1 | 570 | 7(3) |
| 47 | cathepsin D precursor | gi|148231809 | 43881 | 5.50 | Xenopus laevis | 84 | 1(1) |
| 48 | cyclophilin type peptidyl-prolyl cis-trans isomerase/cld | gi|770311810 | 18857 | 7.80 | Aspergillus parasiticus SU-1 | 92 | 1(1) |
| 49 | hypothetical protein fgsg_02523 | gi|758192157 | 18958 | 4.43 | F.graminearum PH-1 | 241 | 3(2) |
| 50 | heat shock protein 70-1 | gi|38325811 | 71195 | 5.02 | Nicotiana tabacum | 554 | 6(4) |
| 52 | FAD-binding monooxygenase | gi|703062904 | 43658 | 6.56 | Catenuloplanes japonicus | 75 | 1(1) |
| 54 | elongation factor 2 | gi|758211940 | 93309 | 6.34 | F. graminearum PH-1 | 645 | 6(4) |
| 57 | aldehyde dehydrogenase | gi|589108195 | 54084 | 5.93 | Trichoderma reesei QM6a | 246 | 3(3) |
| 58 | six-bladed beta-propeller | gi|573988893 | 86498 | 6.10 | Cordyceps militaris CM01 | 64 | 1(1) |
| 59 | 2,3-bisphosphoglycerate mutase | gi|189194371 | 57827 | 5.36 | PyrenopHora tritici-repentis Pt-1C-BFP | 87 | 1(1) |
| 60 | hypothetical protein fg05_02937 | gi|596543275 | 64906 | 5.67 | F. graminearum | 661 | 8(4) |
| 61 | peroxidase/catalase 2 | gi|758200916 | 88543 | 6.49 | F. graminearum PH-1 | 661 | 8(4) |
| 66 | regenerating islet-derived protein 3 gamma | gi|351710837 | 19837 | 6.70 | HeterocepHalus glaber | 89 | 2(0) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Zhang, X.; Zhao, L.; Apaliya, M.T.; Yang, Q.; Sun, W.; Zhang, X.; Zhang, H. Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism. Toxins 2017, 9, 184. https://doi.org/10.3390/toxins9060184
Zheng X, Zhang X, Zhao L, Apaliya MT, Yang Q, Sun W, Zhang X, Zhang H. Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism. Toxins. 2017; 9(6):184. https://doi.org/10.3390/toxins9060184
Chicago/Turabian StyleZheng, Xiangfeng, Xiaoli Zhang, Lina Zhao, Maurice T. Apaliya, Qiya Yang, Wei Sun, Xiaoyun Zhang, and Hongyin Zhang. 2017. "Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism" Toxins 9, no. 6: 184. https://doi.org/10.3390/toxins9060184
APA StyleZheng, X., Zhang, X., Zhao, L., Apaliya, M. T., Yang, Q., Sun, W., Zhang, X., & Zhang, H. (2017). Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism. Toxins, 9(6), 184. https://doi.org/10.3390/toxins9060184