
Development of a LC-MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. LC-MS/MS Optimization
2.2. Optimization of the Extraction Step
2.3. Method Performance
2.3.1. Linearity
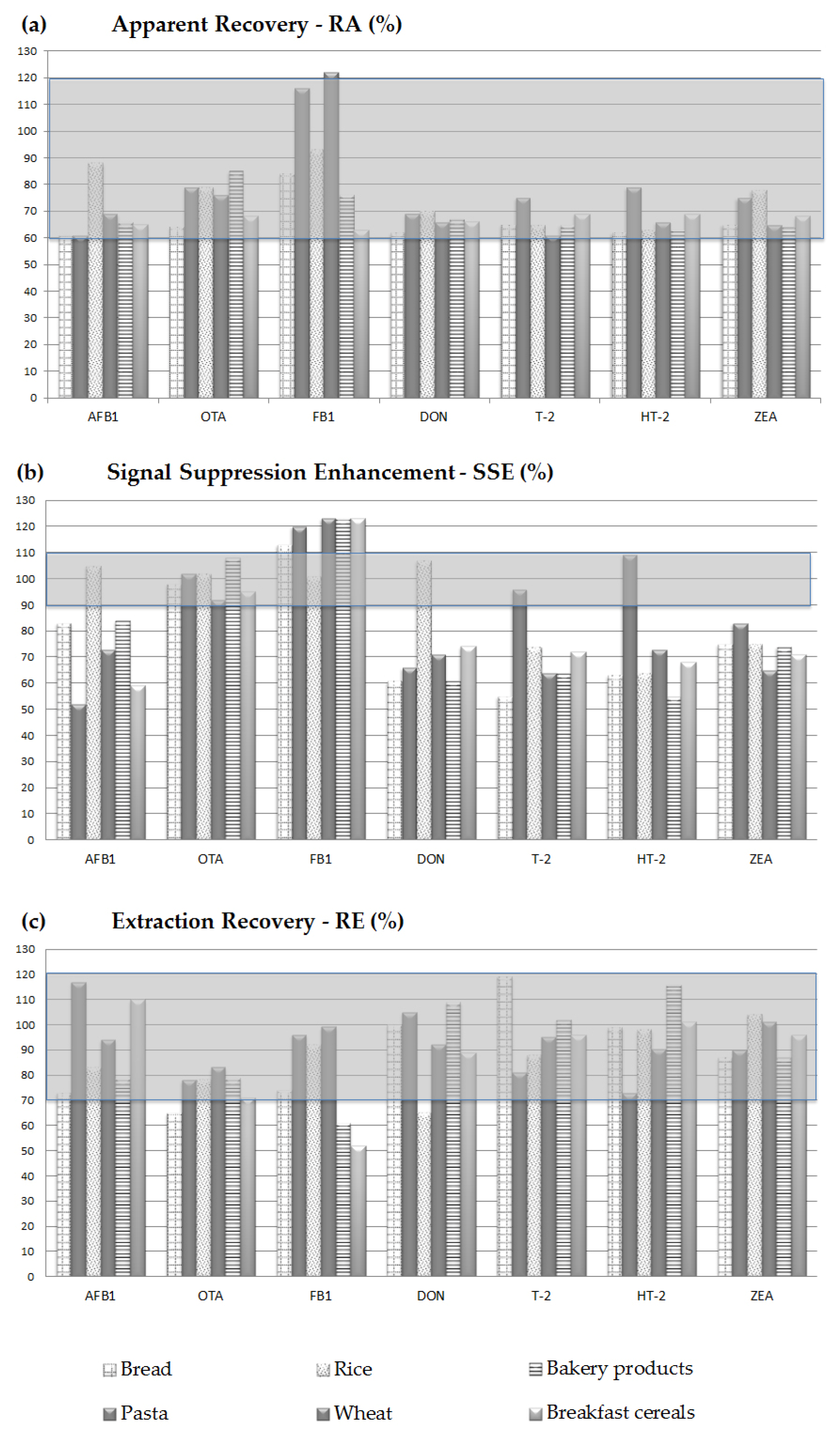
2.3.2. Apparent Recovery, Matrix Effect, and Extraction Recovery
2.3.3. Application to TDS Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Samples
3.3. Sample Preparation
3.4. LC-MS/MS Analysis
3.5. Method Performance
3.5.1. Linearity
3.5.2. Apparent Recovery, Matrix Effect, Recovery of Extraction
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miraglia, M.; De Santis, B.; Brera, C. Climate change: Implications for mycotoxin contamination of foods. J. Biotechnol. 2008, 136, S715. [Google Scholar] [CrossRef]
- Miraglia, M.; Marvin, H.; Kleter, G.; Battilani, P.; Brera, C.; Coni, E.; Cubadda, F.; Croci, L.; De Santis, B.; Dekkers, S. Climate change and food safety: An emerging issue with special focus on Europe. Food Chem. Toxic. 2009, 47, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Tirado, M.; Clarke, R.; Jaykus, L.A.; McQuatters-Gollop, A.; Frank, J. Climate change and food safety: A review. Food Res. Intern. 2010, 43, 1745–1765. [Google Scholar] [CrossRef]
- Van der Fels-Klerx, H.J.; Liu, C.; Battilani, P. Modelling climate change impacts on mycotoxin contamination. World Mycotoxin J. 2009, 9, 717–726. [Google Scholar] [CrossRef]
- Paterson, R.R.M.; Lima, N. How will climate change affect mycotoxins in food? Food Res. Int. 2010, 43, 1902–1914. [Google Scholar] [CrossRef]
- Battilani, P.; Toscano, P.; Van der Fels-Klerx, H.J.; Moretti, A.; Camardo Leggieri, M.; Brera, C.; Rortais, A.; Goumperis, T.; Robinson, T. Aflatoxin B1 contamination in maize in Europe increases due to climate change. Sci. Rep. 2016, 6, 24328. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Hajslova, J.; Zachariasova, M.; Cajka, T. Analysis of multiple mycotoxins in food. Methods Mol. Biol. 2011, 747, 233–258. [Google Scholar] [PubMed]
- Grenier, B.; Loureiro-Bracarense, A.P.; Lucioli, J.; Pacheco, G.D.; Cossalter, A.M.; Moll, W.D.; Schatzmayr, G.; Oswald, I.P. Individual and combined effects of subclinical doses of deoxynivalenol and fumonisins in piglets. Mol. Nutr. Food Res. 2011, 55, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.B.; Edrington, T.S.; Kubena, L.F.; Elissalde, M.H.; Rottinghaus, G.E. Influence of aflatoxin and fumonisin B1-containing culture material on growing barrows. Am. J. Vet. Res. 1995, 56, 1668–1672. [Google Scholar] [PubMed]
- Lee, H.J.; Ryu, D. Worldwide Occurrence of Mycotoxins in Cereals and Cereal-Derived Food Products: Public Health Perspectives of Their Co-occurrence. J. Agric. Food Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Boeira, L.S.; Bryce, J.H.; Stewart, G.G.; Flannigan, B. The effect of combinations of Fusarium mycotoxins (deoxynivalenol, zearalenone and fumonisin B1) on growth of brewing yeasts. J. Appl. Microbiol. 2000, 88, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Johri, T.S.; Swain, B.K.; Ameena, S. Effect of graded levels of aflatoxin, ochratoxin and their combinations on the performance and immune response of broilers. Br. Poult. Sci. 2004, 45, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.B.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a Rapid LC-MS/MS method for the determination of emerging Fusarium mycotoxins enniatins and beauvericin in human biological fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Caruso, G.; Cavaliere, C.; Foglia, P.; Samperi, R.; Laganà, A. Multiclass mycotoxin analysis in food, environmental and biological matrices with chromatography/mass spectrometry. Mass Spectrom. Rev. 2012, 31, 466–503. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.B.; Font, G.; Mañes, J.; Ferrer, E. Comparative assessment of three extraction procedures for determination of emerging Fusarium mycotoxins in pasta by LC-MS/MS. Food Control. 2013, 32, 105–114. [Google Scholar] [CrossRef]
- Krska, R.; Schubert-Ullrich, P.; Molinelli, A.; Sulyok, M.; Macdonald, S.; Crews, C. Mycotoxin analysis: An update. Food Addit. Contam. Part A 2008, 25, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Brera, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; Stranska-Zachariasova, M.; et al. Developments in mycotoxin analysis: An update for 2015–2016. World Mycotoxin J. 2016, 9, 5–30. [Google Scholar] [CrossRef]
- Zhang, K.; Wong, J.W.; Krynitsky, A.J.; Trucksess, M.W. Perspective on advancing FDA regulatory monitoring for mycotoxins in foods using liquid chromatography and mass spectrometry. J. AOAC Int. 2016, 99, 890–894. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, M.; Turrini, A.; Aureli, F.; Moracci, G.; Raggi, A.; Chiaravalle, E.; Mangiacotti, M.; Cenci, T.; Orletti, R.; Candela, L.; et al. Dietary exposure to trace elements and radionuclides: the methodology of the Italian Total Diet Study 2012–2014. Annali dell’Istituto Super. di Sanità 2013, 49, 272–280. [Google Scholar]
- Cubadda, F.; D’Amato, M.; Aureli, F.; Raggi, A.; Mantovani, A. Dietary exposure of the Italian population to inorganic arsenic: The 2012–2014 Total Diet Study. Food Chem. Toxicol. 2016, 98, 148–158. [Google Scholar] [CrossRef] [PubMed]
- EN 14123:2007-Foodstuffs-Determination of Aflatoxin B1 and the Sum of Aflatoxins B1, B2, G1 and G2 in Hazelnuts, Peanuts, Pistachios, Figs and Paprika Powder-High Performance Liquid Chromatographic Method with Post-Column Derivatisation and Immunoaffinity Column Cleanup; BSI: London, UK, 2008; ISBN 978-0-580-58514-2.
- Breidbach, A.; Bouten, K.; Kroeger-Negoita, K.; Stroka, J.; Ulberth, F. LC-MS Based Method of Analysis for the Simultaneous Determination of Four Mycotoxins in Cereals and Feed: Results of a Collaborative Study. 2013. Available online: http://publications.jrc.ec.europa.eu/repository/handle/JRC80176 (accessed on 15 May 2017).
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chrom. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Commission Regulation (EC) No 401/2006. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, L70, 12–34. [Google Scholar]
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; 2014; ISBN 978-91-87461-59-0. Available online: www.eurachem.org (accessed on 15 May 2017).


{kind=link}
{kind=link}
| Mycotoxin | Retention Time (min) | Precursor Ion (m/z) | Product Ions (m/z) a | Collision Energy (V) a | Cone Voltage (V) |
|---|---|---|---|---|---|
| AFB1 | 8.15 | 313.2 [M + H]+ | 285.3/241 | 21/35 | 45 |
| OTA | 7.35 | 404.1 [M + H]+ | 238.8/358.1 | 25/14 | 25 |
| DON | 3.89 | 297.3 [M + H]+ | 203.5/249.5 | 15/10 | 22 |
| FB1 | 5.55 | 723.3 [M + H]+ | 335.1/353.5 | 40/30 | 50 |
| T-2 | 6.24 | 489.6 [M + Na]+ | 245.1/327.1 | 30/20 | 30 |
| HT-2 | 5.77 | 447.2 [M + NH4]+ | 285.1/345.1 | 15/10 | 30 |
| ZEA | 6.79 | 319.3 [M + H]+ | 282.9/301.1 | 10/10 | 22 |
| Extraction Solvent | Recovery Factors (RSDr a) | |
|---|---|---|
| AFB1, OTA | FB1, DON, T-2 and HT-2, ZEA | |
| CH3OH:H2O 80:20 | 75 (20) | 30 (19) |
| CH3CN:H2O:CH3COOH 79:20:1 | 65 (15) | 80 (15) |
| H2O 4mL + EtOH 8mL | 25 (20) | 80 (19) |
| Mycotoxin | R2 | ||||||
|---|---|---|---|---|---|---|---|
| Neat Solvent | Bread | Pasta | Rice | Wheat | Bakery Products | Breakfast Cereal | |
| AFB1 | 0.9995 | 0.9956 | 0.9997 | 0.9901 | 0.9913 | 0.9974 | 0.9995 |
| OTA | 0.9976 | 0.9958 | 0.9995 | 0.9994 | 0.9903 | 0.9965 | 0.9989 |
| FB1 | 0.9975 | 0.9982 | 0.9964 | 0.9968 | 0.9982 | 0.9993 | 0.9969 |
| DON | 0.9941 | 0.9993 | 0.9997 | 0.9993 | 0.9978 | 0.9927 | 0.9914 |
| T-2 | 0.9937 | 0.9983 | 0.9925 | 0.9929 | 0.9969 | 0.9996 | 0.9906 |
| HT-2 | 0.9973 | 0.9909 | 0.9927 | 0.9952 | 0.9919 | 0.9935 | 0.9950 |
| ZEA | 0.9926 | 0.9963 | 0.9937 | 0.9939 | 0.9902 | 0.9942 | 0.9902 |
| Matrix | AFB1 | OTA | FB1 | DON | T-2 | HT-2 | ZEA | |
|---|---|---|---|---|---|---|---|---|
| Bread | LoQ (μg/kg) | 0.13 | 0.8 | 16 | 20 | 8 | 20 | 3.2 |
| RA (%) | 61 | 64 | 84 | 62 | 65 | 62 | 65 | |
| SSE (%) | 83 | 98 | 113 | 61 | 55 | 63 | 75 | |
| RE (%) | 73 | 65 | 74 | 100 | 119 | 99 | 87 | |
| RSDr (%) | 14 | 10 | 15 | 18 | 17 | 15 | 20 | |
| Pasta | LoQ (μg/kg) | 0.06 | 0.4 | 8 | 20 | 4 | 20 | 1.6 |
| RA (%) | 61 | 79 | 116 | 69 | 75 | 79 | 75 | |
| SSE (%) | 52 | 102 | 120 | 66 | 96 | 109 | 83 | |
| RE (%) | 117 | 78 | 96 | 105 | 81 | 73 | 90 | |
| RSDr (%) | 11 | 12 | 11 | 12 | 16 | 15 | 16 | |
| Rice | LoQ (μg/kg) | 0.06 | 0.4 | 8 | 20 | 4 | 20 | 1.6 |
| RA (%) | 88 | 79 | 93 | 70 | 65 | 63 | 78 | |
| SSE (%) | 105 | 102 | 101 | 107 | 74 | 64 | 75 | |
| RE (%) | 83 | 78 | 92 | 65 | 88 | 98 | 104 | |
| RSDr (%) | 11 | 10 | 10 | 15 | 14 | 13 | 15 | |
| Wheat | LoQ (μg/kg) | 0.06 | 0.4 | 8 | 20 | 4 | 20 | 1.6 |
| RA (%) | 69 | 76 | 122 | 66 | 61 | 66 | 65 | |
| SSE (%) | 73 | 92 | 123 | 71 | 64 | 73 | 65 | |
| RE (%) | 94 | 83 | 99 | 92 | 95 | 90 | 101 | |
| RSDr (%) | 12 | 10 | 12 | 15 | 15 | 15 | 15 | |
| Backery products | LoQ (μg/kg) | 0.13 | 0.8 | 16 | 20 | 8 | 20 | 3.2 |
| RA (%) | 66 | 85 | 76 | 67 | 65 | 63 | 64 | |
| SSE (%) | 84 | 108 | 123 | 61 | 64 | 55 | 74 | |
| RE (%) | 79 | 79 | 61 | 109 | 102 | 116 | 87 | |
| RSDr (%) | 13 | 10 | 16 | 18 | 15 | 15 | 18 | |
| Breakfast cereals | LoQ (μg/kg) | 0.13 | 0.8 | 16 | 20 | 8 | 20 | 3.2 |
| RA (%) | 65 | 68 | 63 | 66 | 69 | 69 | 68 | |
| SSE (%) | 59 | 95 | 123 | 74 | 72 | 68 | 71 | |
| RE (%) | 110 | 71 | 52 | 89 | 96 | 101 | 96 | |
| RSDr (%) | 16 | 6 | 15 | 14 | 19 | 11 | 20 |
| Mycotoxin | Concentration Ranges | |
|---|---|---|
| ng/mL | ng/g | |
| AFB1 | 0.075–2.000 | 0.06–1.60 |
| OTA | 0.5–12.5 | 0.4–10.0 |
| FB1 | 10–250 | 8–200 |
| DON | 25–625 | 20–500 |
| T-2 | 5–125 | 4–100 |
| HT-2 | 25–625 | 20–500 |
| ZEA | 2–50 | 1.6–40 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Santis, B.; Debegnach, F.; Gregori, E.; Russo, S.; Marchegiani, F.; Moracci, G.; Brera, C. Development of a LC-MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples. Toxins 2017, 9, 169. https://doi.org/10.3390/toxins9050169
De Santis B, Debegnach F, Gregori E, Russo S, Marchegiani F, Moracci G, Brera C. Development of a LC-MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples. Toxins. 2017; 9(5):169. https://doi.org/10.3390/toxins9050169
Chicago/Turabian StyleDe Santis, Barbara, Francesca Debegnach, Emanuela Gregori, Simona Russo, Francesca Marchegiani, Gabriele Moracci, and Carlo Brera. 2017. "Development of a LC-MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples" Toxins 9, no. 5: 169. https://doi.org/10.3390/toxins9050169
APA StyleDe Santis, B., Debegnach, F., Gregori, E., Russo, S., Marchegiani, F., Moracci, G., & Brera, C. (2017). Development of a LC-MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples. Toxins, 9(5), 169. https://doi.org/10.3390/toxins9050169