
Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
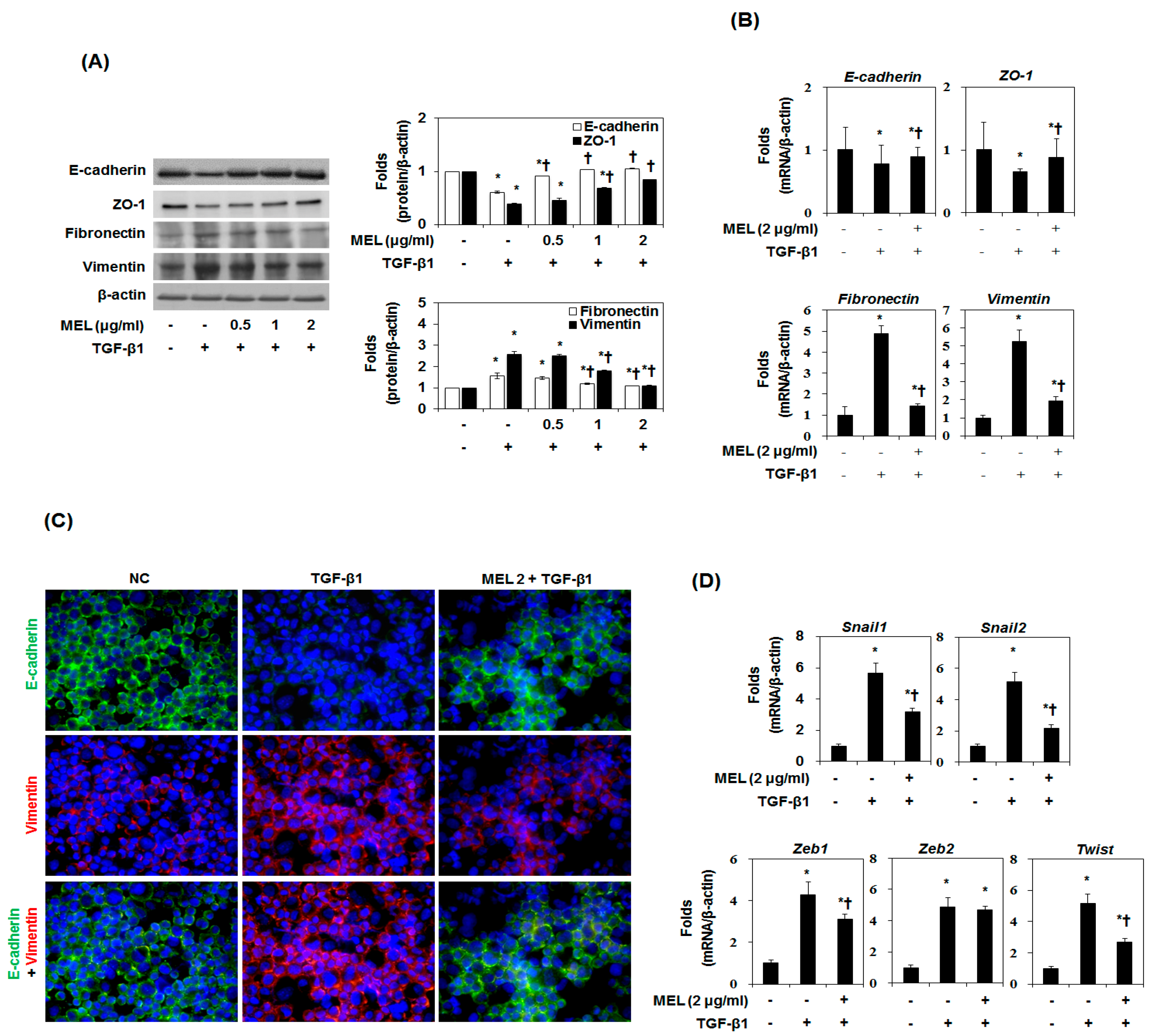
2.1. Effects of MEL on TGF-β1-Induced EMT In Vitro
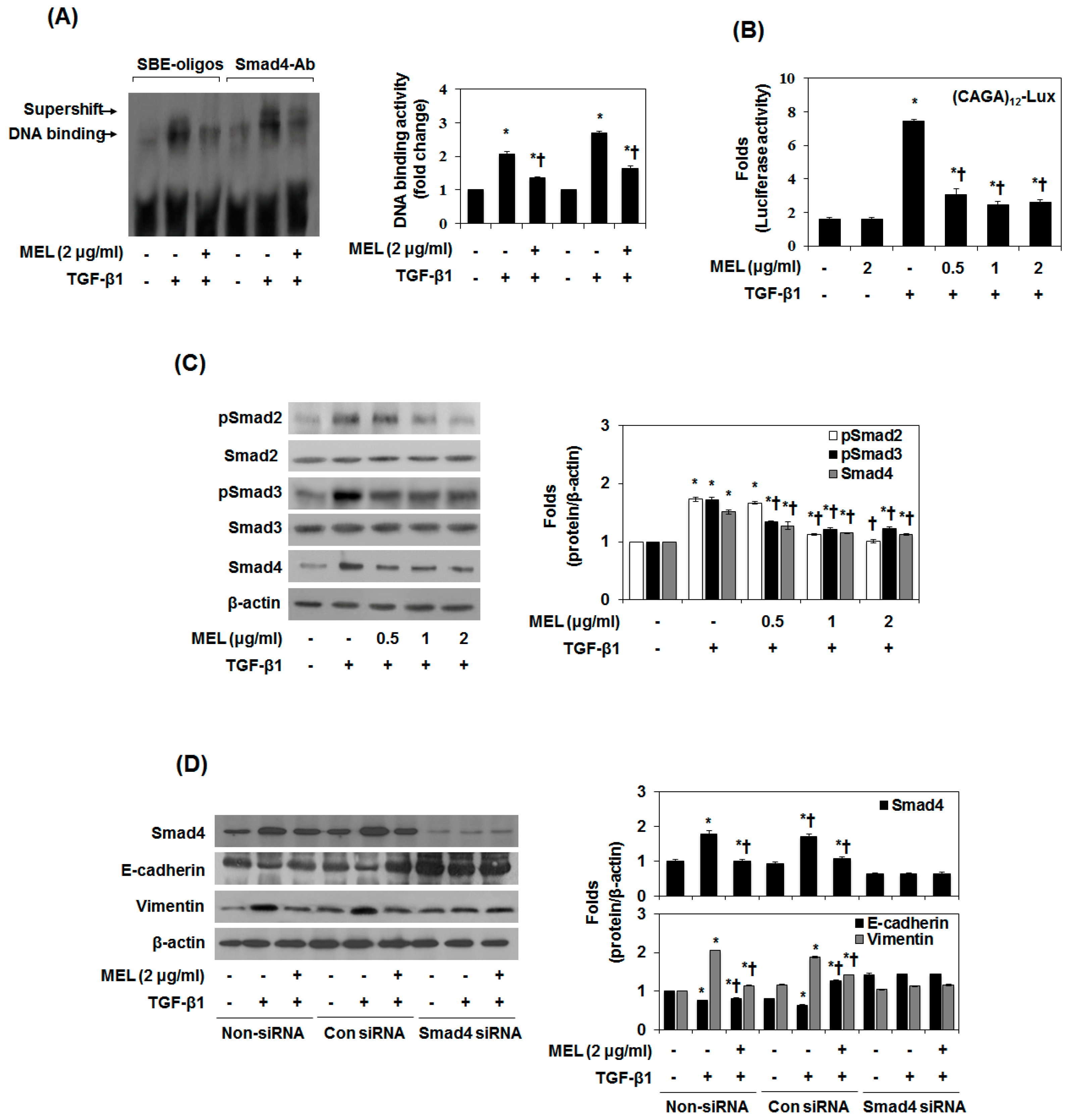
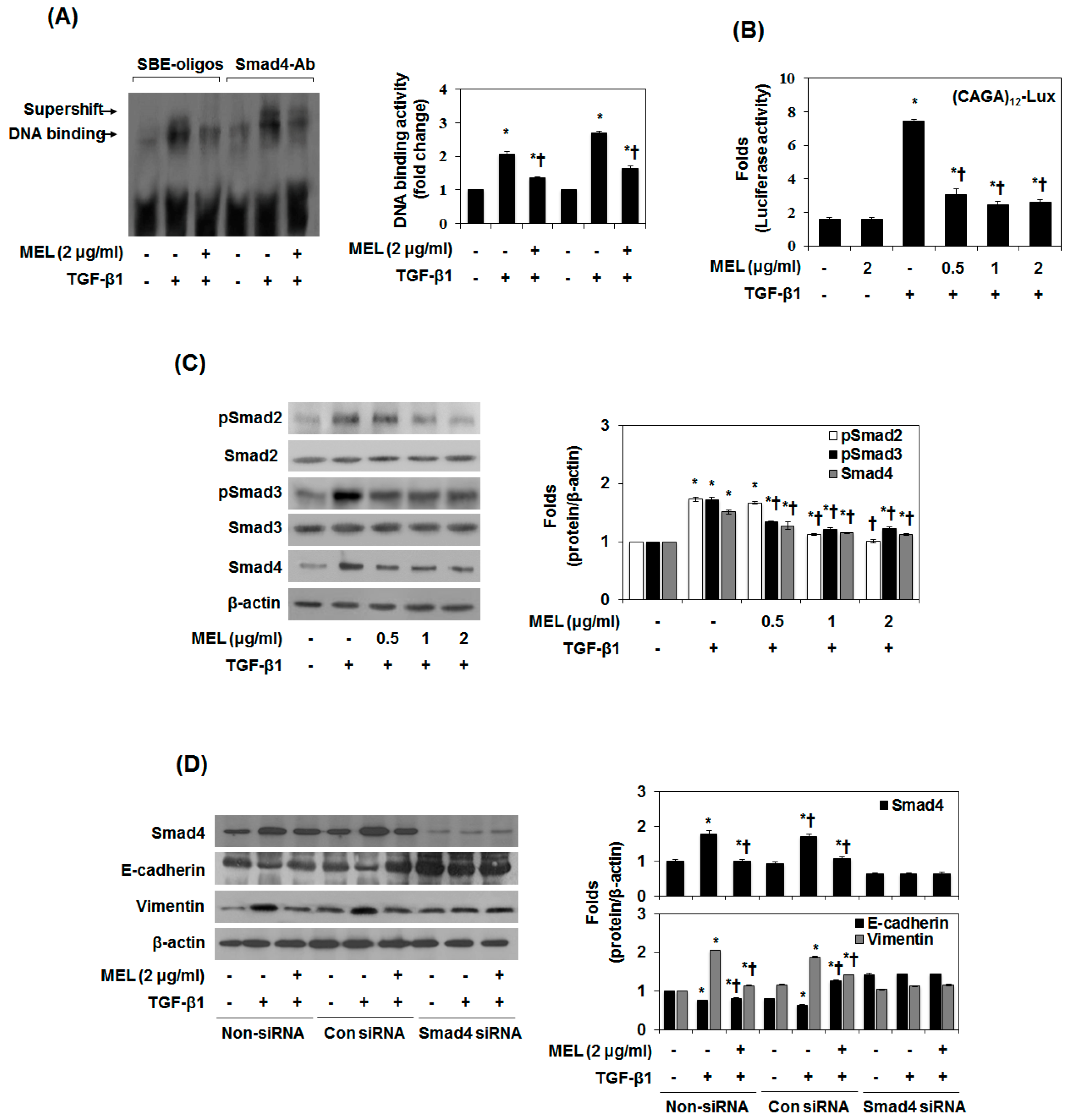
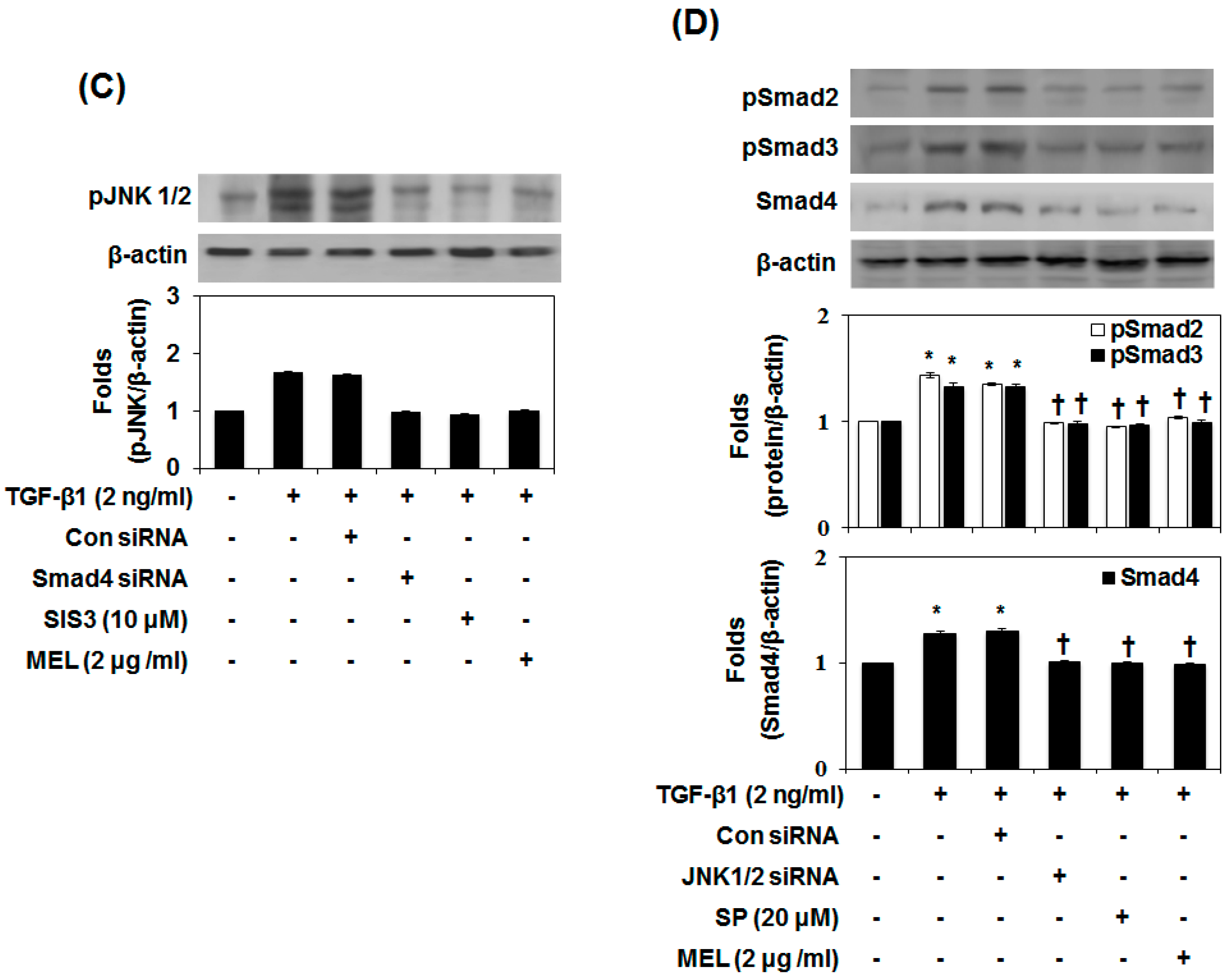
2.2. MEL Prevents EMT via TGF-β/Smad Signal Transduction Pathway
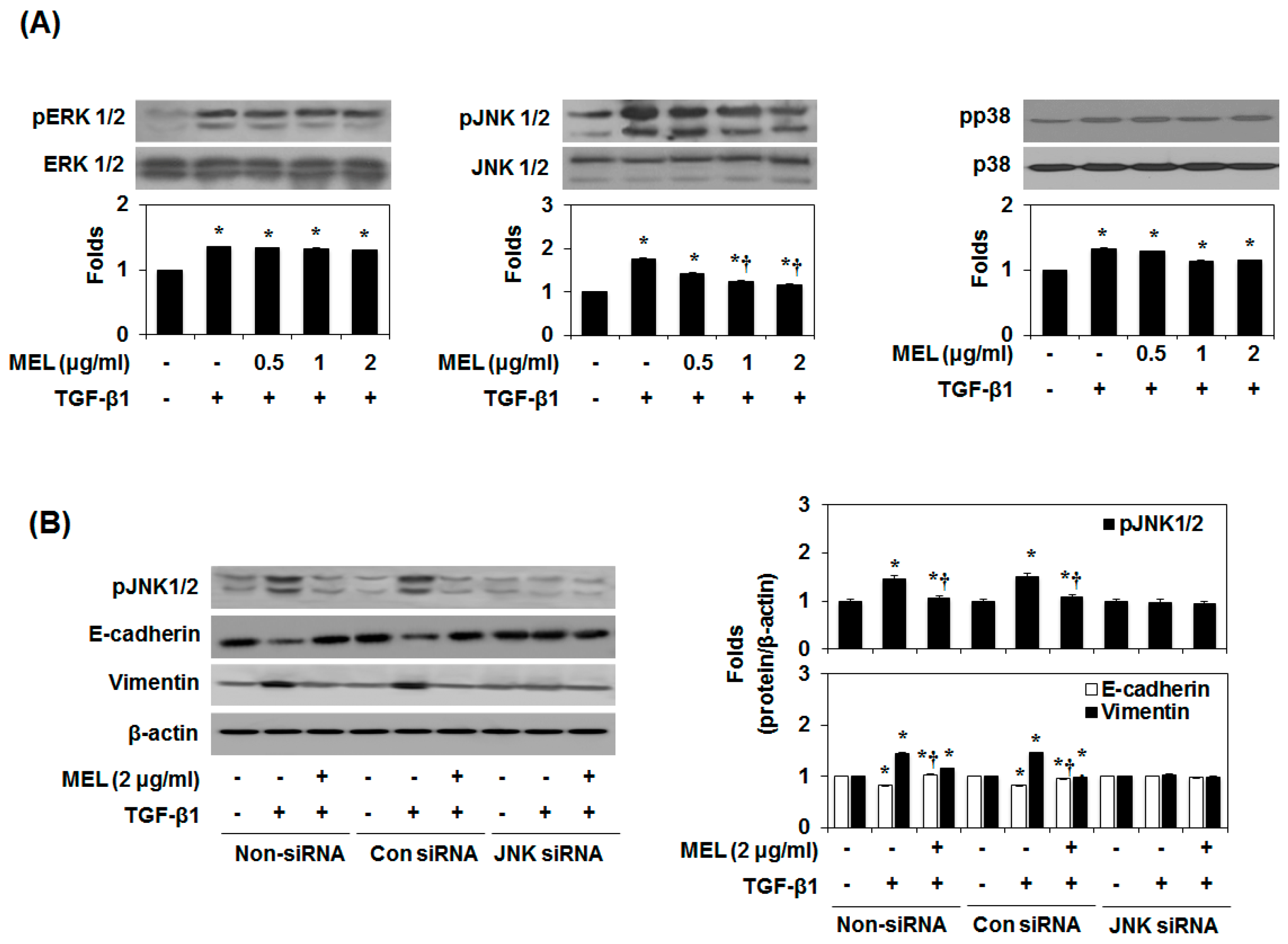
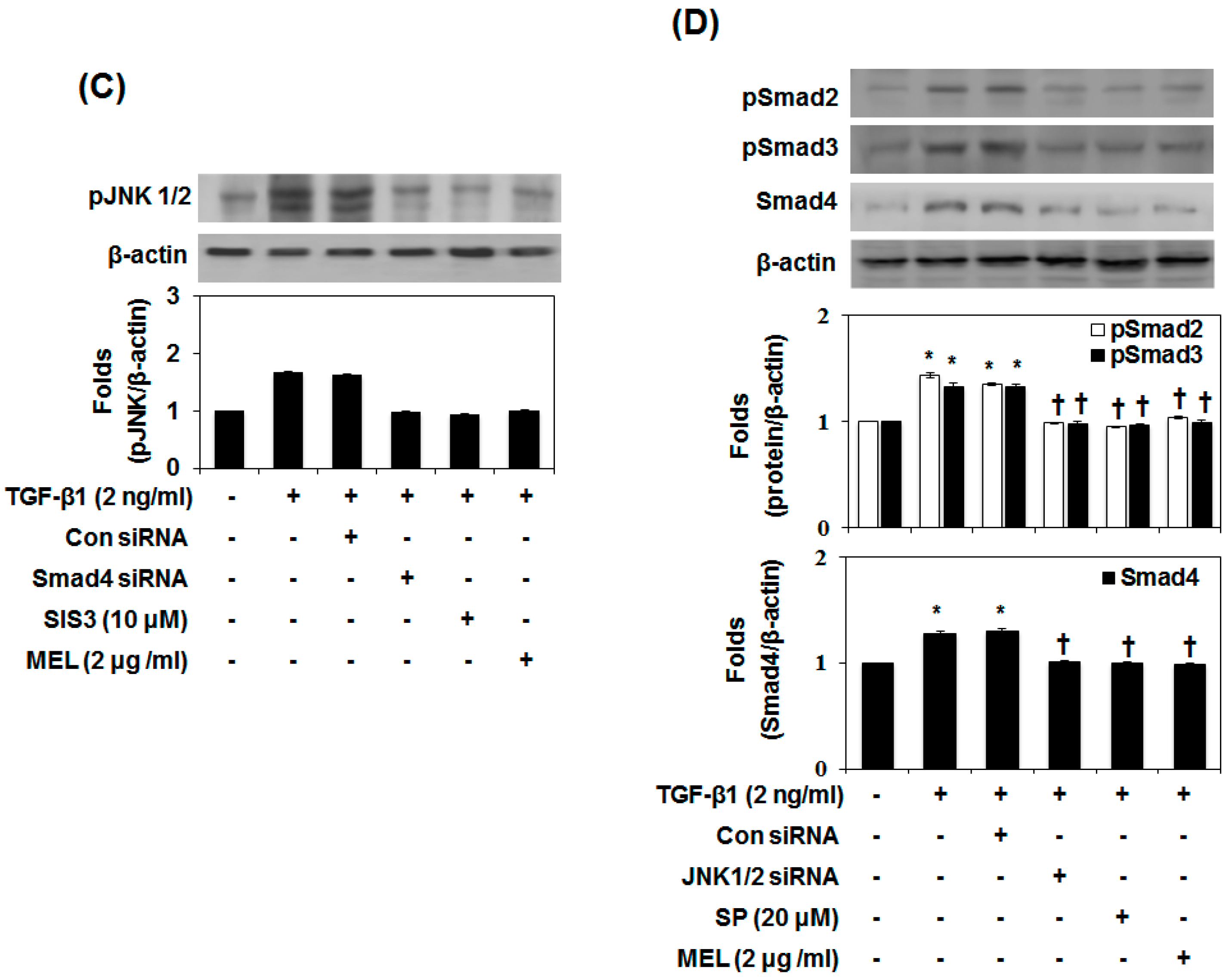
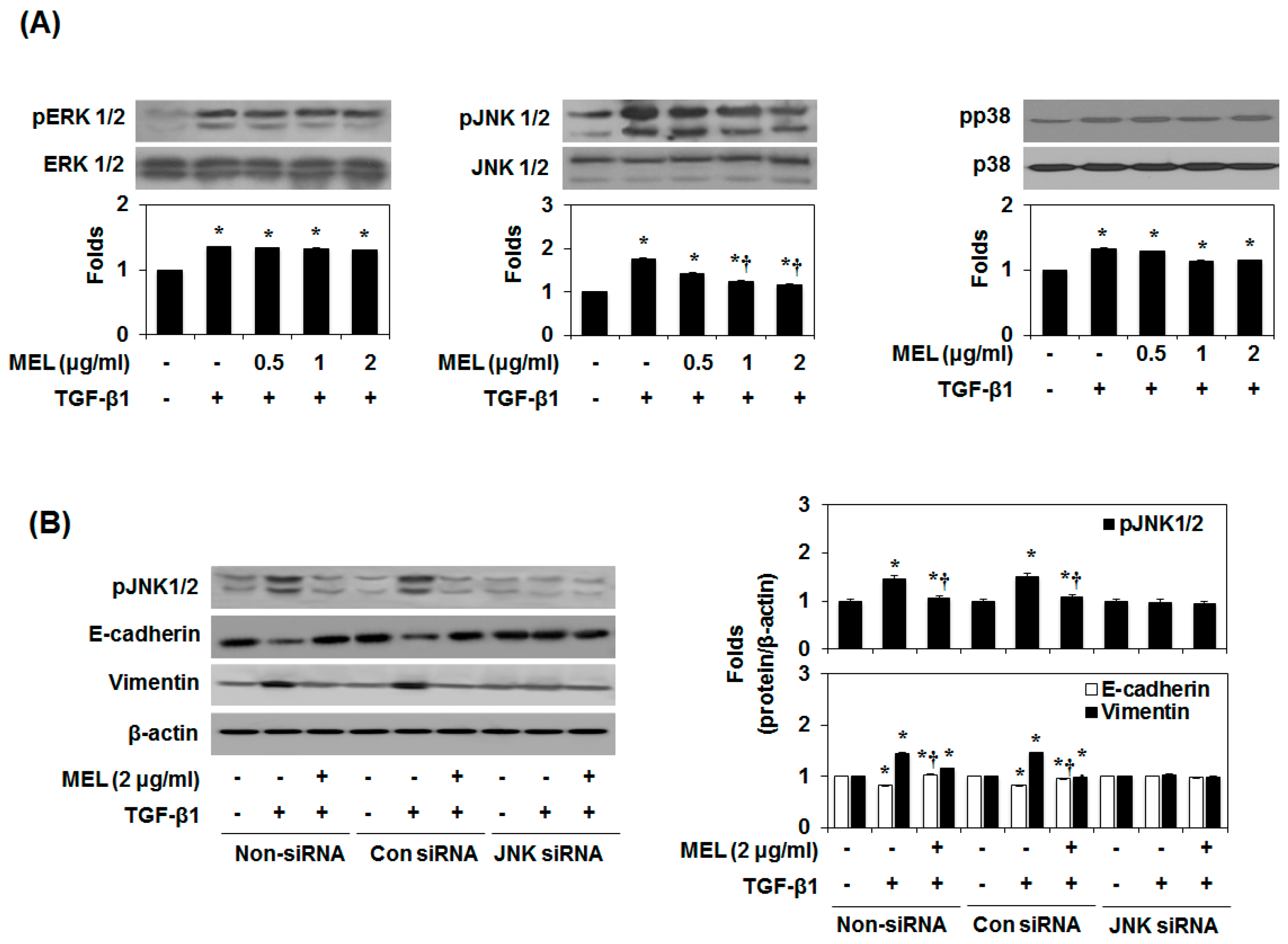
2.3. MEL Prevents EMT via a TGF-β/Non-Smad-Dependent Pathway
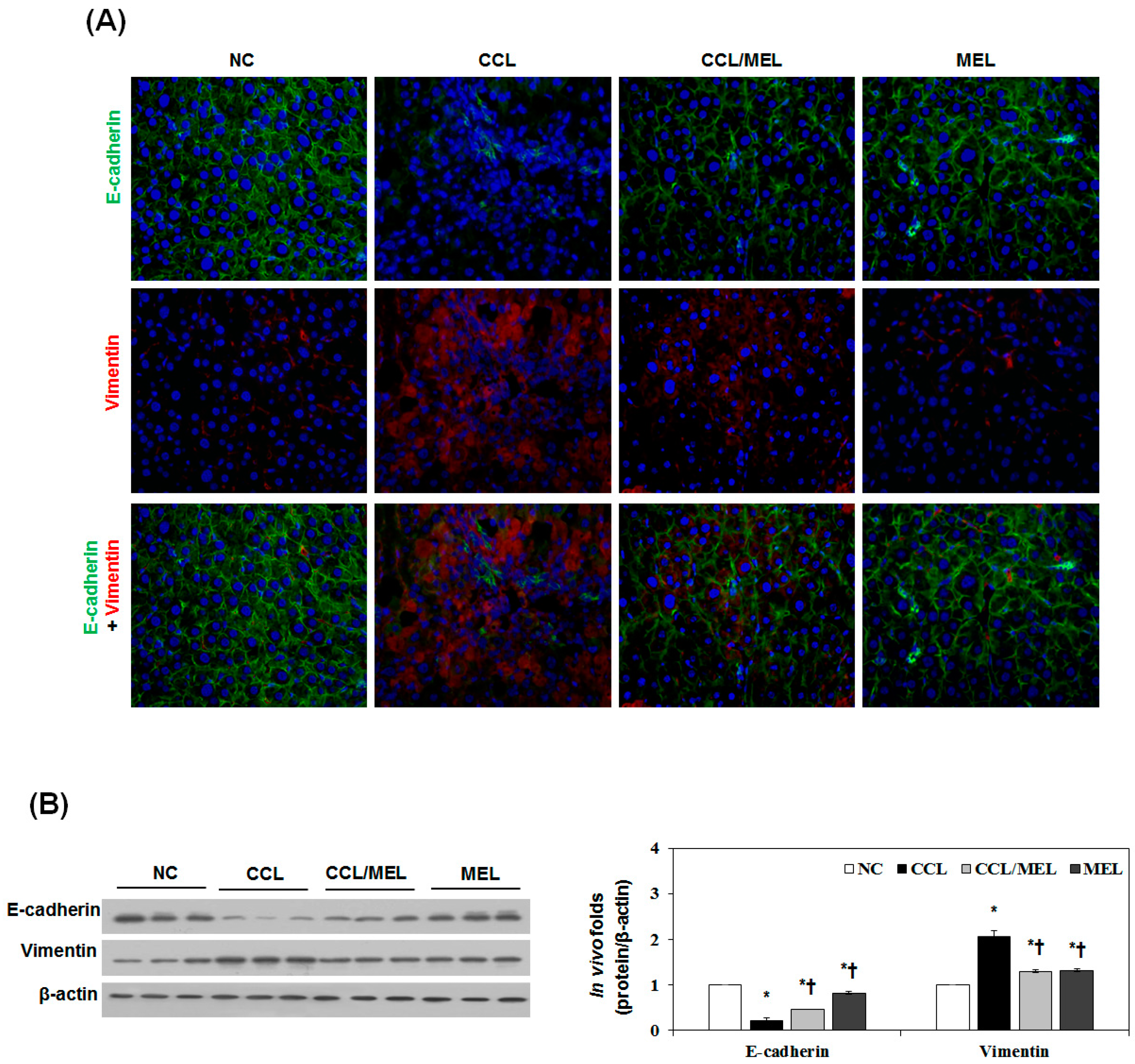
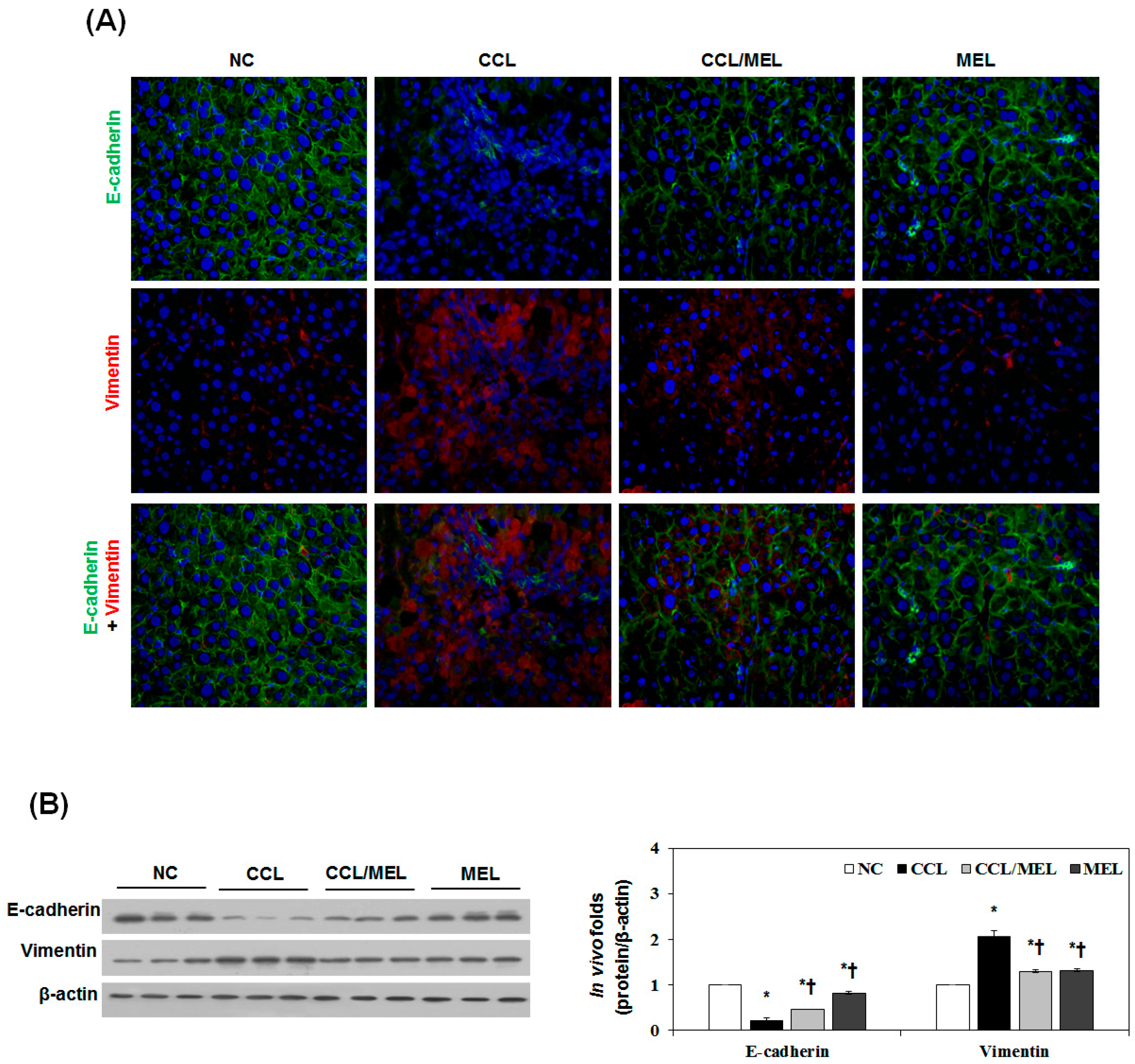
2.4. MEL Ameliorates Liver EMT Induced by CCl4
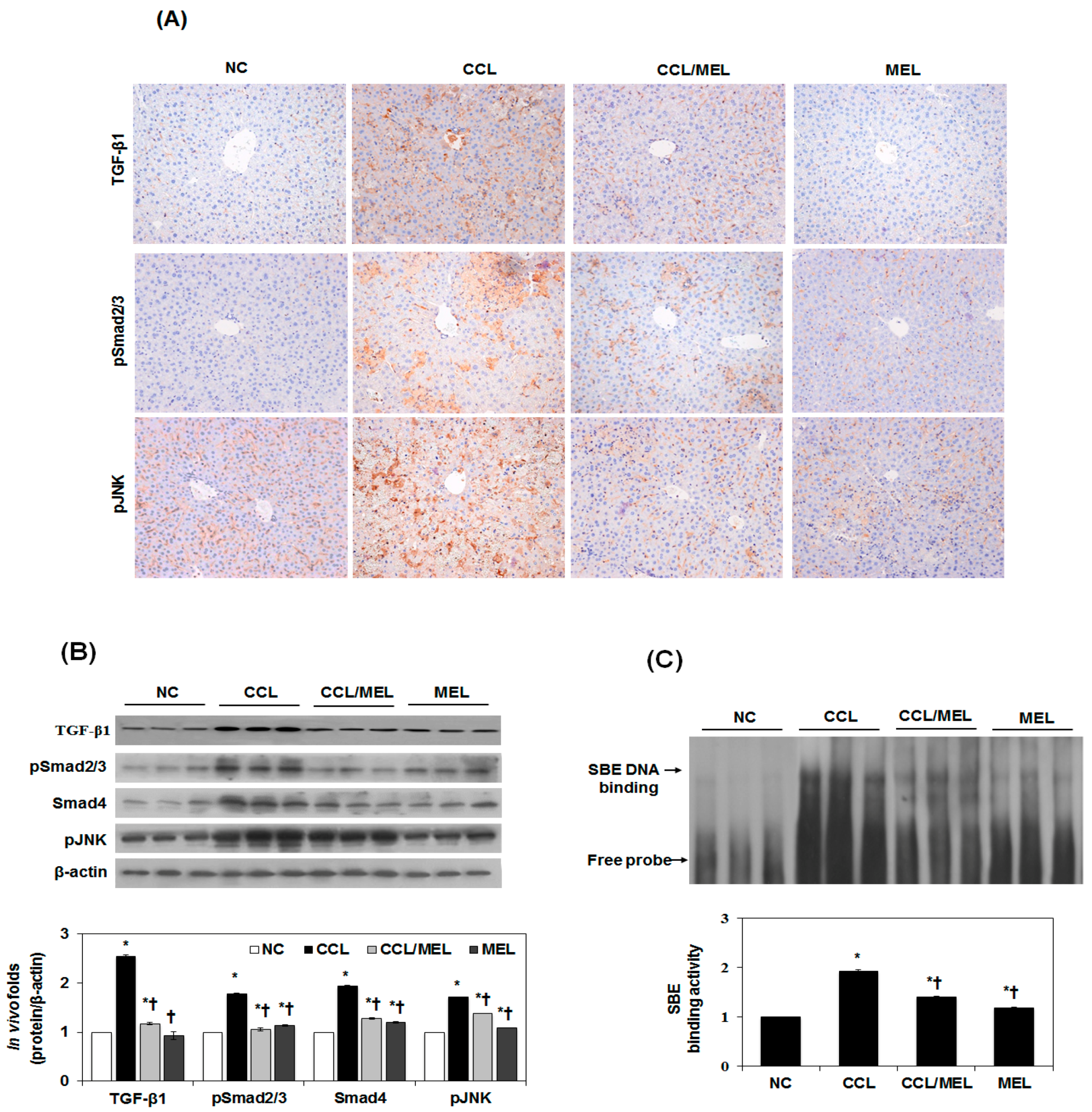
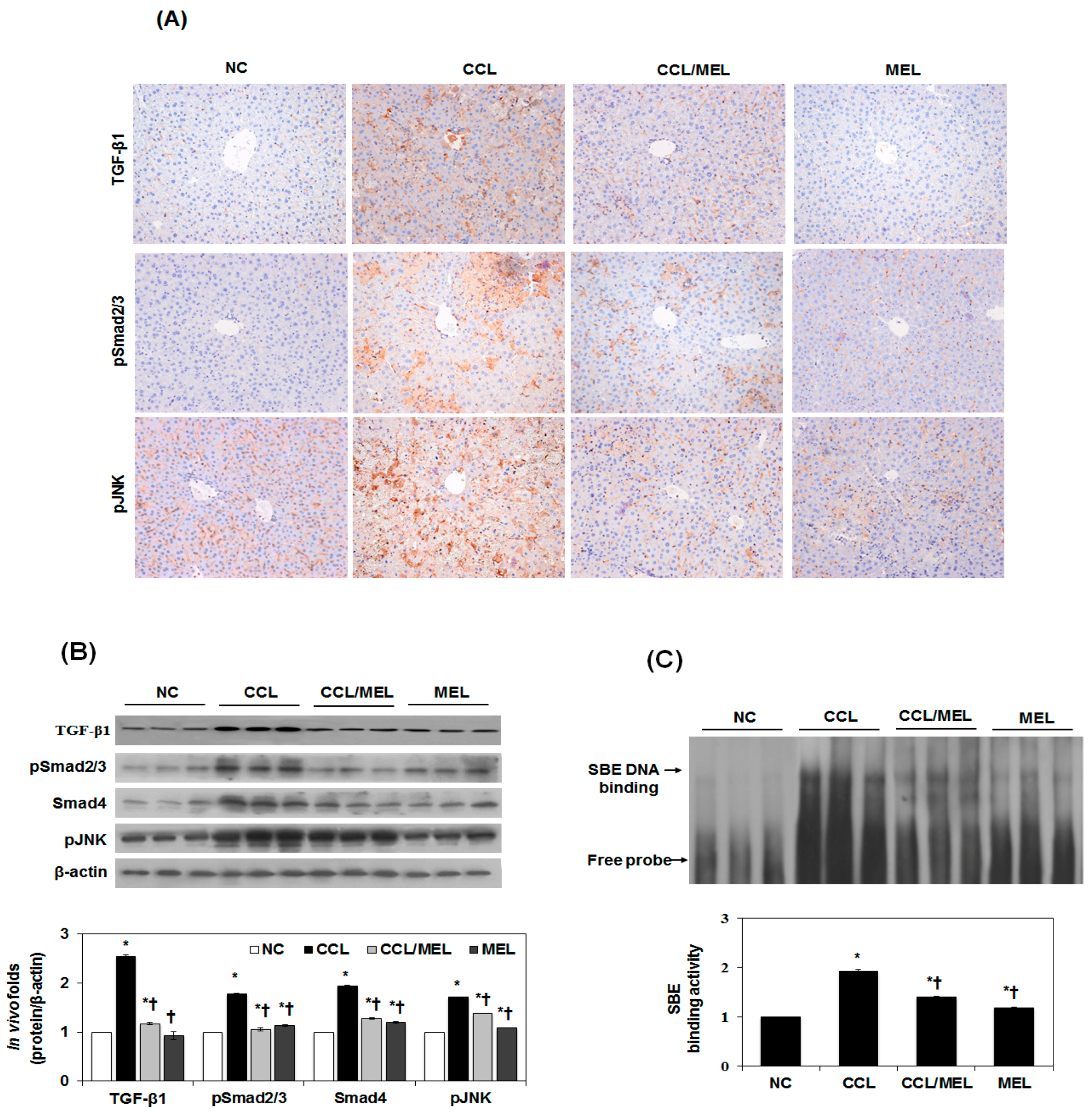
2.5. MEL Ameliorates EMT through TGFβ/Smad and JNK-MAPK In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Reagents
4.2. Animal and Induction of Liver Injury
4.3. Morphological Examination
4.4. Protein Isolation and Immunoblot Analysis
4.5. Reverse-Transcription and Real-Time PCR
4.6. DNA Transfection and Luciferase Assay
4.7. Transient Transfection with Small Interfering RNA
4.8. DNA Binding Activity of SBE and Smad4 Antibody
4.9. Histopathological Investigation and Immunofluorescence Staining
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ECM | Excessive extracellular matrix |
| HSC | Hepatic stellate cells |
| EMT | Epithelial-to-mesenchymal transition |
| TGF | Transforming growth factor |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen-activated protein kinase |
| LPS | Lipopolysaccharide |
| TβR | TGFβ receptor |
References
- Kaimori, A.; Potter, J.J.; Choti, M.; Ding, Z.; Mezey, E.; Koteish, A.A. Histone deacetylase inhibition suppresses the transforming growth factor beta1-induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology 2010, 52, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and tgf-beta as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sevigny, J.; Wells, R.G. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.J.; Takashima, M.; Rippe, R.A. Molecular mechanisms of hepatic fibrogenesis. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S79–S84. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.Y.; Yin, C.; Hou, J.L.; Zeng, X.; Chen, Y.X.; Zhong, W.; Hu, P.F.; Deng, X.; Tan, Y.X.; Zhang, J.P.; et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 2010, 59, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Lv, J.; Ye, X.L.; Sun, M.Y.; Xu, Q.; Liu, C.H.; Min, L.H.; Li, H.P.; Liu, P.; Ding, X. Sorafenib inhibits transforming growth factor beta1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2011, 53, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Furuya, M.H.; Wolfraim, L.A.; Nguyen, A.P.; Holdren, M.S.; Campbell, J.S.; Knight, B.; Yeoh, G.C.; Fausto, N.; Parks, W.T. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology 2007, 45, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Meindl-Beinker, N.M.; Dooley, S. Transforming growth factor-beta and hepatocyte transdifferentiation in liver fibrogenesis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S122–S127. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Kim, J.S.; Mohuczy, D.; Behrns, K.E. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology 2008, 48, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Omenetti, A.; Witek, R.P.; Moylan, C.A.; Syn, W.K.; Jung, Y.; Yang, L.; Sudan, D.L.; Sicklick, J.K.; Michelotti, G.A.; et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1093–G1106. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J. Defining therapeutic targets for liver fibrosis: Exploiting the biology of inflammation and repair. Pharmacol. Res. 2008, 58, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Asahina, K. Hepatic stellate cell progenitor cells. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 80–84. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 65–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.G.; Cho, H.J.; Bae, Y.S.; Park, K.K.; Choe, J.Y.; Chung, I.K.; Kim, M.; Yeo, J.H.; Park, K.H.; Lee, Y.S.; et al. Bee venom suppresses lps-mediated no/inos induction through inhibition of pkc-alpha expression. J. Ethnopharmacol. 2009, 123, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Kim, J.Y.; Chang, Y.C.; Chung, H.; Park, Y.Y.; Lee, M.L.; Park, K.K. The protective effects of melittin on propionibacterium acnes-induced inflammatory responses in vitro and in vivo. J. Investig. Dermatol. 2014, 134, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, K.H.; Lee, W.R.; Han, S.M.; Park, K.K. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis 2012, 17, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kum, Y.S.; Lee, T.I.; Kim, S.J.; Lee, W.R.; Kim, B.I.; Kim, H.S.; Kim, K.H.; Park, K.K. Melittin attenuates liver injury in thioacetamide-treated mice through modulating inflammation and fibrogenesis. Exp. Biol. Med. 2011, 236, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Kim, K.S.; Park, K.K. Melittin inhibits atherosclerosis in lps/high-fat treated mice through atheroprotective actions. J. Atheroscler. Thromb. 2011, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Sung, H.J.; Lee, W.R.; An, H.J.; Kim, J.Y.; Pak, S.C.; Han, S.M.; Park, K.K. Effects of melittin treatment in cholangitis and biliary fibrosis in a model of xenobiotic-induced cholestasis in mice. Toxins 2015, 7, 3372–3387. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Park, J.H.; Kim, K.H.; Park, Y.Y.; Han, S.M.; Park, K.K. Protective effects of melittin on transforming growth factor-beta1 injury to hepatocytes via anti-apoptotic mechanism. Toxicol. Appl. Pharmacol. 2011, 256, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. Mir-30 inhibits tgf-beta1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, M.K.; Yoon, J. Gamma-linolenic acid inhibits hepatic pai-1 expression by inhibiting p38 mapk-dependent activator protein and mitochondria-mediated apoptosis pathway. Apoptosis 2015, 20, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Kim, J.Y.; Lee, S.J.; Han, S.M.; Pak, S.C.; Park, K.K. Apamin inhibits hepatic fibrosis through suppression of transforming growth factor beta1-induced hepatocyte epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 450, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the erk pathway is required for tgf-beta1-induced emt in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jo, J.H.; Kim, K.H.; Kim, S.J.; Lee, W.R.; Park, K.K.; Park, J.B. Antifibrotic effect through the regulation of transcription factor using ring type-sp1 decoy oligodeoxynucleotide in carbon tetrachloride-induced liver fibrosis. J. Gene Med. 2009, 11, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Chen, J.; Gao, W.; Zhou, P.; Ma, X.; Tschudy-Seney, B.; Liu, C.; Zern, M.A.; Liu, P.; Duan, Y. Enhancement of hepatocyte differentiation from human embryonic stem cells by chinese medicine fuzhenghuayu. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wang, X.; Lei, W.; Min, L.; Yang, Y.; Wang, X.; Song, J. Nitric oxide suppresses transforming growth factor-beta1-induced epithelial-to-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2009, 50, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Capelo, A. Dual role for tgf-beta1 in apoptosis. Cytokine Growth Factor Rev. 2005, 16, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Park, K.G.; Min, A.K.; Koh, E.H.; Kim, H.S.; Kim, M.O.; Park, H.S.; Kim, Y.D.; Yoon, T.S.; Jang, B.K.; Hwang, J.S.; et al. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (ampk)-dependent and ampk-independent pathways. Hepatology 2008, 48, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.J.; Yoo, J. Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating pi3 kinase/akt signal pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.F.; Guala, A.S.; van der Velden, J.; McElhinney, B.; Irvin, C.G.; Davis, R.J.; Janssen-Heininger, Y.M. Jun n-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by tgf-beta1. J. Cell Sci. 2008, 121, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, M.S.; Jeong, G.S.; Yoon, J. Xanthii fructus extract inhibits tnf-alpha/ifn-gamma-induced th2-chemokines production via blockade of nf-kappab, stat1 and p38-mapk activation in human epidermal keratinocytes. J. Ethnopharmacol. 2015, 171, 85–93. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-H.; Park, B.; Park, K.-K. Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways. Toxins 2017, 9, 138. https://doi.org/10.3390/toxins9040138
Park J-H, Park B, Park K-K. Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways. Toxins. 2017; 9(4):138. https://doi.org/10.3390/toxins9040138
Chicago/Turabian StylePark, Ji-Hyun, Byoungduck Park, and Kwan-Kyu Park. 2017. "Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways" Toxins 9, no. 4: 138. https://doi.org/10.3390/toxins9040138
APA StylePark, J.-H., Park, B., & Park, K.-K. (2017). Suppression of Hepatic Epithelial-to-Mesenchymal Transition by Melittin via Blocking of TGFβ/Smad and MAPK-JNK Signaling Pathways. Toxins, 9(4), 138. https://doi.org/10.3390/toxins9040138