
Sequence Polymorphism and Intrinsic Structural Disorder as Related to Pathobiological Performance of the Helicobacter pylori CagA Oncoprotein

{kind=link}
{kind=link}
{kind=link}
Abstract
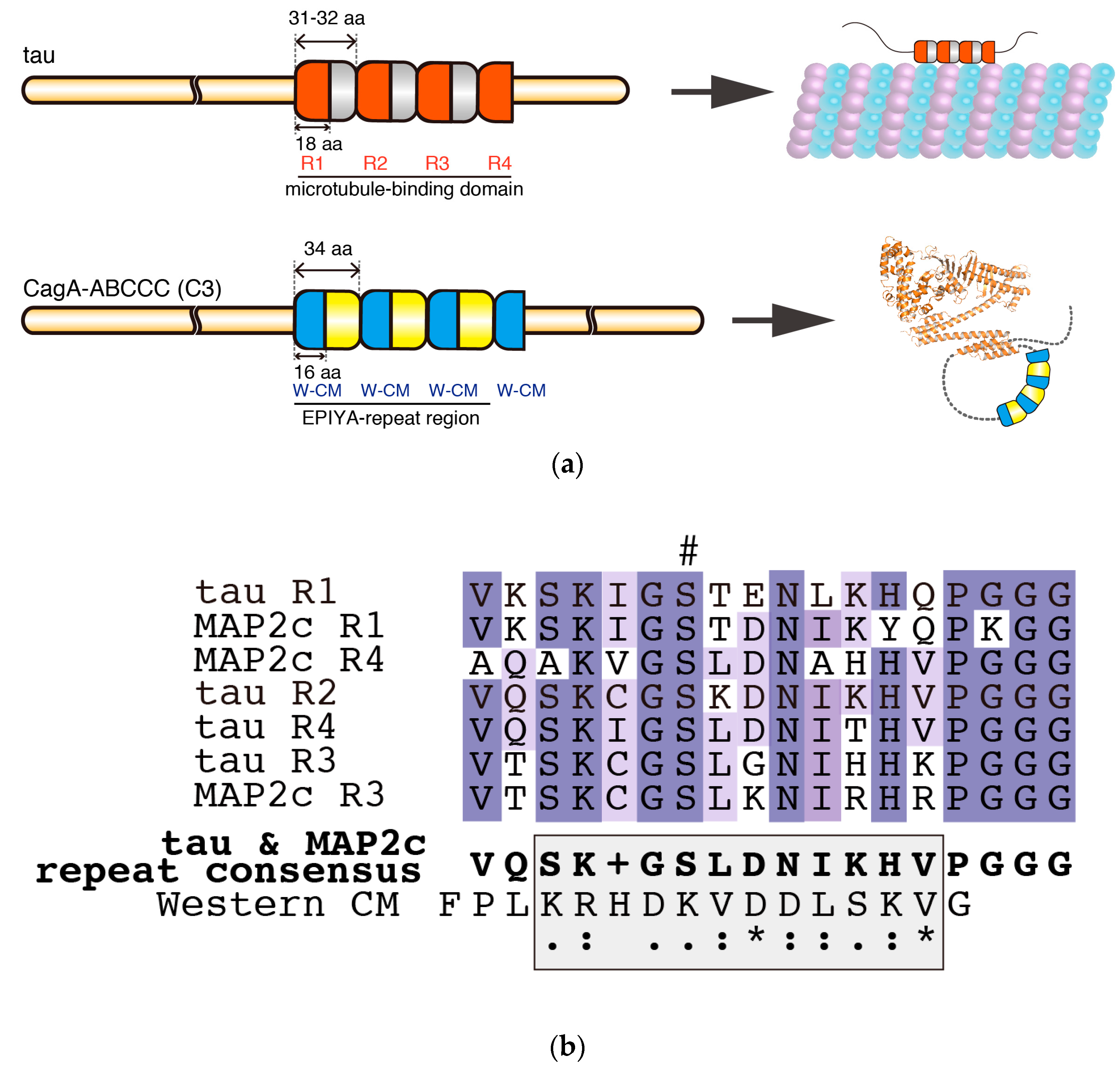
:1. Introduction
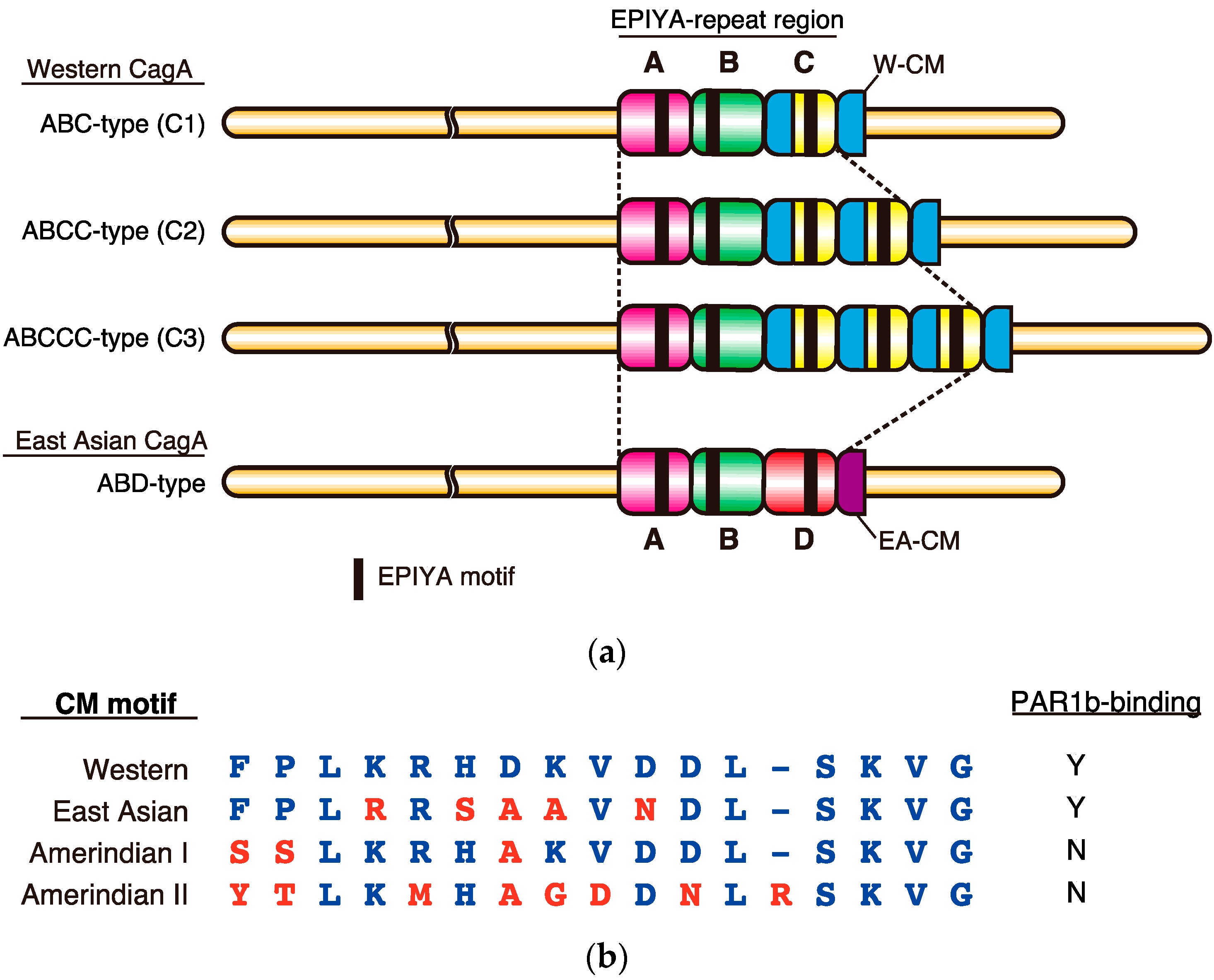
2. Sequence Polymorphism in the EPIYA Motif-Containing Region of CagA
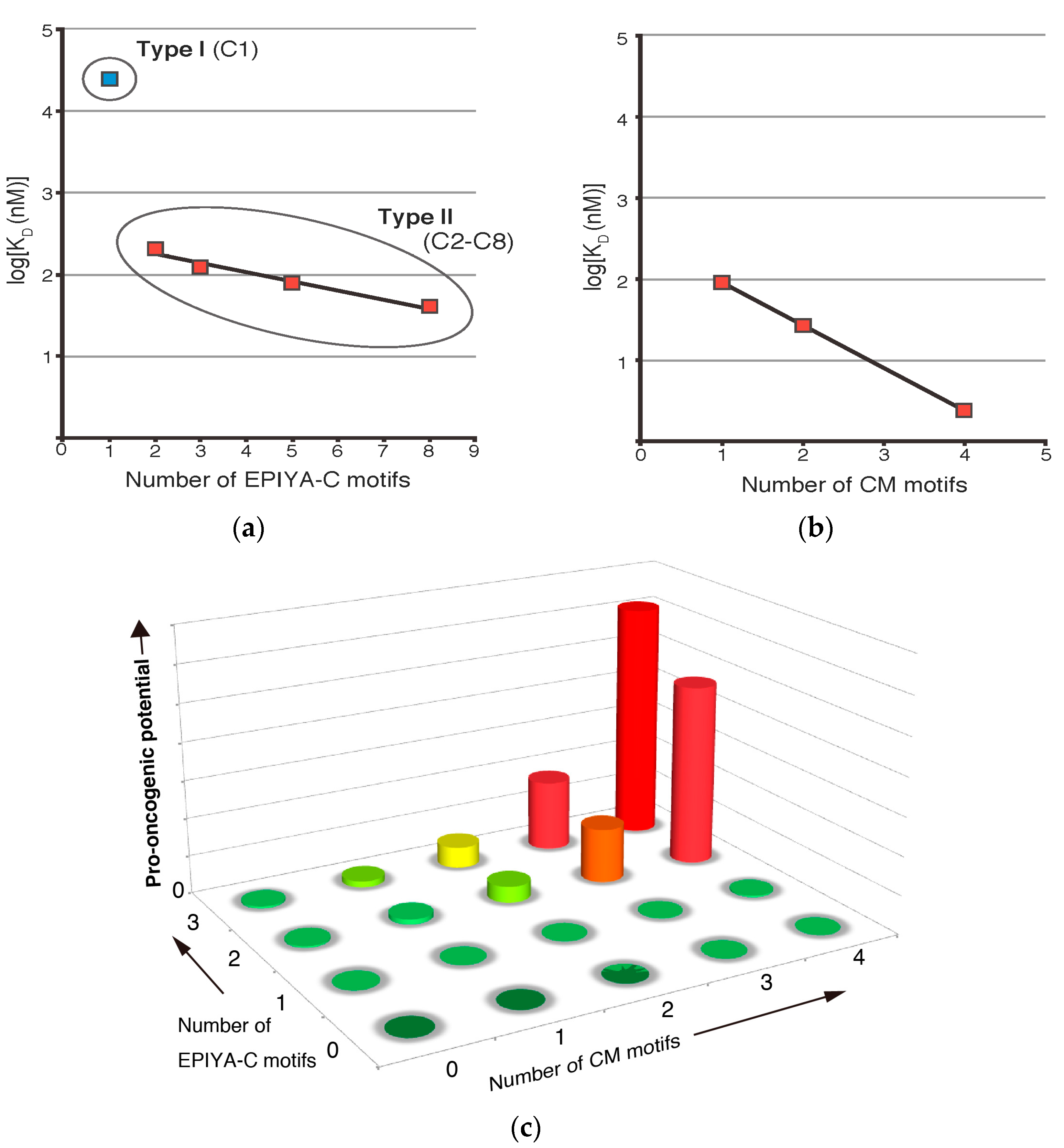
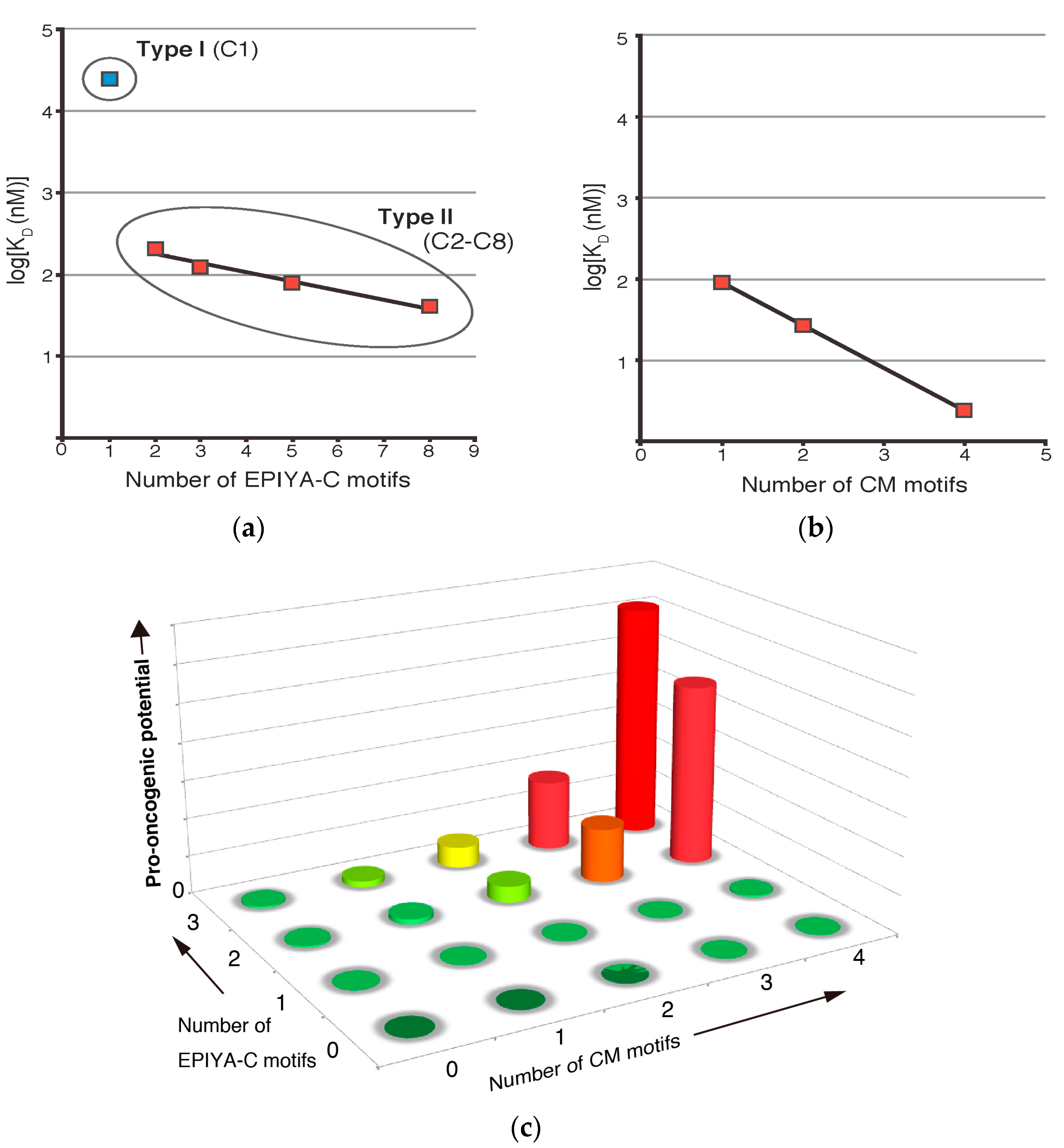
3. Role of EPIYA Polymorphism in SHP2 Binding
4. Role of CM Polymorphism in PAR1 Binding
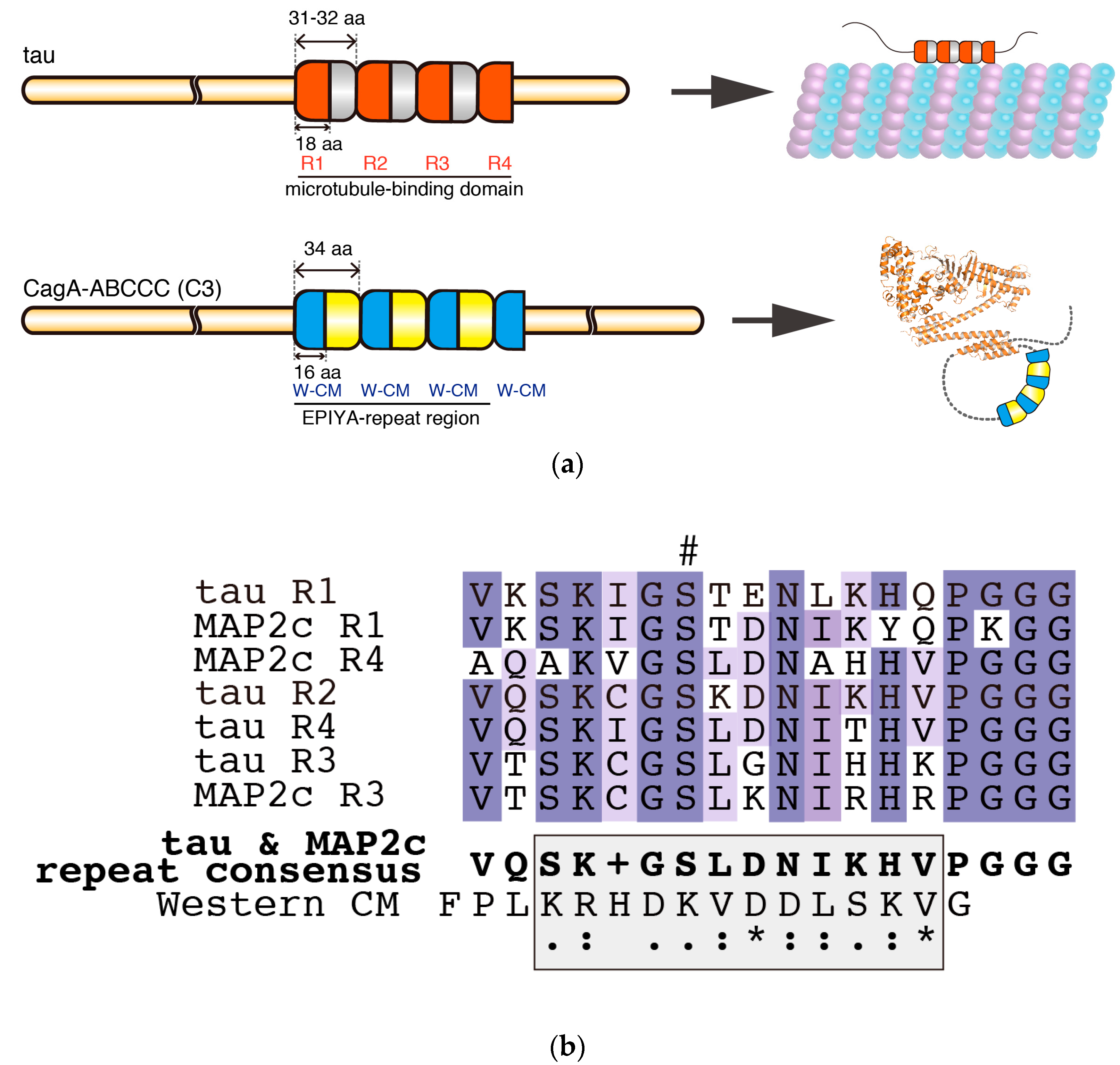
5. Intrinsic Structural Disorder—A Critical Structural Feature for Bacterial Effectors
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Watanabe, H.; Nishikura, K.; Umezawa, H.; Asakura, H. Topographic distribution of Helicobacter pylori in the resected stomach. Eur. J. Gastroenterol. Hepatol. 1998, 10, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Linking epithelial polarity and carcinogenesis by multitasking Helicobacter pylori virulence factor CagA. Oncogene 2008, 27, 7047–7054. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Morohashi, H.; Hatakeyama, M. Bacterial EPIYA effectors—Where do they come from? What are they? Where are they going? Cell. Microbiol. 2013, 15, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [PubMed]
- Akopyants, N.S.; Clifton, S.W.; Kersulyte, D.; Crabtree, J.E.; Youree, B.E.; Reece, C.A.; Bukanov, N.O.; Drazek, E.S.; Roe, B.A.; Berg, D.E. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 1998, 28, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Senda, M.; Morohashi, H.; Higashi, H.; Horio, M.; Kashiba, Y.; Nagase, L.; Sasaya, D.; Shimizu, T.; Venugopalan, N.; et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 2012, 12, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Türköz, B.; Jiménez-Soto, L.F.; Dian, C.; Ertl, C.; Remaut, H.; Louche, A.; Tosi, T.; Haas, R.; Terradot, L. Structural insights into Helicobacter pylori oncoprotein CagA interaction with β1 integrin. Proc. Natl. Acad. Sci. USA 2012, 109, 14640–14645. [Google Scholar] [CrossRef] [PubMed]
- Woon, A.P.; Tohidpour, A.; Alonso, H.; Saijo-Hamano, Y.; Kwok, T.; Roujeinikova, A. Conformational analysis of isolated domains of Helicobacter pylori CagA. PLoS ONE 2013, 8, e79367. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Guharoy, M.; Pauwels, K.; Tompa, P. SnapShot: Intrinsic structural disorder. Cell 2015, 161, 1230–1230.e1. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2016, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Bagnoli, F.; Halenbeck, R.; Rappuoli, R.; Fantl, W.J.; Covacci, A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 2002, 43, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Higashi, H.; Lu, H.; Azuma, T.; Hatakeyama, M. Structural basis and functional consequence of Helicobacter pylori CagA multimerization in cells. J. Biol. Chem. 2006, 281, 32344–32352. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N.; Selbach, M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter 2010, 15, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kikuchi, K.; Hayashi, T.; Higashi, H.; Hatakeyama, M. Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe 2010, 7, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, R.; Higashi, H.; Higuchi, M.; Okada, M.; Hatakeyama, M. Attenuation of Helicobacter pylori CagA·SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J. Biol. Chem. 2003, 278, 3664–3670. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mimuro, H.; Suzuki, T.; Park, M.; Yamamoto, T.; Sasakawa, C. Interaction of CagA with Crk plays an important role in Helicobacter pylori–induced loss of gastric epithelial cell adhesion. J. Exp. Med. 2005, 202, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [PubMed]
- Nešić, D.; Miller, M.C.; Quinkert, Z.T.; Stein, M.; Chait, B.T.; Stebbins, C.E. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat. Struct. Mol. Biol. 2010, 17, 130–132. [Google Scholar] [PubMed]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Yahara, K.; Hatakeyama, M.; Kobayashi, I. Evolution of cagA oncogene of Helicobacter pylori through recombination. PLoS ONE 2011, 6, e23499. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The “Shp”ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Xiao, S.; Rose, D.W.; Sasaoka, T.; Maegawa, H.; Burke, T.R.; Roller, P.P.; Shoelson, S.E.; Olefsky, J.M. Syp (SH-PTP2) is a positive mediator of growth factor-stimulated mitogenic signal transduction. J. Biol. Chem. 1994, 269, 21244–21248. [Google Scholar] [PubMed]
- Yamauchi, K.; Milarski, K.L.; Saltiel, A.R.; Pessin, J.E. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc. Natl. Acad. Sci. USA 1995, 92, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Choong, K.; Freedman, M.H.; Chitayat, D.; Kelly, E.N.; Taylor, G.; Zipursky, A. Juvenile myelomonocytic leukemia and Noonan syndrome. J. Pediatr. Hematol. Oncol. 1999, 21, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lieuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Ohnishi, N.; Tanaka, S.; Yanagiya, K.; Hatakeyama, M. Differential oncogenic potential of geographically distinct Helicobacter pylori CagA isoforms in mice. Int. J. Cancer 2009, 125, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yamaoka, Y.; Zhu, Q.; Matha, I.; Gao, X. A comprehensive sequence and disease correlation analyses for the C-terminal region of CagA protein of Helicobacter pylori. PLoS ONE 2009, 4, e7736. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Ryberg, A.; Dehnoei, M.; Borch, K.; Monstein, H.-J. Association between cagA and vacA genotypes and pathogenesis in a Helicobacter pylori infected population from South-eastern Sweden. BMC Microbiol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Nagase, L.; Hayashi, T.; Senda, T.; Hatakeyama, M. Dramatic increase in SHP2 binding activity of Helicobacter pylori Western CagA by EPIYA-C duplication: Its implications in gastric carcinogenesis. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Hayashi, T.; Arisaka, F.; Senda, T.; Hatakeyama, M. Impact of structural polymorphism for the Helicobacter pylori CagA oncoprotein on binding to polarity-regulating kinase PAR1b. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Kemphues, K.J.; Priess, J.R.; Morton, D.G.; Cheng, N. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell 1988, 52, 311–320. [Google Scholar] [CrossRef]
- Drewes, G.; Trinczek, B.; Illenberger, S.; Biernat, J.; Schmitt-Ulms, G.; Meyer, H.E.; Mandelkow, E.-M.; Mandelkow, E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark): A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 1995, 270, 7679–7688. [Google Scholar] [PubMed]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.-M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Böhm, H.; Brinkmann, V.; Drab, M.; Henske, A.; Kurzchalia, T.V. Mammalian homologues of C. elegans PAR-1 are asymmetrically localized in epithelial cells and may influence their polarity. Curr. Biol. 1997, 7, 603–606. [Google Scholar] [CrossRef]
- Hurov, J.B.; Watkins, J.L.; Piwnica-Worms, H. Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr. Biol. 2004, 14, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hirata, M.; Kamimura, K.; Maniwa, R.; Yamanaka, T.; Mizuno, K.; Kishikawa, M.; Hirose, H.; Amano, Y.; Izumi, N.; et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr. Biol. 2004, 14, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-S.; Saito, Y.; Umeda, M.; Murata-Kamiya, N.; Zhang, H.-M.; Higashi, H.; Hatakeyama, M. Structural and functional diversity in the PAR1b/MARK2-binding region of Helicobacter pylori CagA. Cancer Sci. 2008, 99, 2004–2011. [Google Scholar] [PubMed]
- Zeaiter, Z.; Cohen, D.; Müsch, A.; Bagnoli, F.; Covacci, A.; Stein, M. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cell. Microbiol. 2008, 10, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Yamahashi, Y.; Saito, Y.; Murata-Kamiya, N.; Hatakeyama, M. Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J. Biol. Chem. 2011, 286, 44576–44584. [Google Scholar] [CrossRef] [PubMed]
- Kersulyte, D.; Kalia, A.; Gilman, R.H.; Mendez, M.; Herrera, P.; Cabrera, L.; Velapatiño, B.; Balqui, J.; Paredes Puente de la Vega, F.; Rodriguez Ulloa, C.A.; et al. Helicobacter pylori from Peruvian Amerindians: Traces of human migrations in strains from remote Amazon, and genome sequence of an Amerind strain. PLoS ONE 2010, 5, e15076. [Google Scholar] [CrossRef] [PubMed]
- Mane, S.P.; Dominguez-Bello, M.G.; Blaser, M.J.; Sobral, B.W.; Hontecillas, R.; Skoneczka, J.; Mohapatra, S.K.; Crasta, O.R.; Evans, C.; Modise, T.; et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their old world homologs and mediate inflammatory responses. J. Bacteriol. 2010, 192, 3078–3092. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kiga, K.; Kersulyte, D.; Cok, J.; Hooper, C.C.; Mimuro, H.; Sanada, T.; Suzuki, S.; Oyama, M.; Kozuka-Hata, H.; et al. Attenuated CagA oncoprotein in Helicobacter pylori from Amerindians in Peruvian Amazon. J. Biol. Chem. 2011, 286, 29964–29972. [Google Scholar] [CrossRef] [PubMed]
- Hashi, K.; Murata-Kamiya, N.; Varon, C.; Mégraud, F.; Dominguez-Bello, M.G.; Hatakeyama, M. Natural variant of the Helicobacter pylori CagA oncoprotein that lost the ability to interact with PAR1. Cancer Sci. 2014, 105, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Hamada, D.; Hamaguchi, M.; Suzuki, K.N.; Sakata, I.; Yanagihara, I. Cytoskeleton-modulating effectors of enteropathogenic and enterohemorrhagic Escherichia coli: A case for EspB as an intrinsically less-ordered effector. FEBS J. 2010, 277, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Cortese, M.S.; Uversky, V.N.; Keith Dunker, A. Intrinsic disorder in scaffold proteins: Getting more from less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. BioEssays 2003, 25, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Selbach, M. Tyrosine-phosphorylated bacterial effector proteins: The enemies within. Trends Microbiol. 2005, 13, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Race, P.R.; Solovyova, A.S.; Banfield, M.J. Conformation of the EPEC Tir protein in solution: Investigating the impact of serine phosphorylation at positions 434/463. Biophys. J. 2007, 93, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Simister, P.C.; Feller, S.M. Order and disorder in large multi-site docking proteins of the Gab family—Implications for signalling complex formation and inhibitor design strategies. Mol. BioSyst. 2012, 8, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Keppel, T.R.; Sarpong, K.; Murray, E.M.; Monsey, J.; Zhu, J.; Bose, R. Biophysical evidence for intrinsic disorder in the C-terminal tails of the epidermal growth factor receptor (EGFR) and HER3 receptor tyrosine kinases. J. Biol. Chem. 2017, 292, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Lippens, G.; Landrieu, I.; Smet, C.; Huvent, I.; Gandhi, N.S.; Gigant, B.; Despres, C.; Qi, H.; Lopez, J. NMR meets tau: Insights into its function and pathology. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Crowther, R.A.; Garner, C.C. Molecular characterization of microtubule-associated proteins tau and MAP2. Trends Neurosci. 1991, 14, 193–199. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6. [Google Scholar] [CrossRef]
- Schwalbe, M.; Biernat, J.; Bibow, S.; Ozenne, V.; Jensen, M.R.; Kadavath, H.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Phosphorylation of human tau protein by microtubule affinity-regulating kinase 2. Biochemistry 2013, 52, 9068–9079. [Google Scholar] [CrossRef] [PubMed]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.-M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Mawal-Dewan, M.; Barbour, R.; Jakes, R.; Goedert, M.; Johnson, G.V.W.; Litersky, J.M.; Schenk, D.; Lieberburg, I.; Trojanowski, J.Q.; et al. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J. Biol. Chem. 1995, 270, 18917–18922. [Google Scholar] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.; Sawada, S.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Pasceri, V.; Cammarota, G.; Patti, G.; Cuoco, L.; Gasbarrini, A.; Grillo, R.L.; Fedeli, G.; Gasbarrini, G.; Maseri, A. Association of virulent Helicobacter pylori strains with ischemic heart disease. Circulation 1998, 97, 1675–1679. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yujiri, T.; Shinohara, K.; Inoue, Y.; Sato, Y.; Fujii, Y.; Okubo, M.; Zaitsu, Y.; Ariyoshi, K.; Nakamura, Y.; et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br. J. Haematol. 2004, 124, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, A.; Cardaropoli, S.; Piccoli, E.; Rolfo, A.; Gennero, L.; Kanduc, D.; Todros, T. Pre-eclampsia is associated with Helicobacter pylori seropositivity in Italy. J. Hypertens. 2006, 24, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Ghabaee, M.; Ghanbarian, D.; Brujeni, G.N.; Bokaei, S.; Siavoshi, F.; Gharibzadeh, S. Could Helicobacter pylori play an important role in axonal type of Guillain-Barré Syndrome pathogenesis? Clin. Neurol. Neurosurg. 2010, 112, 193–198. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikawa, H.; Hatakeyama, M. Sequence Polymorphism and Intrinsic Structural Disorder as Related to Pathobiological Performance of the Helicobacter pylori CagA Oncoprotein. Toxins 2017, 9, 136. https://doi.org/10.3390/toxins9040136
Nishikawa H, Hatakeyama M. Sequence Polymorphism and Intrinsic Structural Disorder as Related to Pathobiological Performance of the Helicobacter pylori CagA Oncoprotein. Toxins. 2017; 9(4):136. https://doi.org/10.3390/toxins9040136
Chicago/Turabian StyleNishikawa, Hiroko, and Masanori Hatakeyama. 2017. "Sequence Polymorphism and Intrinsic Structural Disorder as Related to Pathobiological Performance of the Helicobacter pylori CagA Oncoprotein" Toxins 9, no. 4: 136. https://doi.org/10.3390/toxins9040136
APA StyleNishikawa, H., & Hatakeyama, M. (2017). Sequence Polymorphism and Intrinsic Structural Disorder as Related to Pathobiological Performance of the Helicobacter pylori CagA Oncoprotein. Toxins, 9(4), 136. https://doi.org/10.3390/toxins9040136