
Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing

Abstract
:1. Introduction
2. Results
2.1. Next Generation Sequencing
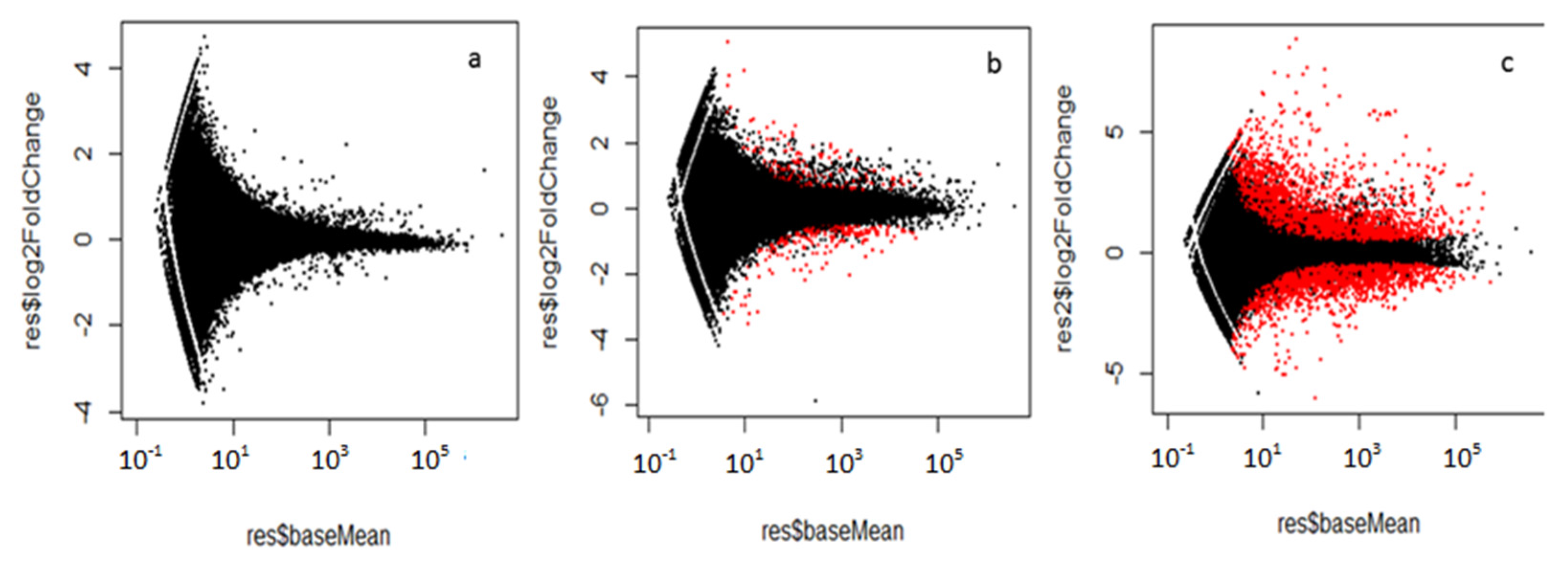
2.2. Mapping and Differential Expression Gene Analysis
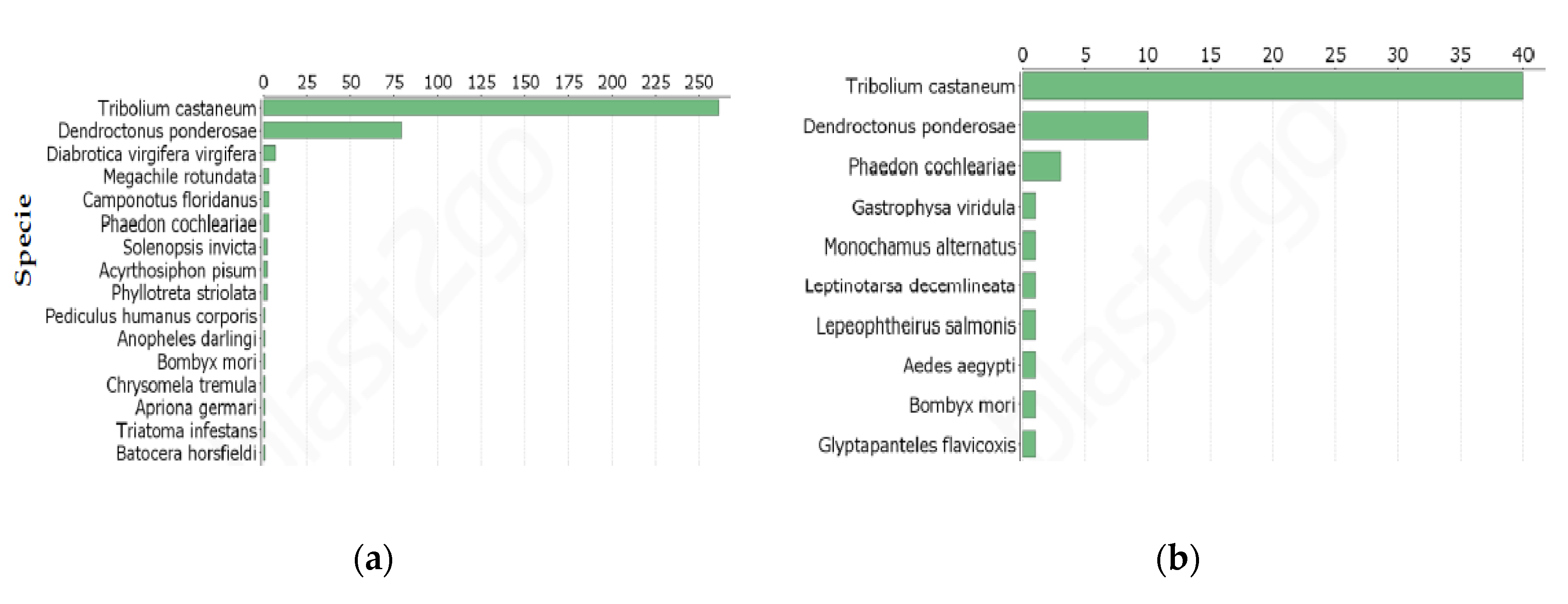
Annotation of DETs
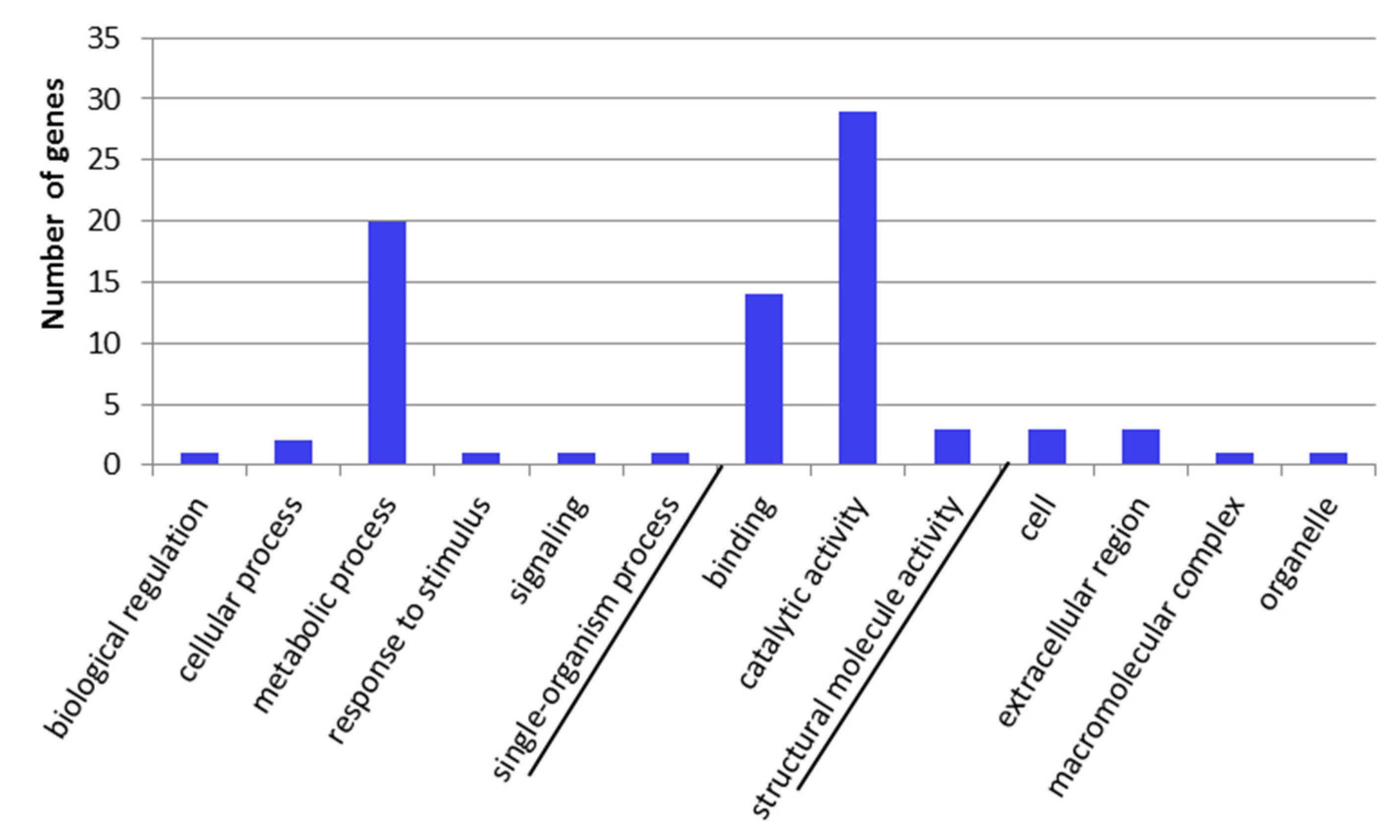
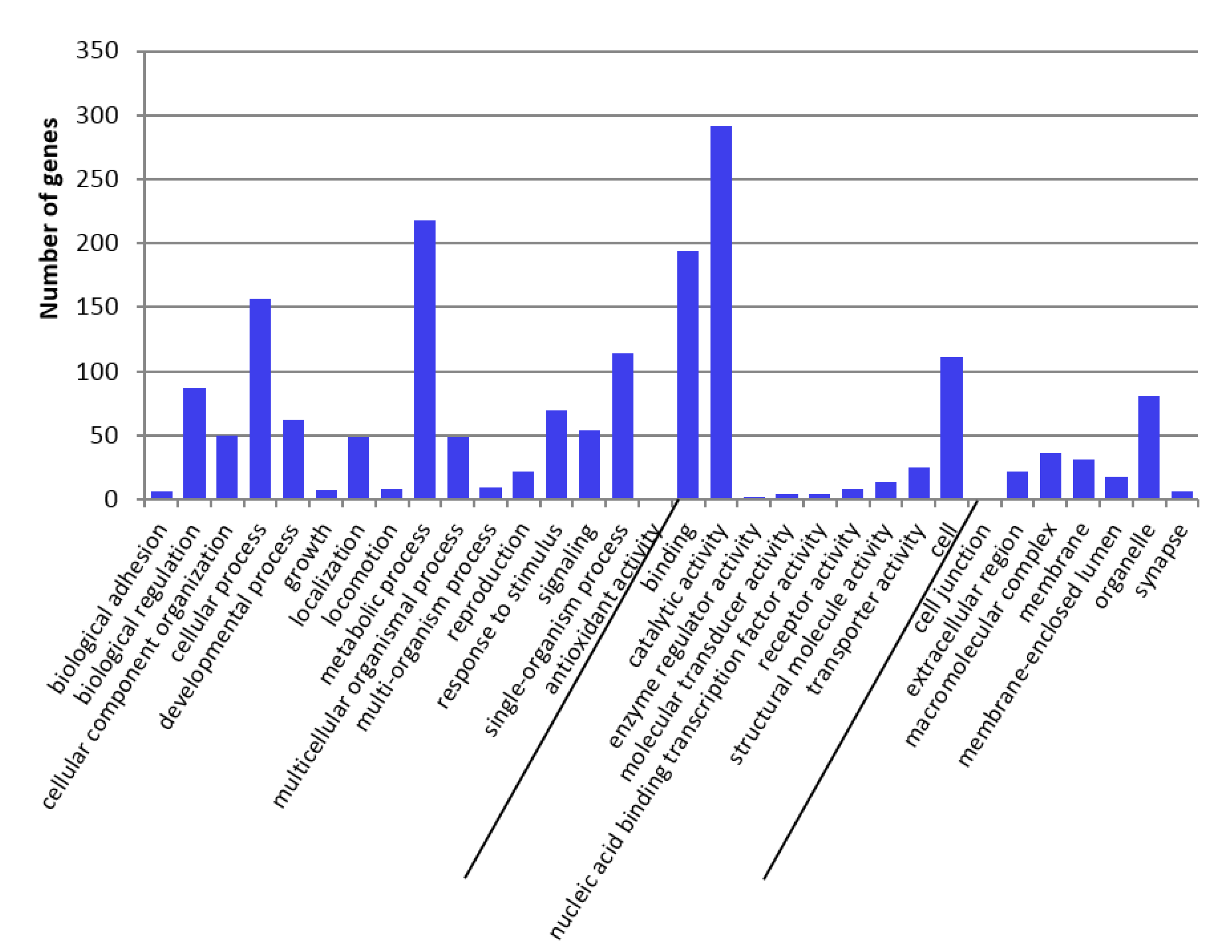
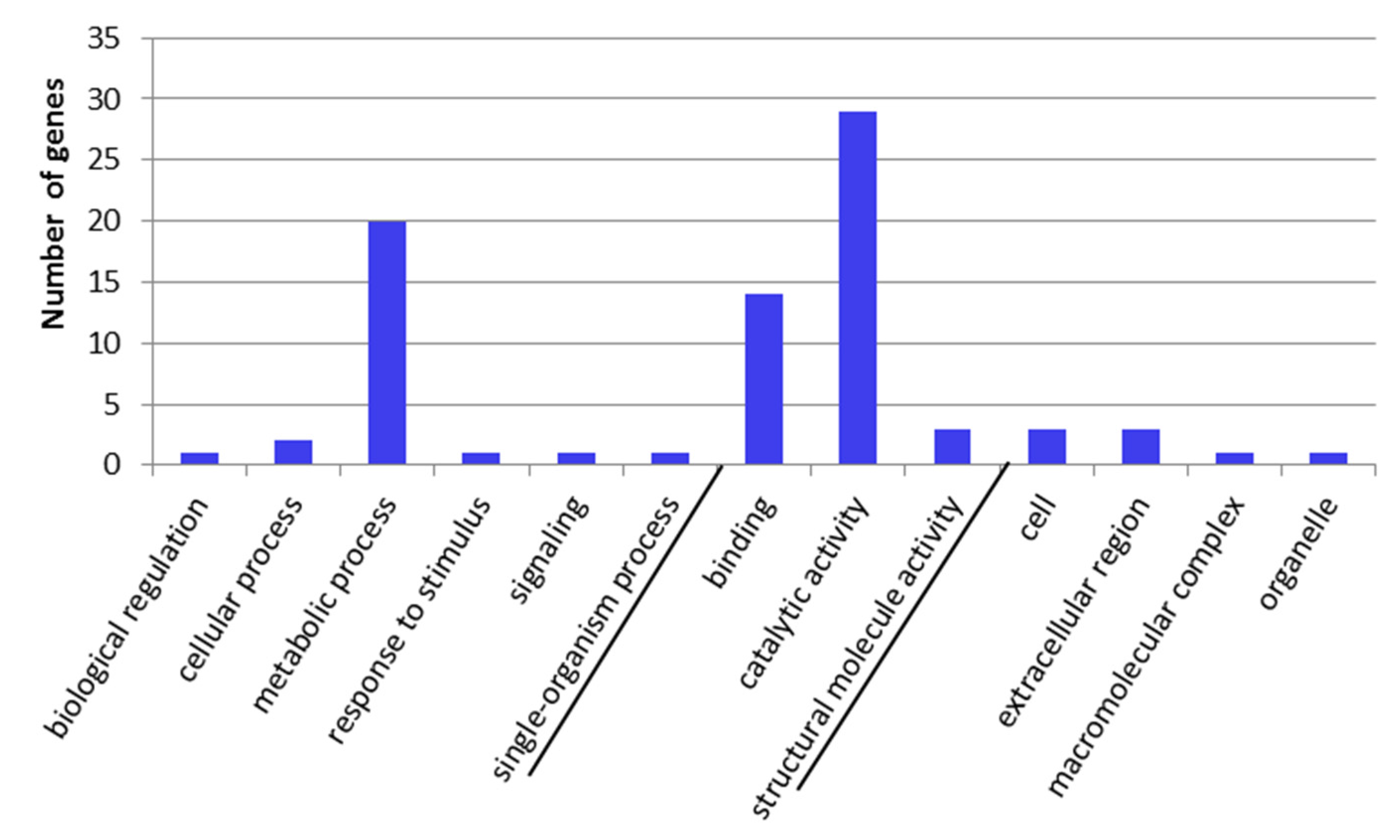
2.3. GO-Term Enrichment and Pathway Analysis
2.4. Validation with RT-qPCR
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Insects
5.2. Bt Proteins
5.3. Exposure
5.4. RNA Isolation
5.5. Next Generation Sequencing
5.6. Read Mapping and Differential Expression Analysis
5.7. Homology Searches and Gene Ontology Analysis
5.8. Validation of Gene Expression via qPCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levine, E.; Oloumisadeghi, H. Management of diabroticite rootworms in corn. Annu. Rev. Entomol. 1991, 36, 229–255. [Google Scholar] [CrossRef]
- Sappington, T.W.; Siegfried, B.D.; Guillemaud, T. Coordinated diabrotica genetics research: Accelerating progress on an urgent insect pest problem. Am. Entomol. 2006, 52, 90–97. [Google Scholar] [CrossRef]
- Miller, N.J.; Guillemaud, T.; Giordano, R.; Siegfried, B.D.; Gray, M.E.; Meinke, L.J.; Sappington, T.W. Genes, gene flow and adaptation of diabrotica virgifera virgifera. Agric. For. Entomol. 2009, 11, 47–60. [Google Scholar] [CrossRef]
- Metcalf, R.L. Methods for the study of pest diabroticai. In Methods for the Study of Pest Diabrotica; Krysan, J.L., Miller, T.A., Eds.; Springer: New York, NY, USA, 1986; pp. 7–15. [Google Scholar]
- Meinke, L.J.; Siegfried, B.D.; Wright, R.J.; Chandler, L.D. Adult susceptibility of nebraska western corn rootworm (coleoptera: Chrysomelidae) populations to selected insecticides. J. Econ. Entomol. 1998, 91, 594–600. [Google Scholar] [CrossRef]
- Parimi, S.; Meinke, L.J.; French, B.W.; Chandler, L.D.; Siegfried, B.D. Stability and persistence of aldrin and methyl-parathion resistance in western corn rootworm populations (coleoptera: Chrysomelidae). Crop Prot. 2006, 25, 269–274. [Google Scholar] [CrossRef]
- Levine, E.; Spencer, J.L.; Isard, S.A.; Onstad, D.W.; Gray, M.E. Adaptation of the western corn rootworm to crop rotation: Evolution of a new strain in response to a management practice. Am. Entomol. 2002, 48, 94–107. [Google Scholar] [CrossRef]
- Gray, M.E.; Sappington, T.W.; Miller, N.J.; Moeser, J.; Bohn, M.O. Adaptation and invasiveness of western corn rootworm: Intensifying research on a worsening pest. Annu. Rev. Entomol. 2009, 54, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, A.J.; Petzold-Maxwell, J.L.; Keweshan, R.S.; Dunbar, M.W. Field-evolved resistance to bt maize by western corn rootworm. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, A.J.; Petzold-Maxwell, J.L.; Clifton, E.H.; Dunbar, M.W.; Hoffmann, A.M.; Ingber, D.A.; Keweshan, R.S. Field-evolved resistance by western corn rootworm to multiple bacillus thuringiensis toxins in transgenic maize. Proc. Natl. Acad. Sci. USA 2014, 111, 5141–5146. [Google Scholar] [CrossRef] [PubMed]
- Wangila, D.S.; Gassmann, A.J.; Petzold-Maxwell, J.L.; French, B.W.; Meinke, L.J. Susceptibility of nebraska western corn rootworm (coleoptera: Chrysomelidae) populations to bt corn events. J. Econ. Entomol. 2015, 108, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Olson, M.; Lin, G.F.; Hey, T.; Tan, S.Y.; Narva, K.E. Bacillus thuringiensis Cry34Ab1/Cry35Ab1 interactions with western corn rootworm midgut membrane binding sites. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Moellenbeck, D.J.; Peters, M.L.; Bing, J.W.; Rouse, J.R.; Higgins, L.S.; Sims, L.; Nevshemal, T.; Marshall, L.; Ellis, R.T.; Bystrak, P.G.; et al. Insecticidal proteins from bacillus thuringiensis protect corn from corn rootworms. Nat. Biotechnol. 2001, 19, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Mamidala, P.; Wijeratne, A.J.; Wijeratne, S.; Kornacker, K.; Sudhamalla, B.; Rivera-Vega, L.J.; Hoelmer, A.; Meulia, T.; Jones, S.C.; Mittapalli, O. RNA-seq and molecular docking reveal multi-level pesticide resistance in the bed bug. BMC Genom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.S.; Jin, F.L.; Hu, Z.D.; Chen, H.Y.; Yin, F.; Li, Z.Y.; Dong, X.L.; Zhang, D.Y.; Ren, S.X.; Feng, X. Transcriptome analysis of chlorantraniliprole resistance development in the diamondback moth Plutella xylostella. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.E.; Blackburn, M.B.; Kuhar, D.; Gundersen-Rindal, D.E. Transcriptome of the lymantria dispar (gypsy moth) larval midgut in response to infection by bacillus thuringiensis. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.Y.; Zhu, X.; Xie, W.; Wu, Q.J.; Wang, S.L.; Guo, Z.J.; Xu, B.Y.; Li, X.C.; Zhou, X.G.; Zhang, Y.J. Midgut transcriptome response to a cry toxin in the diamondback moth, Plutella xylostella (lepidoptera: Plutellidae). Gene 2014, 533, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Canton, P.E.; Cancino-Rodezno, A.; Gill, S.S.; Soberon, M.; Bravo, A. Transcriptional cellular responses in midgut tissue of aedes aegypti larvae following intoxication with cry11aa toxin from Bacillus thuringiensis. BMC Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, A.J.; Shrestha, R.B.; Jakka, S.R.; Dunbar, M.W.; Clifton, E.H.; Paolino, A.R.; Ingber, D.A.; French, B.W.; Masloski, K.E.; Dounda, J.W.; et al. Evidence of resistance to cry34/35ab1 corn by western corn rootworm (coleoptera: Chrysomelidae): Root injury in the field and larval survival in plant-based bioassays. J. Econ. Entomol. 2016, 109, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Eyun, S.I.; Wang, H.C.; Pauchet, Y.; Ffrench-Constant, R.H.; Benson, A.K.; Valencia-Jimenez, A.; Moriyama, E.N.; Siegfried, B.D. Molecular evolution of glycoside hydrolase genes in the western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Schnepf, H.E.; Lee, S.; Dojillo, J.; Burmeister, P.; Fencil, K.; Morera, L.; Nygaard, L.; Narva, K.E.; Wolt, J.D. Characterization of Cry34/Cry35 binary insecticidal proteins from diverse bacillus thuringiensis strain collections. Appl. Environ. Microbiol. 2005, 71, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.T.; Stockhoff, B.A.; Stamp, L.; Schnepf, H.E.; Schwab, G.E.; Knuth, M.; Russell, J.; Cardineau, G.A.; Narva, K.E. Novel bacillus thuringiensis binary insecticidal crystal proteins active on western corn rootworm, Diabrotica virgifera virgifera leconte. Appl. Environ. Microbiol. 2002, 68, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Herman, R.A.; Scherer, P.N.; Young, D.L.; Mihaliak, C.A.; Meade, T.; Woodsworth, A.T.; Stockhoff, B.A.; Narva, K.E. Binary insecticidal crystal protein from bacillus thuringiensis, strain PS149B1: Effects of individual protein components and mixtures in laboratory bioassays. J. Econ. Entomol. 2002, 95, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Oppert, B.; Dowd, S.E.; Bouffard, P.; Li, L.; Conesa, A.; Lorenzen, M.D.; Toutges, M.; Marshall, J.; Huestis, D.L.; Fabrick, J.; et al. Transcriptome profiling of the intoxication response of tenebrio molitor larvae to bacillus thuringiensis Cry3Aa protoxin. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Derecka, K.; Blythe, M.J.; Malla, S.; Genereux, D.P.; Guffanti, A.; Pavan, P.; Moles, A.; Snart, C.; Ryder, T.; Ortori, C.A.; et al. Transient exposure to low levels of insecticide affects metabolic networks of honeybee larvae. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.; Shen, G.M.; Niu, J.Z.; Ding, T.B.; Wei, D.D.; Wang, J.J. Mining genes involved in insecticide resistance of liposcelis bostrychophila badonnel by transcriptome and expression profile analysis. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Vellichirammal, N.N.; Wang, H.C.; Eyun, S.; Moriyama, E.N.; Coates, B.S.; Miller, N.J.; Siegfried, S.D. Transcriptional analysis of susceptible and resistant european corn borer strains and their response to Cry1f protoxin. BMC Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Dierking, K.; Esser, D.; Tholey, A.; Leippe, M.; Rosenstiel, P.; Schulenburg, H. Overlapping and unique signatures in the proteomic and transcriptomic responses of the nematode caenorhabditis elegans toward pathogenic bacillus thuringiensis. Dev. Comp. Immunol. 2015, 52, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.N.; Wang, Y.Q.; Wang, Z.Y.; Hu, B.J.; Ling, Y.H.; He, K.L. Transcriptome differences between Cry1Ab resistant and susceptible strains of asian corn borer. BMC Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.; Wiechman, B.; Struewing, I.; Smith, M.; French, W.; Nielsen, C.; Bagley, M. Isolation of transcripts from diabrotica virgifera virgifera leconte responsive to the bacillus thuringiensis toxin Cry3Bb1. Insect Mol. Biol. 2010, 19, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wang, Q.Y.; Nangong, Z.Y.; Su, J.P.; Ge, D.H. Identification of henosepilachna vigintioctomaculata (coleoptera: Coccinellidae) midgut putative receptor for bacillus thuringiensis insecticidal Cry7Ab3 toxin. J. Invertebr. Pathol. 2012, 109, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Campuzano, C.; Real, M.D.; Martinez-Ramirez, A.C.; Bravo, A.; Rausell, C. An adam metalloprotease is a Cry3Aa bacillus thuringiensis toxin receptor. Biochem. Biophys. Res. Commun. 2007, 362, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Bando, H.; Asano, S. Identification of a bacillus thuringiensis Cry8Da toxin-binding glucosidase from the adult japanese beetle, popillia japonica. J. Invertebr. Pathol. 2013, 113, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Gill, S.S.; Soberon, M. Mode of action of bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 2007, 49, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.J.; Kang, S.; Chen, D.F.; Wu, Q.J.; Wang, S.L.; Xie, W.; Zhu, X.; Baxter, S.W.; Zhou, X.G.; Jurat-Fuentes, J.L.; et al. Mapk signaling pathway alters expression of midgut alp and abcc genes and causes resistance to bacillus thuringiensis cry1ac toxin in diamondback moth. PLoS Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pigott, C.R.; Ellar, D.J. Role of receptors in bacillus thuringiensis crystal toxin activity. Microbiol. Mol. Biol. Res. 2007, 71, 255–281. [Google Scholar] [CrossRef] [PubMed]
- Enayati, A.A.; Ranson, H.; Hemingway, J. Insect glutathione transferases and insecticide resistance. Insect Mol. Biol. 2005, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Teets, N.M.; Peyton, J.T.; Colinet, H.; Renault, D.; Kelley, J.L.; Kawarasaki, Y.; Lee, R.E.; Denlinger, D.L. Gene expression changes governing extreme dehydration tolerance in an antarctic insect. Proc. Natl. Acad. Sci. USA 2012, 109, 20744–20749. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.W.; Wright, A.E.; Mank, J.E. The evolution of gene expression and the transcriptome-phenotype relationship. Semin. Cell Dev. Biol. 2012, 23, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Flagel, L.E.; Bansal, R.; Kerstetter, R.A.; Chen, M.; Carroll, M.; Flannagan, R.; Clark, T.; Goldman, B.S.; Michel, A.P. Western corn rootworm (diabrotica virgifera virgifera) transcriptome assembly and genomic analysis of population structure. BMC Genom. 2014. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.L.; Spencer, T.A.; Nekl, E.; Pusztai-Carey, M.; Moar, W.J.; Siegfried, B.D. Comparison and validation of methods to quantify Cry1Ab toxin from bacillus thuringiensis for standardization of insect bioassays. Appl. Environ. Microbiol. 2008, 74, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for Fastq Files. Available online: https://github.com/najoshi/sickle (accessed on 29 March 2017).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of software packages for detecting differential expression in rna-seq studies. Brief. Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3plus, an enhanced web interface to primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.B.; Khajuria, C.; Wang, H.C.; Matz, N.; Cardoso, D.C.; Valicente, F.H.; Zhou, X.G.; Siegfried, B. Validation of reference housekeeping genes for gene expression studies in western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-delta delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis Method | Cry34Ab1 vs. Buffer | Cry35Ab1 vs. Buffer | Cry34Ab1 + Cry35Ab1 vs. Buffer | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| up | down | total | Shared * | up | down | total | up | down | total | shared | |
| DESeq | 44 | 72 | 116 | 114 | 0 | 0 | 0 | 992 | 1223 | 2215 | 1300 |
| edgeR | 49 | 83 | 132 | 0 | 0 | 0 | 647 | 1026 | 1673 | ||
| limma | 48 | 87 | 135 | 0 | 0 | 0 | 1093 | 1243 | 2336 | ||
| Category | Category | # Contigs | Digital Expression in Fold Change * | ||
|---|---|---|---|---|---|
| up-regulated | shared | 31 | 2.55 (2–4.66) a | 4.5 (2.3–12.5) b | |
| unique | 12 a | 534 b | 2.36 (2.14–2.9) a | 2.69 (2–64) b | |
| down-regulated | Shared | 61 | 2.64 (2–5.6) a | 10.6 (2.49–194) b | |
| unique | 10 a | 674 b | 3.16 (2–1) a | 3.3 (2–88) b | |
| Category | Gene | Fold in RNAseq Analysis | Fold Change in qPCR |
|---|---|---|---|
| up-regulated | GSC | 12.55 * | 91.01 * |
| PAT | 8.69 * | 3.44 * | |
| down-regulated | GH45 | 32 * | 0.31 * |
| ALP | 7.4 * | 0.16 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Eyun, S.-i.; Arora, K.; Tan, S.Y.; Gandra, P.; Moriyama, E.; Khajuria, C.; Jurzenski, J.; Li, H.; Donahue, M.; et al. Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing. Toxins 2017, 9, 124. https://doi.org/10.3390/toxins9040124
Wang H, Eyun S-i, Arora K, Tan SY, Gandra P, Moriyama E, Khajuria C, Jurzenski J, Li H, Donahue M, et al. Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing. Toxins. 2017; 9(4):124. https://doi.org/10.3390/toxins9040124
Chicago/Turabian StyleWang, Haichuan, Seong-il Eyun, Kanika Arora, Sek Yee Tan, Premchand Gandra, Etsuko Moriyama, Chitvan Khajuria, Jessica Jurzenski, Huarong Li, Maia Donahue, and et al. 2017. "Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing" Toxins 9, no. 4: 124. https://doi.org/10.3390/toxins9040124
APA StyleWang, H., Eyun, S.-i., Arora, K., Tan, S. Y., Gandra, P., Moriyama, E., Khajuria, C., Jurzenski, J., Li, H., Donahue, M., Narva, K., & Siegfried, B. (2017). Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing. Toxins, 9(4), 124. https://doi.org/10.3390/toxins9040124