
Human scFvs That Counteract Bioactivities of Staphylococcus aureus TSST-1

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
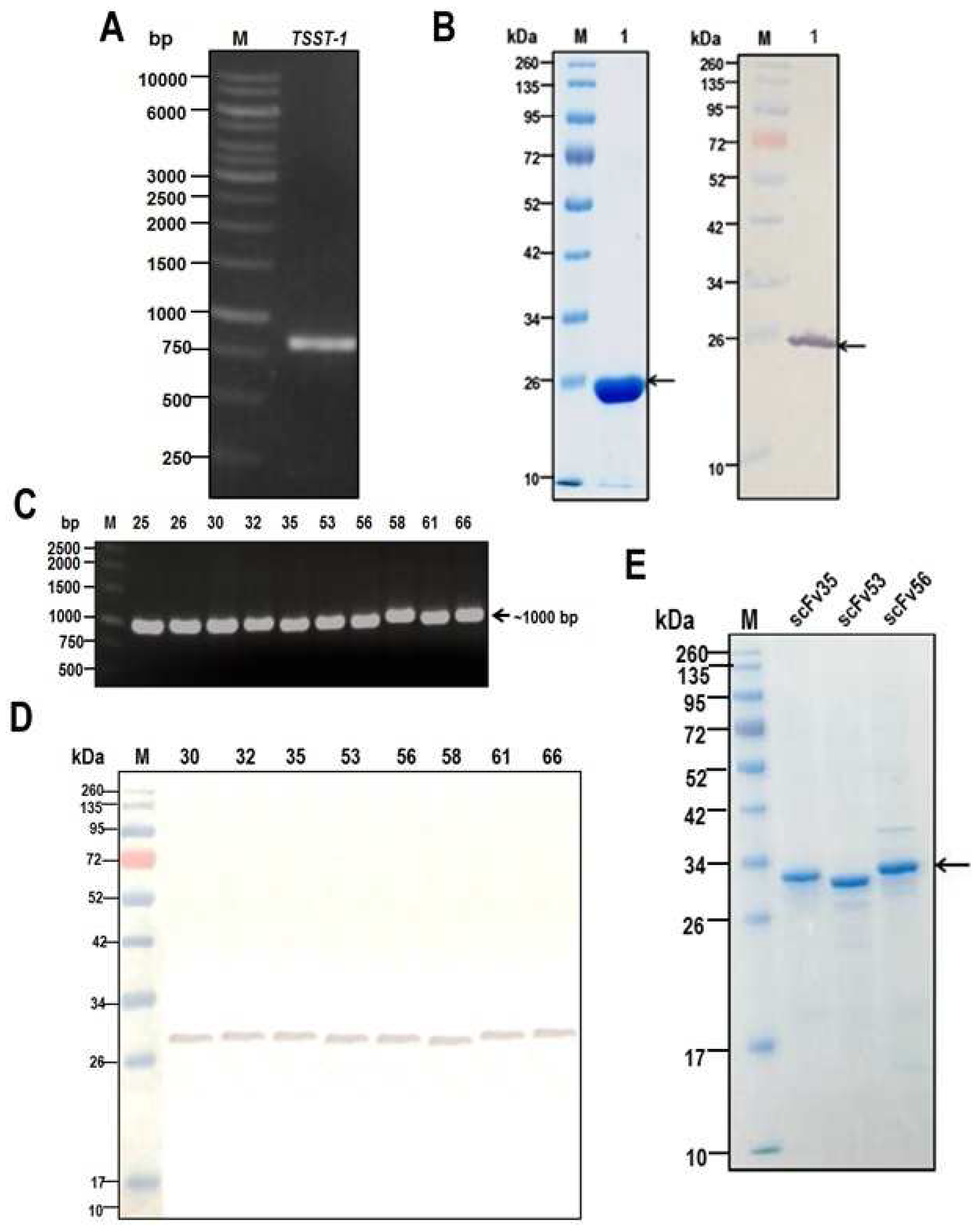
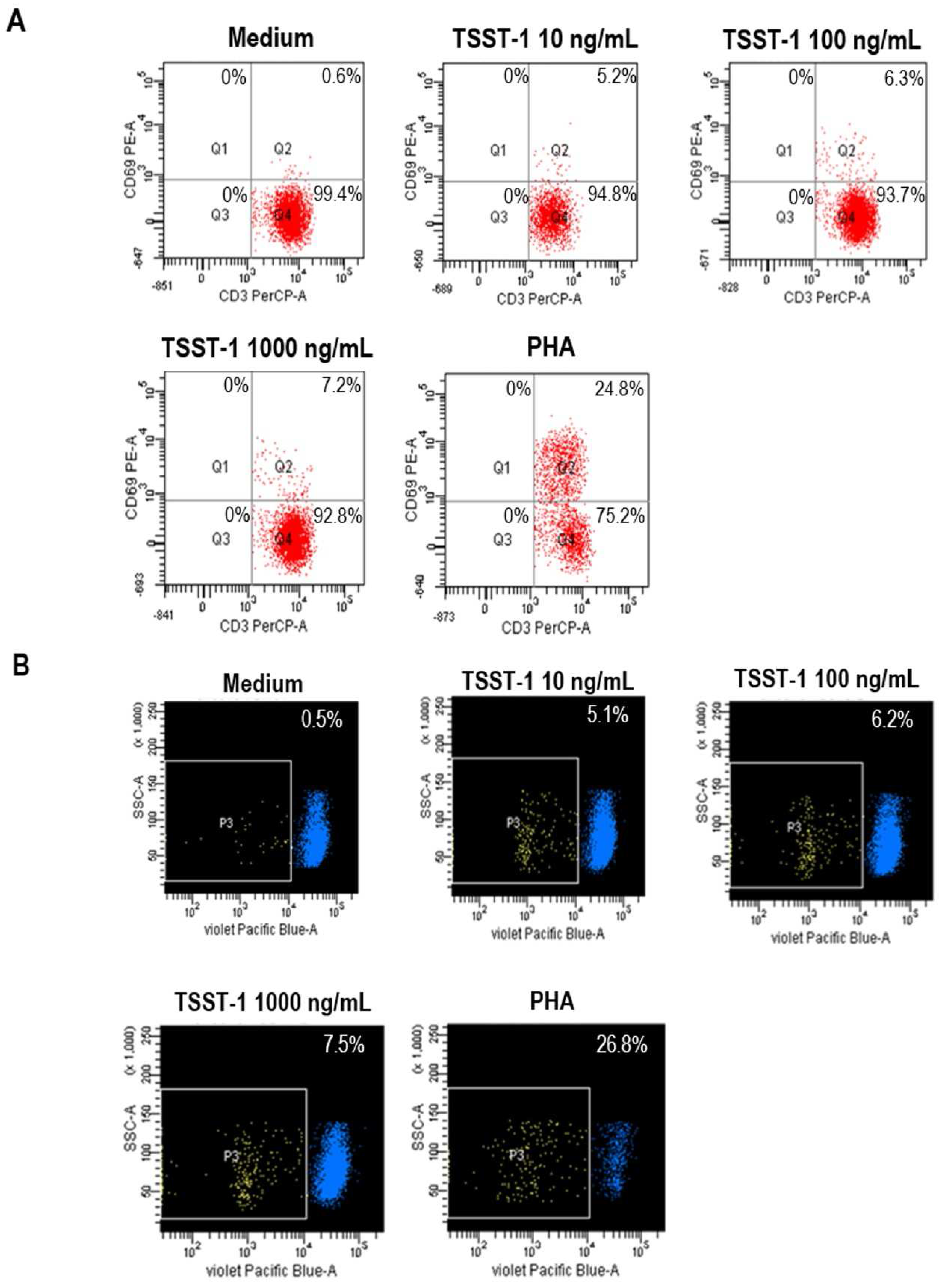
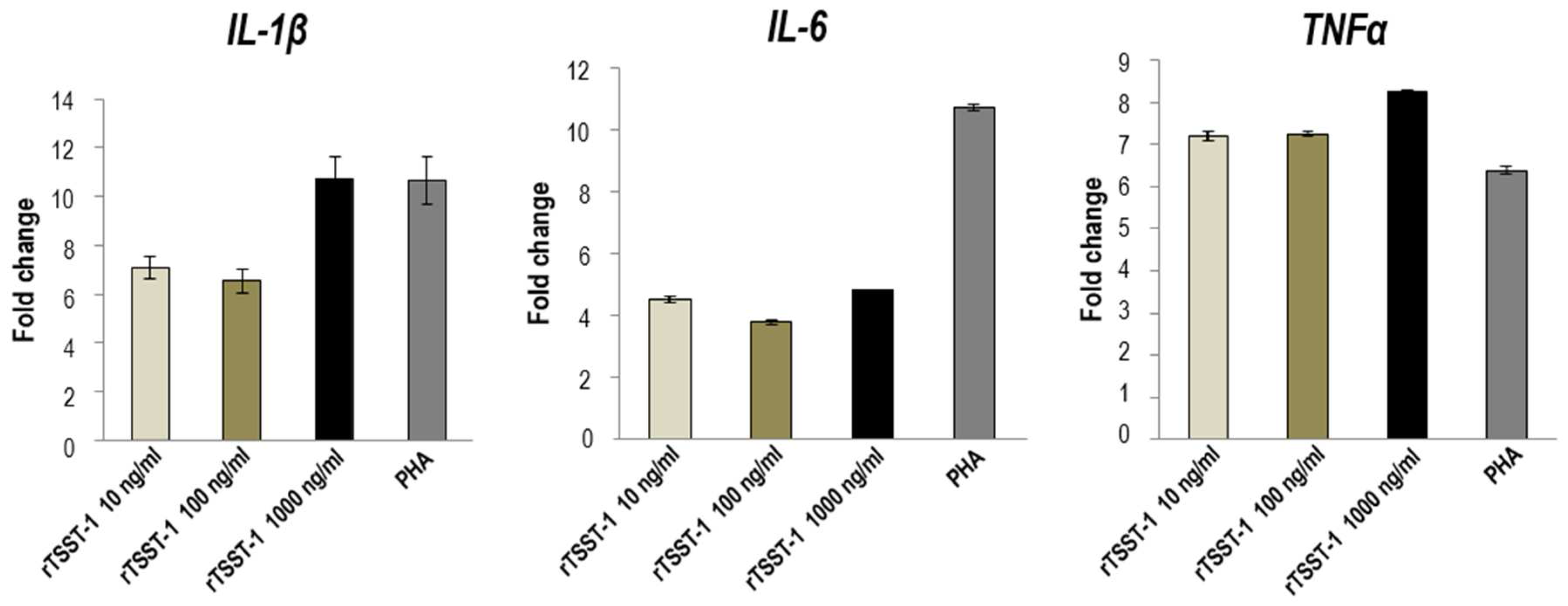
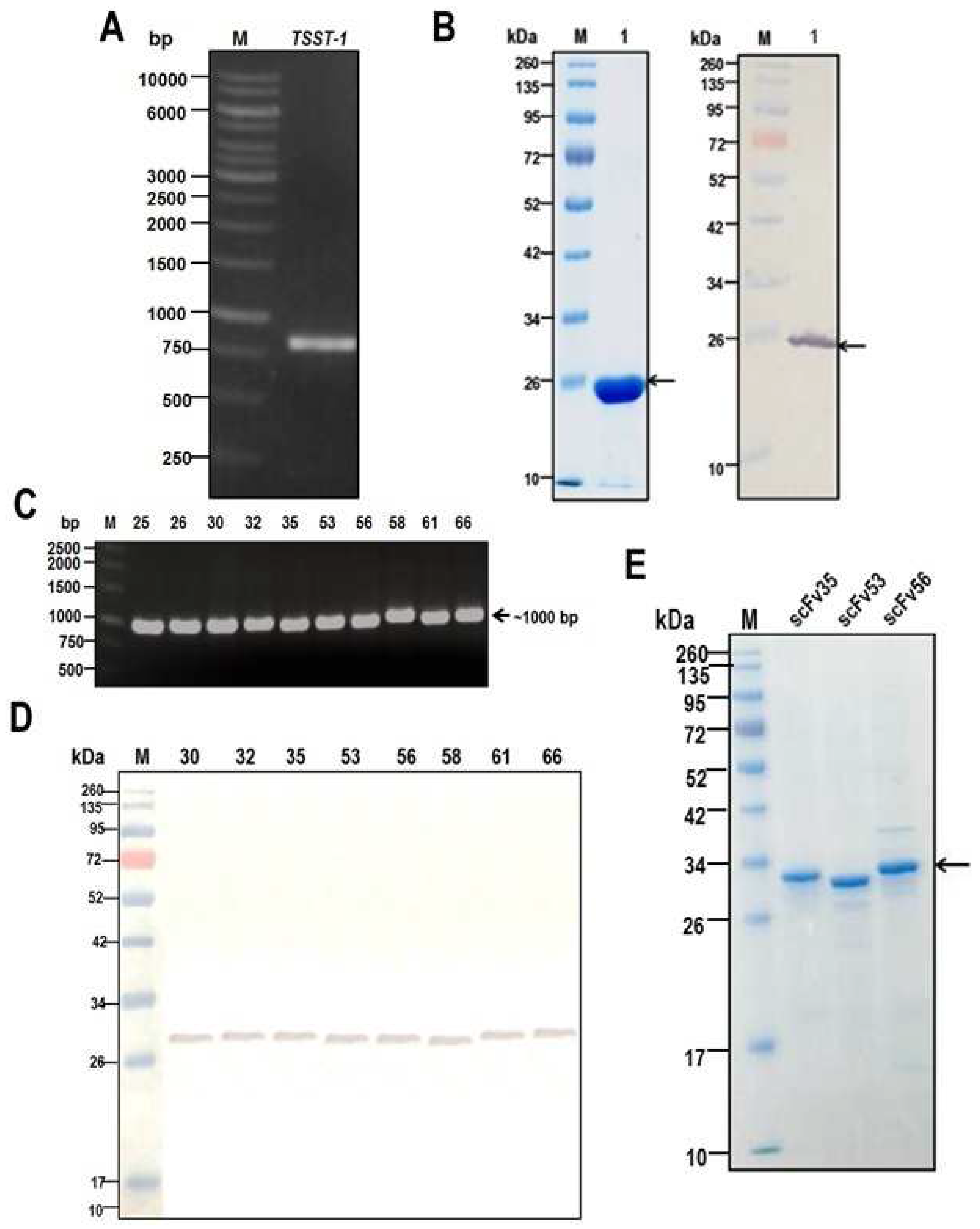
2.1. Recombinant TSST-1 and Activities
2.2. Production of HuscFvs
2.3. Presumptive Residues and Regions of TSST-1 Bound by the HuscFvs
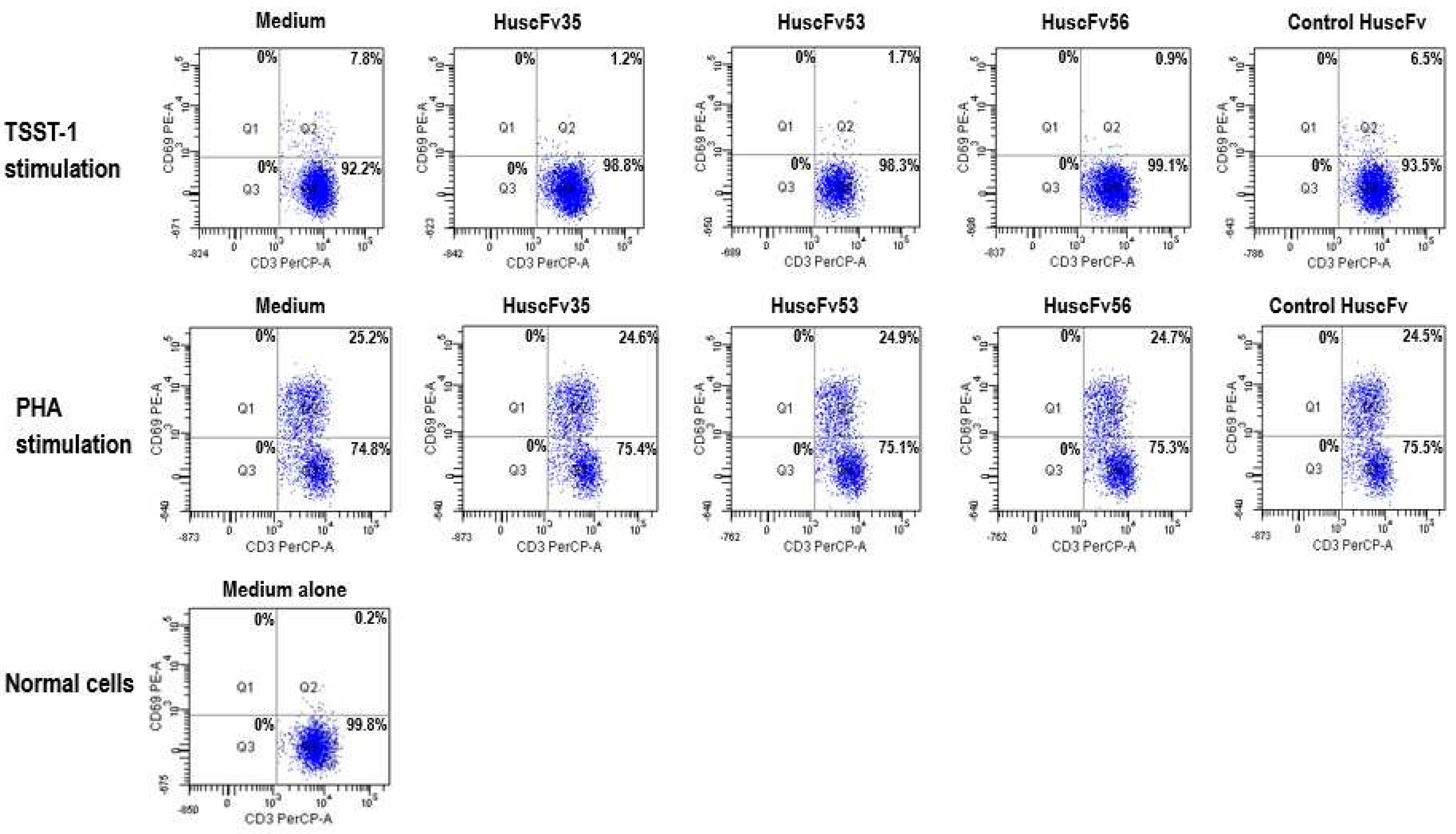
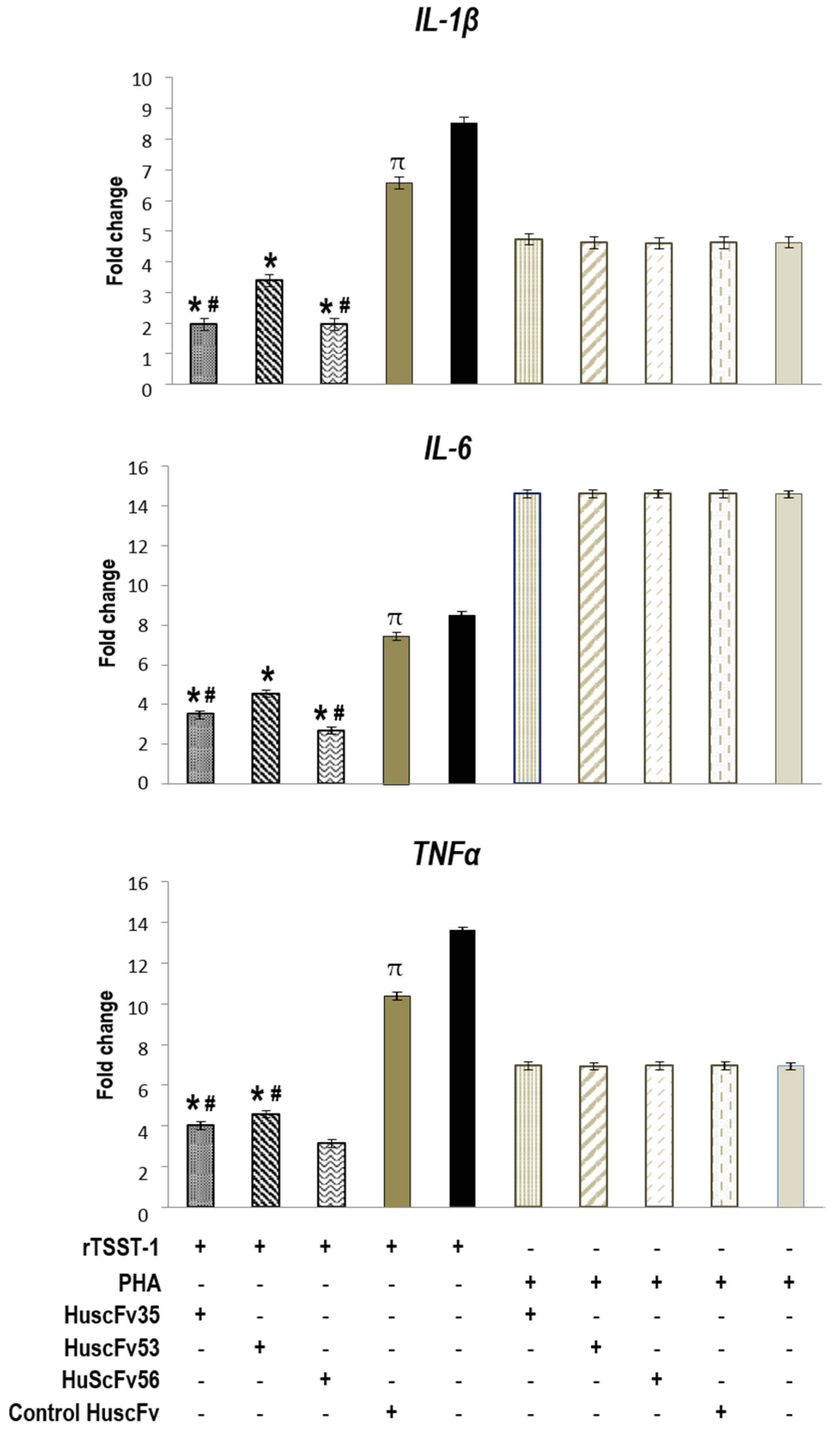
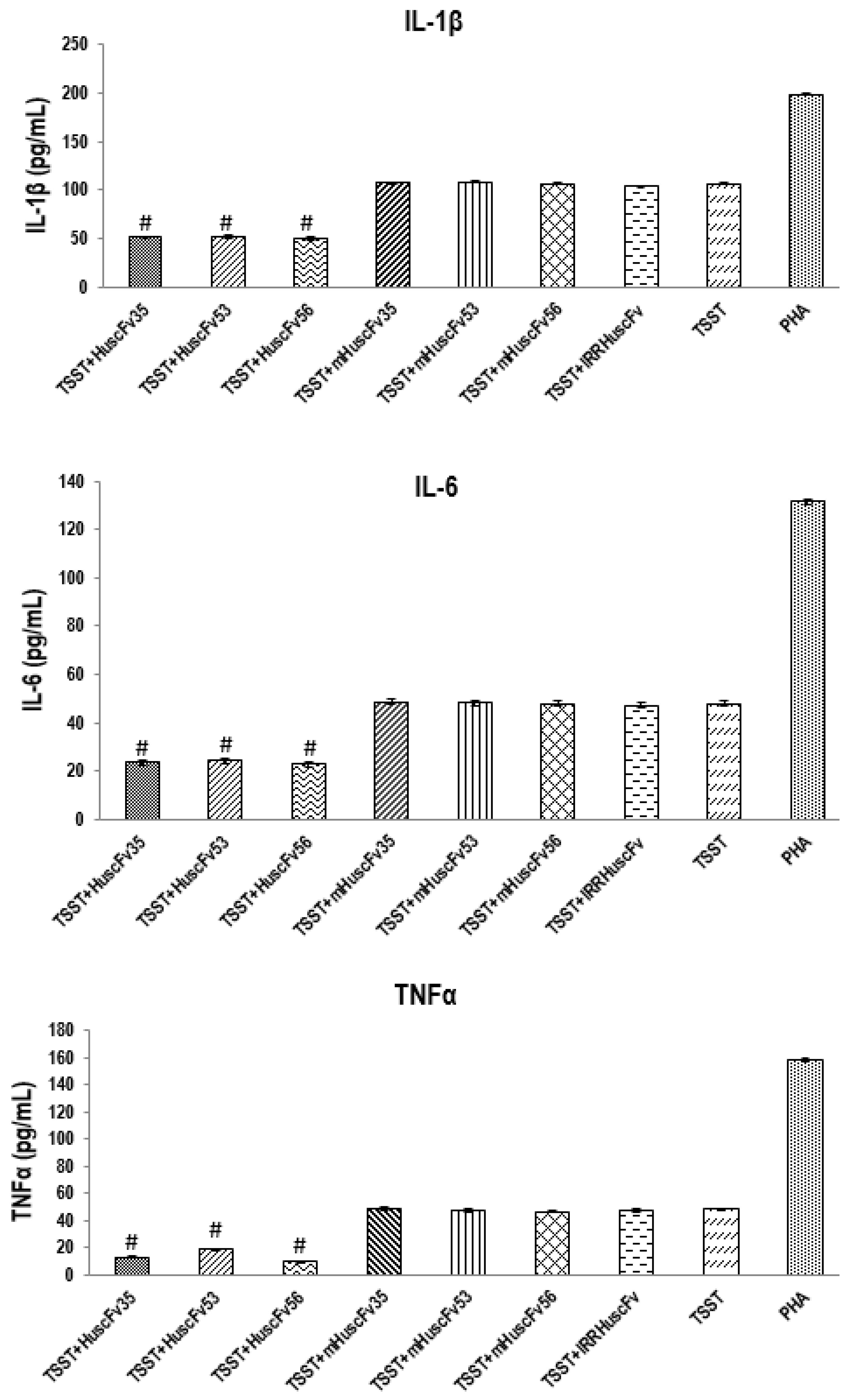
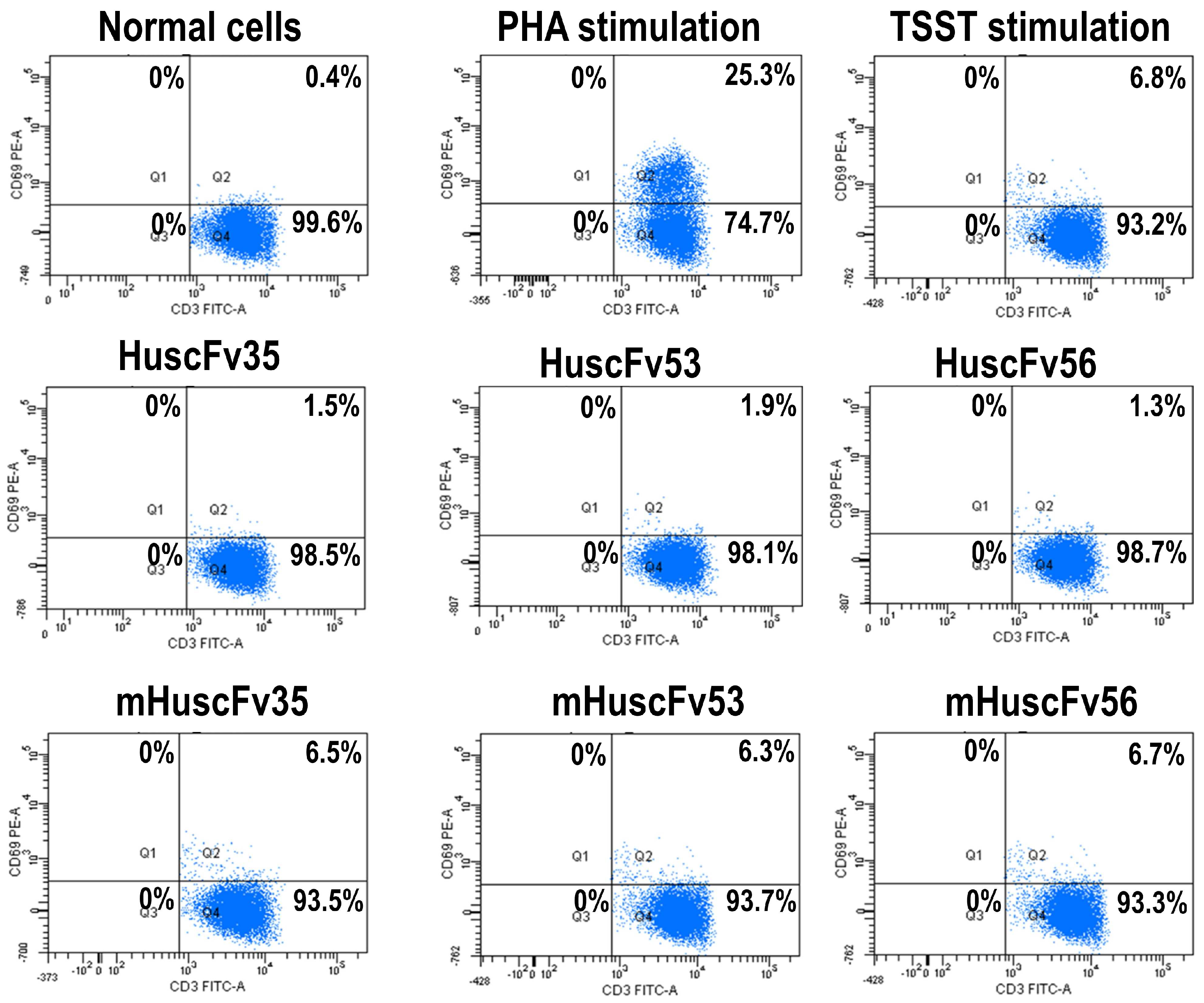
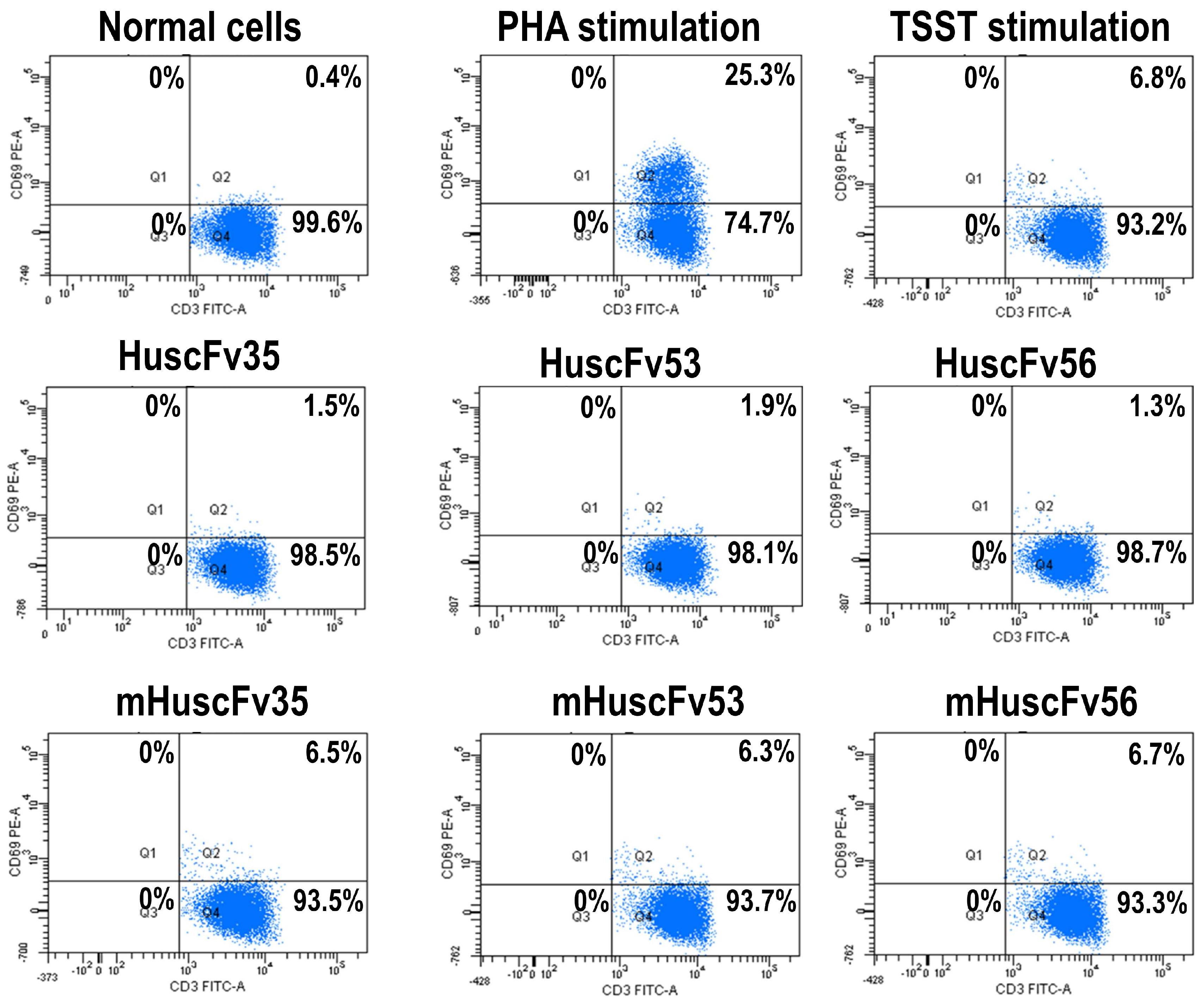
2.4. Inhibition of TSST-1 Activities by HuscFvs
3. Materials and Methods
3.1. Recombinant TSST-1 (rTSST-1) Production
3.2. Mitogenic and Pyrogenic Activities of rTSST-1
3.3. Production of TSST-1-bound HuscFvs
3.4. Computerized Simulation for Determining Interactive Residues between TSST-1 and HuscFvs
3.5. Preparation of Mutated-HuscFvs (mHuscFvs)
3.6. HuscFvs-mediated Inhibition of TSST-1 Activities
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kotb, M. Bacterial pyrogenic exotoxins as superantigens. Clin. Microbiol. Rev. 1995, 8, 411–426. [Google Scholar] [PubMed]
- Todd, J.; Fishaut, M.; Kapral, F.; Welch, T. Toxic-shock syndrome associated with phage-group-I Staphylococci. Lancet 1978, 2, 1116–1118. [Google Scholar] [CrossRef]
- Bueno, C.; Lemke, C.D.; Criado, G.; Baroja, M.L.; Ferguson, S.S.; Rahman, A.K.; Tsoukas, C.D.; McCormick, J.K.; Madrenas, J. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Gα11-dependent, PLC-β-mediated pathway. Immunity 2006, 25, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Tofte, R.W.; Williams, D.N. Toxic shock syndrome: Clinical and laboratory features in 15 patients. Ann. Intern. Med. 1981, 94, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Priest, B.P.; Schlievert, P.M.; Dunn, D.L. Treatment of toxic shock syndrome with endotoxin-neutralizing antibody. J. Surg. Res. 1989, 46, 527–531. [Google Scholar] [CrossRef]
- Kulhankova, K.; King, J.; Salgado-Pabón, W. Staphylococcal toxic shock syndrome: Superantigen-mediated enhancement of endotoxin shock and adaptive immune suppression. Immunol. Rev. 2014, 59, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Proft, T.; Frazer, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.H.; Parsonnet, J. On the pathogenesis of toxic shock syndrome. Rev. Infect. Dis. 1987, 9, S482–S489. [Google Scholar] [CrossRef] [PubMed]
- Shands, K.N.; Schmid, G.P.; Dan, B.B.; Blum, D.; Guidotti, R.J.; Hargrett, N.T.; Anderson, R.L.; Hill, D.L.; Broome, C.V.; Band, J.D.; et al. Toxic shock syndrome in menstruating women: Association with tampon use and Staphylococcus aureus and clinical features in 52 cases. N. Engl. J. Med. 1980, 303, 1438–1442. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecules HLA-DR1. Science 1994, 266, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Petersson, K.; Håkansson, M.; Nilsson, H.; Forsberg, G.; Anders Svensson, L.; Liljas, A.; Walse, B. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 2001, 20, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Llera, A.; Tsuchiya, D.; Leder, L.; Ysern, X.; Schlievert, P.M.; Karjalalnen, K.; Mariuzza, R.A. Three dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity 1998, 9, 807–816. [Google Scholar] [CrossRef]
- Li, H.; Llera, A.; Malchiodi, E.L.; Mariuzza, R.A. The structural basis of T cell activation by superantigens. Annu. Rev. Imunol. 1999, 17, 435–466. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Varma, A.K.; Moza, B.; Kasper, K.J.; Wyatt, A.W.; Zhu, P.; Nur-urRahman, A.K.M.; Li, Y.; Mariuzza, R.A.; McCormick, J.K.; et al. A novel loop domain in superantigens extends their T cell receptor recognition site. J. Mol. Biol. 2007, 371, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.L.; Earhart, C.A.; Mitchell, D.T.; Ohlendorf, D.H.; Norvick, R.P.; Schlievert, P.M. Localization of biologically important regions on toxic shock syndrome toxin 1. Infect. Immun. 1996, 64, 371–374. [Google Scholar] [PubMed]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Mocrobiol. 2001, 55, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A.; Ruzin, A.; Ross, H.F.; Kurepina, N.; Novick, R.P. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 1998, 29, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Passalacqua, E.F.; Jones, F.Y.; Harlos, K.; Stuart, D.I.; Brehm, R.D.; Tranter, H.S. Structural basis of superantigen action inferred from crystal structure of toxic shock syndrome toxin-1. Nature 1994, 367, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.S.; Radhakrishnan, R.; Mitchell, D.T.; Earhart, C.A.; Dinges, M.M.; Cook, W.J.; Schlievert, P.M.; Ohlendorf, D.H. Refined structures of three crystal forms of toxic shock syndrome toxin-1 and of a tetramutant with reduced activity. Prot. Sci. 1997, 6, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Wahlsten, J.L.; Ramakrishnan, S. Separation of function between the domains of toxic shock syndrome toxin-1. J. Immunol. 1998, 160, 854–859. [Google Scholar] [PubMed]
- Wu, C.C. Possible therapies of septic shock: Based on animal studies and clinical trials. Curr. Pharm. Des. 2006, 27, 3535–3541. [Google Scholar] [CrossRef]
- Mellish, M.E.; Cherry, J.D. Staphylococcal infections. In Pediatric Infectious Diseases; Feigin, R.D., Ed.; WB Saunders and Company: Philadelphia, PA, USA, 1992. [Google Scholar]
- Shankar-Hari, M.; Spencer, J.; Sewell, W.A.; Rowan, K.M.; Singer, M. Bench-to-bedside review: Immunoglobulin therapy for sepsis-biological plausibility from a critical perspective. Crit. Care 2002, 16, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, P.F.; Thompson, M.R.; Adinolfi, L.E.; Gillis, Z.A.; Parsonnet, J. Neutralization of toxic shock syndrome toxin-1 by monoclonal antibodies in vitro and in vivo. Infect. Immun. 1988, 56, 135–141. [Google Scholar] [PubMed]
- Stich, N.; Model, N.; Samstag, A.; Gruener, C.S.; Wolf, H.M.; Eibl, M.M. Toxic shock syndrome toxin-1 mediated toxicity inhibited by neutralizing antibodies late in the course of continual in vivo and in vitro exposure. Toxins 2014, 6, 1724–1741. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J. Endotoxin and Cell Culture Technical Bulletin; Corning Incorporated Life-Science: Lowell, MA, USA, 2008. [Google Scholar]
- Stich, N.; Waclavicek, M.; Model, N.; Eibl, M.M. Staphylococcal superantigen (TSST-1) mutant analysis reveals that T cell activation is required for biological effect in the rabbit including the cytokine storm. Toxins 2010, 2, 2272–2288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Chen, Z.; Purcell, RH.; Emerson, S.U. Positive reactions on Western blots do not necessarily indicate the epitopes on antigens are continuous. Immunol. Cell Biol. 2007, 85, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Gampfer, J.M.; Samstag, A.; Waclavicek, M.; Wolf, H.M.; Eibl, M.M.; Gulle, H. Epitope mapping of neutralizing TSST-1 specific antibodies induced by immunization with toxin and toxoid. Vaccine 2002, 20, 3675–3684. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Brehm, R.D.; Leonidas, D.D.; Tranter, H.S.; Acharya, K.R. The refined crystal structure of toxic shock syndrome toxin-1 at 2.07 Å resolution. J. Mol. Biol. 1996, 260, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Earhart, C.A.; Mitchell, D.T.; Murray, D.L.; Pinheiro, D.M.; Matsumura, M.; Schlievert, P.M.; Ohlendorf, D.H. Structures of five mutants of toxic shock syndrome toxin-1 with reduced biological activity. Biochemistry 1998, 37, 7194–7202. [Google Scholar] [CrossRef] [PubMed]
- Deresiewicz, R.L.; Woo, J.; Chan, M.; Finberg, R.W.; Kasper, D.L. Mutations affecting the activity of toxic shock syndrome toxin-1. Biochemistry 1994, 33, 12844–12851. [Google Scholar] [CrossRef] [PubMed]
- Moza, B.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Nur-urRahman, A.K.M.; McCormick, J.K.; Kranz, D.M.; Sundberg, E.J. Long-range cooperative binding effects in a T cell receptor variable domain. Proc. Natl. Acad. Sci. USA 2006, 103, 9867–9872. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, P.F.; Heeg, H.; Cullen, C.; Lian, C.J. Toxicity of recombinant toxic shock syndrome toxin 1 and mutant toxins produced by Staphyloccocus aureus in a rabbit infection model of toxic shock syndrome. Infect. Immun. 1993, 61, 793–799. [Google Scholar] [PubMed]
- Drynda, A.; König, B.; Bonventre, P.F.; König, W. Role of a carboxy-terminal site of toxic shock syndrome toxin 1 in eliciting immune responses of human peripheral blood mononuclear cells. Infect. Immun. 1995, 63, 1095–1101. [Google Scholar] [PubMed]
- McCormick, J.K.; Tripp, T.J.; Llera, A.S.; Sundberg, E.J.; Dinges, M.M.; Mariuzza, R.A.; Schlievert, P.M. Functional analysis of the TCR binding domain of toxic shock syndrome toxin-1 predicts further diversity in MHC class II/superantigen/TCR ternary complexes. J. Immunol. 2003, 171, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Zoccal, K.F.; Roncolato, E.C.; Bertolini, T.B.; Campos, L.B.; Cologna, C.T.; Faccioli, L.H.; Arantes, E.C.; Barbosa, J.E. Serrumab: A human monoclonal antibody that counters the biochemical and immunological effects of Tityusserrulatus venom. J. Immunotoxicol. 2012, 9, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Jittavisutthikul, S.; Seesuay, W.; Thanongsaksrikul, J.; Thueng-in, K.; Srimanote, P.; Werner, R.G.; Chaicumpa, W. Human transbodies to HCV NS3/4A protease inhibit viral replication and restore host innate immunity. Front. Immunol. 2016, 7, 318. [Google Scholar] [CrossRef] [PubMed]
- Lina, G.; Cozon, G.; Ferrandiz, J.; Greenland, T.; Vandenesch, F.; Etienne, J. Detection of staphylococcal superantigenic toxin by a CD69-specific cytofluorimetric assay measuring T-cell activation. J. Clin. Microbiol. 1998, 36, 1042–1045. [Google Scholar] [PubMed]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungthrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Najakaouthia, neurotoxin. J. Proteom. 2009, 72, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Giudicelli, V.; Brochet, X.; Lefranc, M.P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011, 2011, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Andrio, P.; Fenollosa, C.; Cicin-Sain, D.; Orozco, M.; Gelpí, J.L. MDWeb and MDMoby: An integrated web-based platform for molecular dynamics simulations. Bioinformatics 2012, 28, 1278–1279. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Orthologous Protein | Accession No. | Number of Matched Peptides | Score | Matched Peptide Sequence (Score) |
|---|---|---|---|---|---|
| TSST-1 | Toxic shock syndrome toxin-1 of S. aureus | Gi 136457 | 9 | 239 | DSPLKYGPK (44) |
| LPTPIELPLKVK (38) | |||||
| HQLTQIHGLYR (36) | |||||
| ITMNDGSTYQSDLSK (91) | |||||
| ITMNDGSTYQSDLSK (35) | |||||
| ITMNDGSTYQSDLSK (28) | |||||
| ITMNDGSTYQSDLSK (28) | |||||
| NTDGSISLIIFPSPYYSPAFTKGEK (32) | |||||
| NTDGSISLIIFPSPYYSPAFTKGEK (23) |
| TSST-1 | HuscFv35 | Intermolecular Bond | ||
|---|---|---|---|---|
| Residue | Motif | Residue(s) | Domain | |
| S1 | α1-helix | T165 | VL-CDR1 | Van de Waals |
| D4 | Before α1-helix | T165 | VL-CDR1 | Van de Waals |
| D4 | Before α1-helix | N166 | VL-CDR1 | H-bond |
| I6 | α1-helix | N166 | VL-CDR1 | H-bond |
| K7 | α1-helix | Y168 | VL-CDR1 | Water bridge |
| K7 | α1-helix | Y227 | VL-CDR3 | H-bond |
| K7 | α1-helix | D228 | VL-CDR3 | H-bond |
| D8 | α1-helix | Y103 | VL-CDR3 | OH-π |
| D8 | Before α1-helix | N166 | VL-CDR1 | Van de Waals |
| W12 | α1-helix | L102 | VH-CDR3 | CH-π |
| S15 | α1-helix | Q101 | VL-CDR3 | H-bond |
| G16 | Before β1-strand | Q101 | VH-CDR3 | Hydrophobic |
| K67 | β4-strand | Q101 | VH-CDR3 | Water bridge |
| R68 | β4-strand | D31 | VH-CDR1 | H-bond |
| R68 | β4-strand | H100 | VH-CDR3 | CH-π |
| R68 | β4-strand | Q101 | VH-CDR3 | Van de Waals |
| K71 | β4-strand | H100 | VH-CDR3 | CH-π |
| K71 | β4-strand | D108 | VH-CDR3 | H-bond |
| K71 | β4-strand | Y185 | VL-FR2 | H-bond |
| S72 | β4-strand | H100 | VH-CDR3 | H-bond |
| Q73 | β4-strand | T192 | VL-FR3 | H bond |
| Y80 | β5-strand | Y27 | VH-FR1 | H bond |
| Y80 | β5-strand | D31 | VH-CDR1 | OH-π |
| TSST-1 | HuscFv53 | Intermolecular Bond | ||
|---|---|---|---|---|
| Residue | Motif | Residue(s) | Domain | |
| D18 | Before β1-strand | R31 | VH-CDR1 | H-bond |
| D39 | Before β3-strand | R31 | VH-CDR1 | H-bond |
| R68 | β4-strand | T52 | VH-CDR2 | Van de Waals |
| R68 | β4-strand | D57 | VH-CDR2 | H-bond |
| K71 | β4-strand | W33 | VH-CDR1 | NH-π |
| K71 | β4-strand | T52 | VH-CDR2 | H-bond |
| K71 | β4-strand | D57 | VH-CDR2 | H-bond |
| Q73 | β4-strand | W33 | VH-CDR1 | Van de Waals |
| Q73 | β4-strand | W230 | VL-CDR3 | Van de Waals |
| H74 | β4-strand | W33 | VH-CDR3 | π-stacking |
| H74 | β4-strand | R100 | VH-CDR3 | NH-π |
| H74 | β4-strand | F101 | VL-CDR1 | π-stacking |
| S76 | Before β5-strand | D166 | VL-CDR1 | H bond |
| S76 | Before β5-strand | Y168 | VL-CDR1 | H bond |
| S76 | Before β5-strand | K186 | VL-CDR2 | H bond |
| E77 | Before β5-strand | K186 | VL-CDR2 | Water bridge |
| Y80 | β5-strand | W33 | VH-CDR1 | H-bond |
| Y80 | β5-strand | R100 | VH-CDR3 | NH-π |
| TSST-1 | HuscFv56 | Intermolecular Bond | ||
|---|---|---|---|---|
| Residue | Motif | Residue(s) | Domain | |
| K7 | α1-helix | S162 | VL-CDR1 | H-bond |
| K7 | α1-helix | I163 | VL-CDR1 | Van der Waals |
| L10 | α1-helix | I163 | VL-CDR1 | Hydrophobic |
| D11 | α1-helix | I163 | VL-CDR1 | Van de Waals |
| D11 | α1-helix | R164 | VL-CDR1 | H-bond |
| D11 | α1-helix | Y165 | VL-CDR1 | OH-π |
| S15 | α1-helix | Y165 | VL-CDR1 | H-bond |
| S17 | Before β1-strand | Y102 | VL-CDR3 | H-bond |
| D18 | Before β1-strand | R103 | VH-CDR3 | Water bridge |
| T19 | β1-strand | Y102 | VH-CDR3 | CH-π |
| T19 | β1-strand | R103 | VH-CDR3 | Van de Waals |
| F20 | β1-strand | R103 | VH-CDR3 | CH-π |
| D39 | Before β3-strand | Y182 | VL-FR2 | Water bridge |
| D39 | Before β3-strand | P189 | VL-CDR2 | Van de Waals |
| N65 | β4-strand | Y102 | VH-CDR3 | OH-π |
| R68 | β4-strand | Y182 | VL-FR2 | H bond |
| R68 | β4-strand | N186 | VL-CDR2 | Van de Waals |
| K71 | β4-strand | S185 | VL-CDR2 | Van de Waals |
| K71 | β4-strand | N186 | VL-CDR2 | Van de Waals |
| S72 | β4-strand | N186 | VL-CDR2 | H bond |
| H74 | β4-strand | F193 | VL-FR3 | π-stacking |
| Y80 | Β5-strand | Y182 | VL-FR2 | π-stacking |
| Y80 | Β5-strand | N186 | VL-CDR2 | Van de Waals |
| Y80Y80 | Β5-strand | V187 | VL-CDR2 | H bond |
| Β5-strand | F193 | VL-FR3 | π-stacking | |
| K114 | Before β8-strand | S57 | VH-CDR2 | Van de Waals |
| K114K114 | Before β8-strand | T58 | VH-CDR2 | H bond |
| Before β8-strand | E59 | VH-CDR2 | Salt bridge | |
| Y115 | Before β8-strand | W50 | VH-CDR2 | π-stacking |
| Y115Y115 | Before β8-strand | F52 | VH-CDR2 | π-stacking |
| Before β8-strand | Y101 | VH-CDR3 | H-bond | |
| P117 | Before β8-strand | F52 | VH-CDR2 | CH-π |
| P117 | Before β8-strand | Y101 | VH-CDR3 | CH-π |
| P117 | Before β8-strand | Y102 | VH-CDR3 | CH-π |
| K118 | β8-strand | S31 | VH-CDR1 | H bond |
| K118 | β8-strand | E55 | VH-CDR2 | H bond |
| K118 | β8-strand | Y102 | VH-CDR3 | H bond |
| F119 | β8-strand | Y102 | VH-CDR3 | π-stacking |
| E132 | α2-helix | Y101 | VH-CDR3 | OH-π |
| E132 | α2-helix | Y102 | VH-CDR3 | CH-π |
| E132 | α2-helix | R104 | VH-CDR3 | Salt bridge |
| H135 | α2-helix | R104 | VH-CDR3 | NH-π |
| H135 | α2-helix | W224 | VL-CDR3 | π-stacking |
| Q139 | α2-helix | W224 | VL-CDR3 | CH-π |
| Q139 | α2-helix | Y227 | VL-CDR3 | CH-π |
| Q139 | α2-helix | Y229 | VL-CDR3 | H bond |
| I140 | α2-helix | Y227 | VL-CDR3 | CH-π |
| R145 | Before β9-strand | S225 | VL-CDR3 | H-bond |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rukkawattanakul, T.; Sookrung, N.; Seesuay, W.; Onlamoon, N.; Diraphat, P.; Chaicumpa, W.; Indrawattana, N. Human scFvs That Counteract Bioactivities of Staphylococcus aureus TSST-1. Toxins 2017, 9, 50. https://doi.org/10.3390/toxins9020050
Rukkawattanakul T, Sookrung N, Seesuay W, Onlamoon N, Diraphat P, Chaicumpa W, Indrawattana N. Human scFvs That Counteract Bioactivities of Staphylococcus aureus TSST-1. Toxins. 2017; 9(2):50. https://doi.org/10.3390/toxins9020050
Chicago/Turabian StyleRukkawattanakul, Thunchanok, Nitat Sookrung, Watee Seesuay, Nattawat Onlamoon, Pornphan Diraphat, Wanpen Chaicumpa, and Nitaya Indrawattana. 2017. "Human scFvs That Counteract Bioactivities of Staphylococcus aureus TSST-1" Toxins 9, no. 2: 50. https://doi.org/10.3390/toxins9020050
APA StyleRukkawattanakul, T., Sookrung, N., Seesuay, W., Onlamoon, N., Diraphat, P., Chaicumpa, W., & Indrawattana, N. (2017). Human scFvs That Counteract Bioactivities of Staphylococcus aureus TSST-1. Toxins, 9(2), 50. https://doi.org/10.3390/toxins9020050