
Membrane-Active Properties of an Amphitropic Peptide from the CyaA Toxin Translocation Region

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
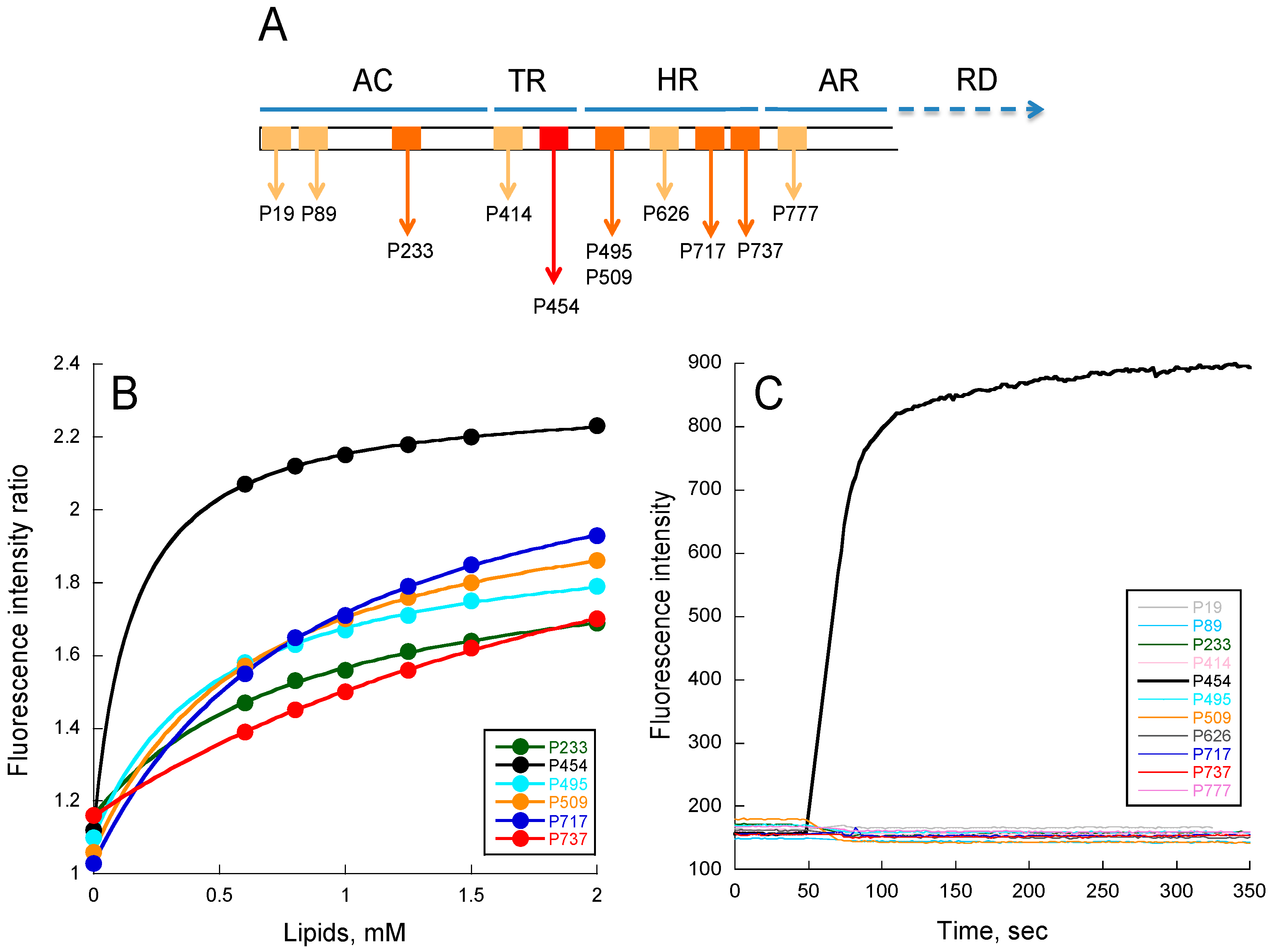
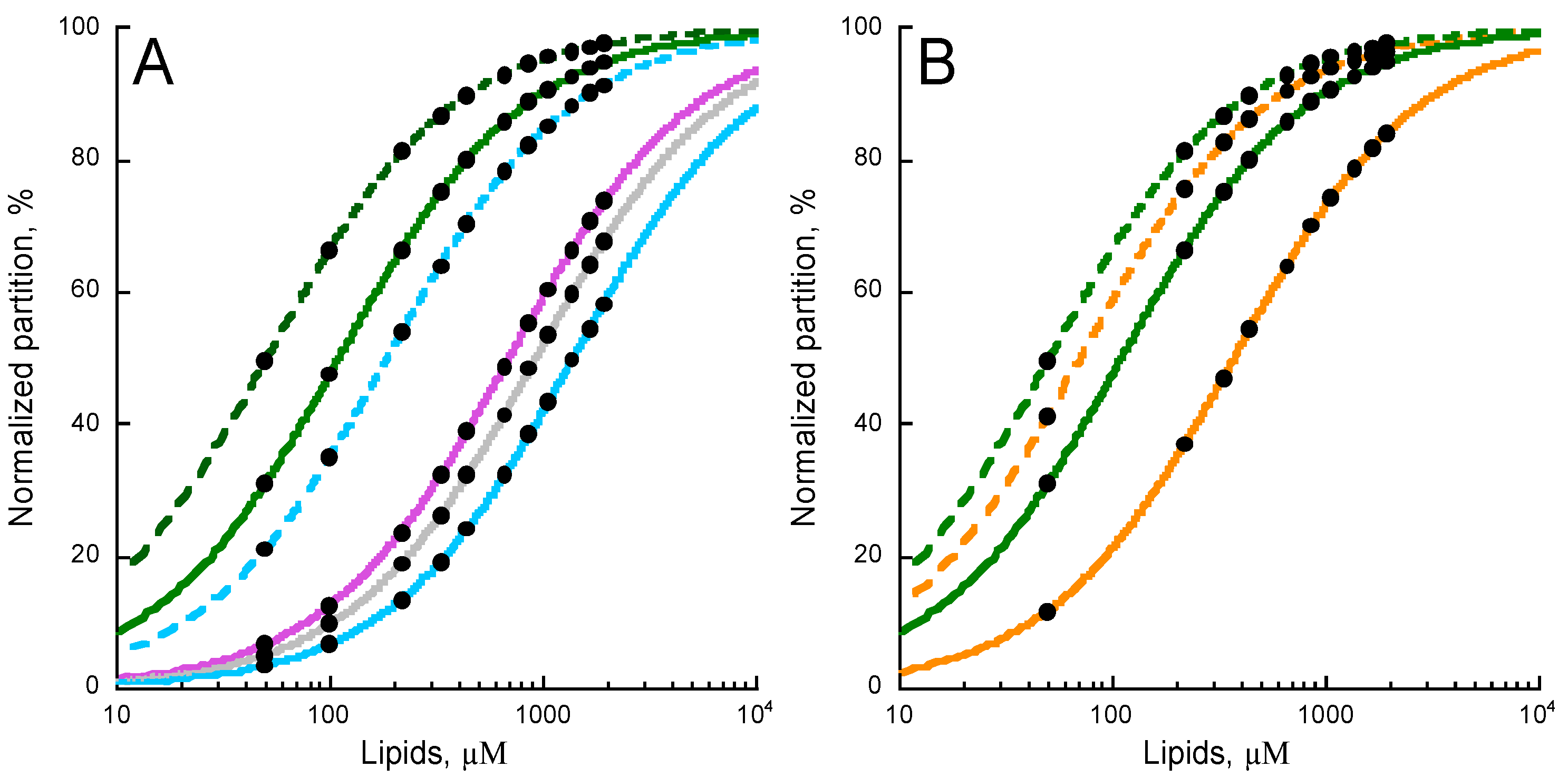
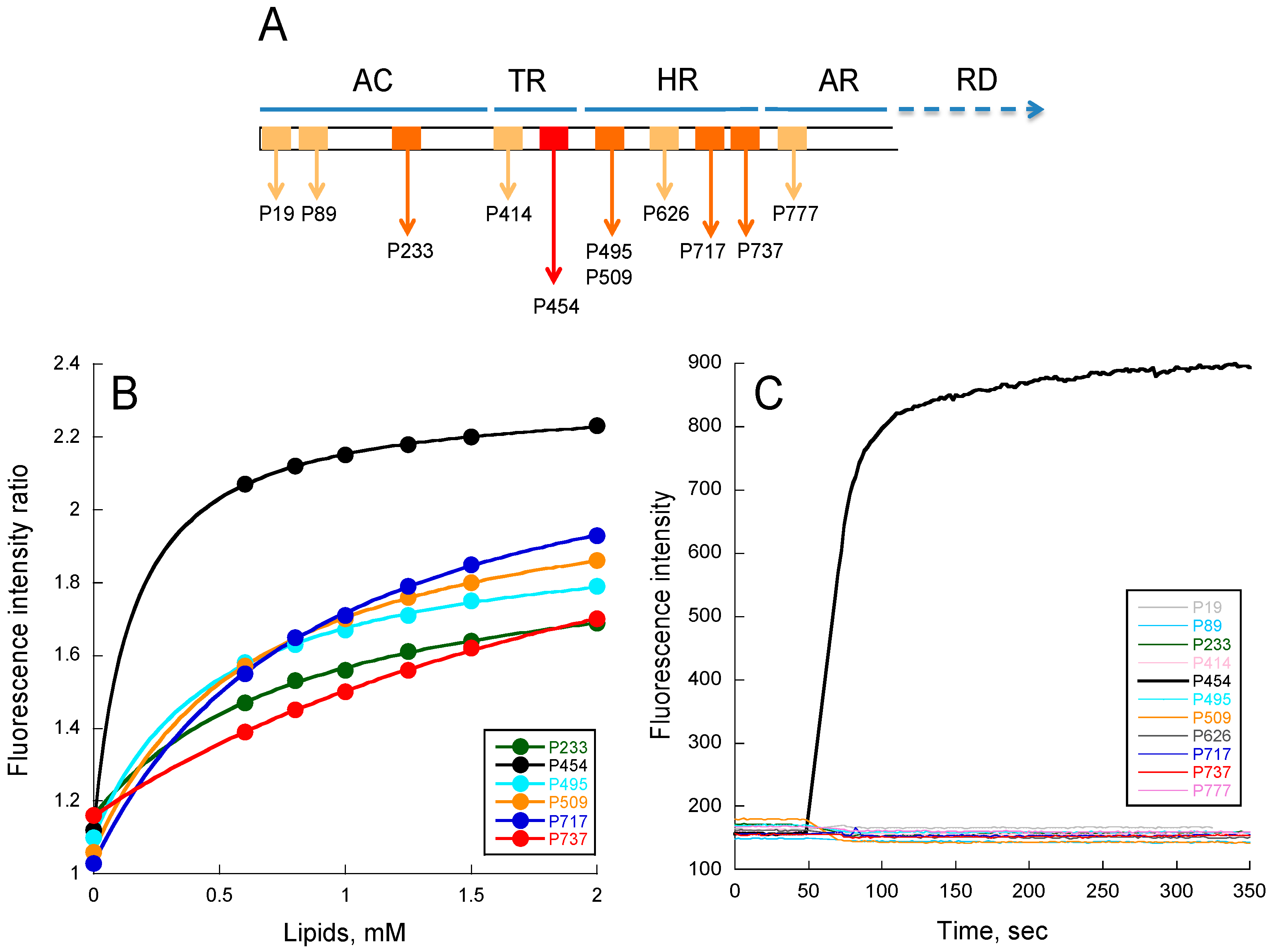
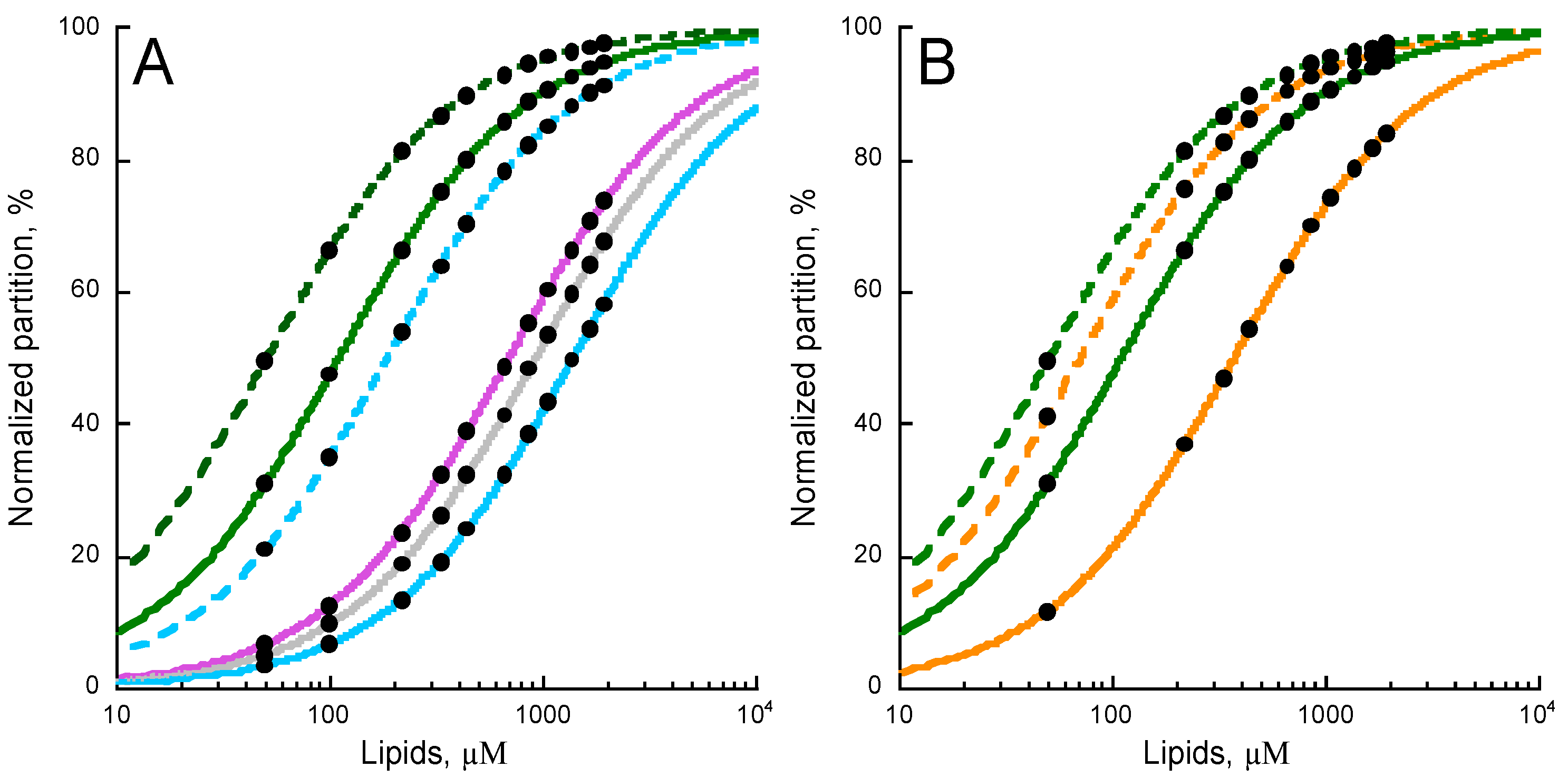
2.1. Experimental Determination of Membrane Partition of CyaA-Derived Peptides
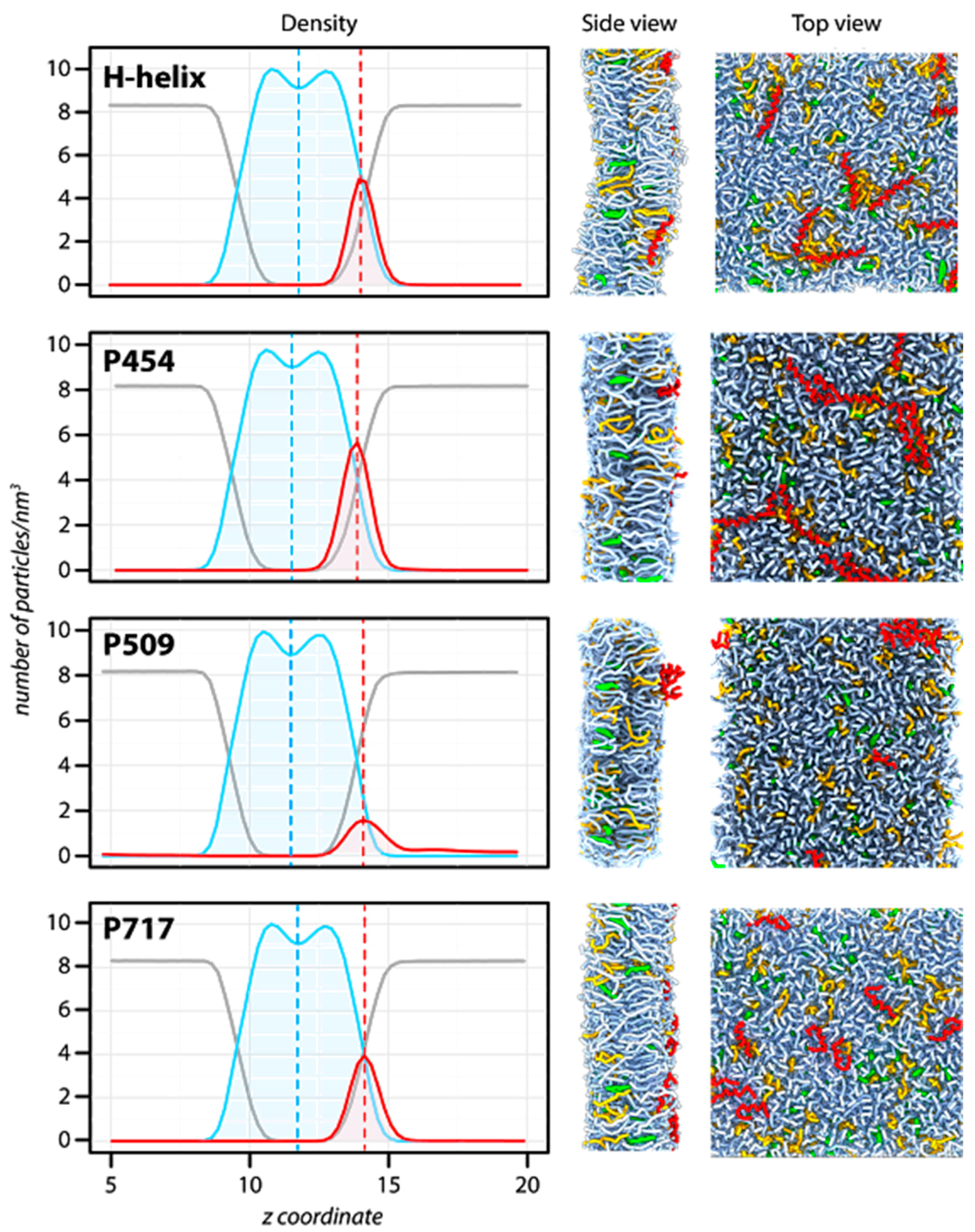
2.2. Molecular Dynamics of CyaA-Derived Peptides in Lipid Bilayers
2.3. Lipid Vesicle Permeabilization
2.4. Effects of Non-Lamellar Lipids, Charge, and Acyl Chains Fluidity on P454 Membrane Partition
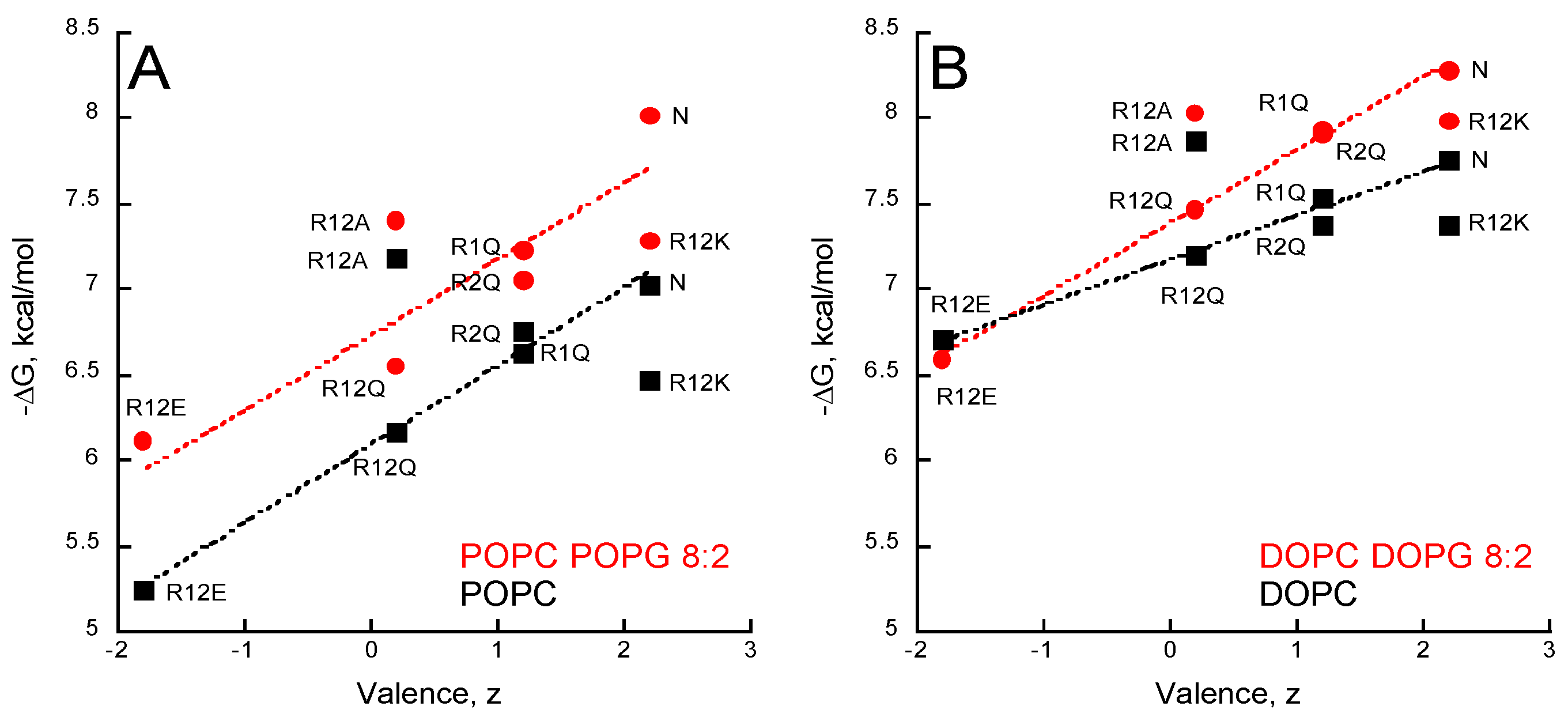
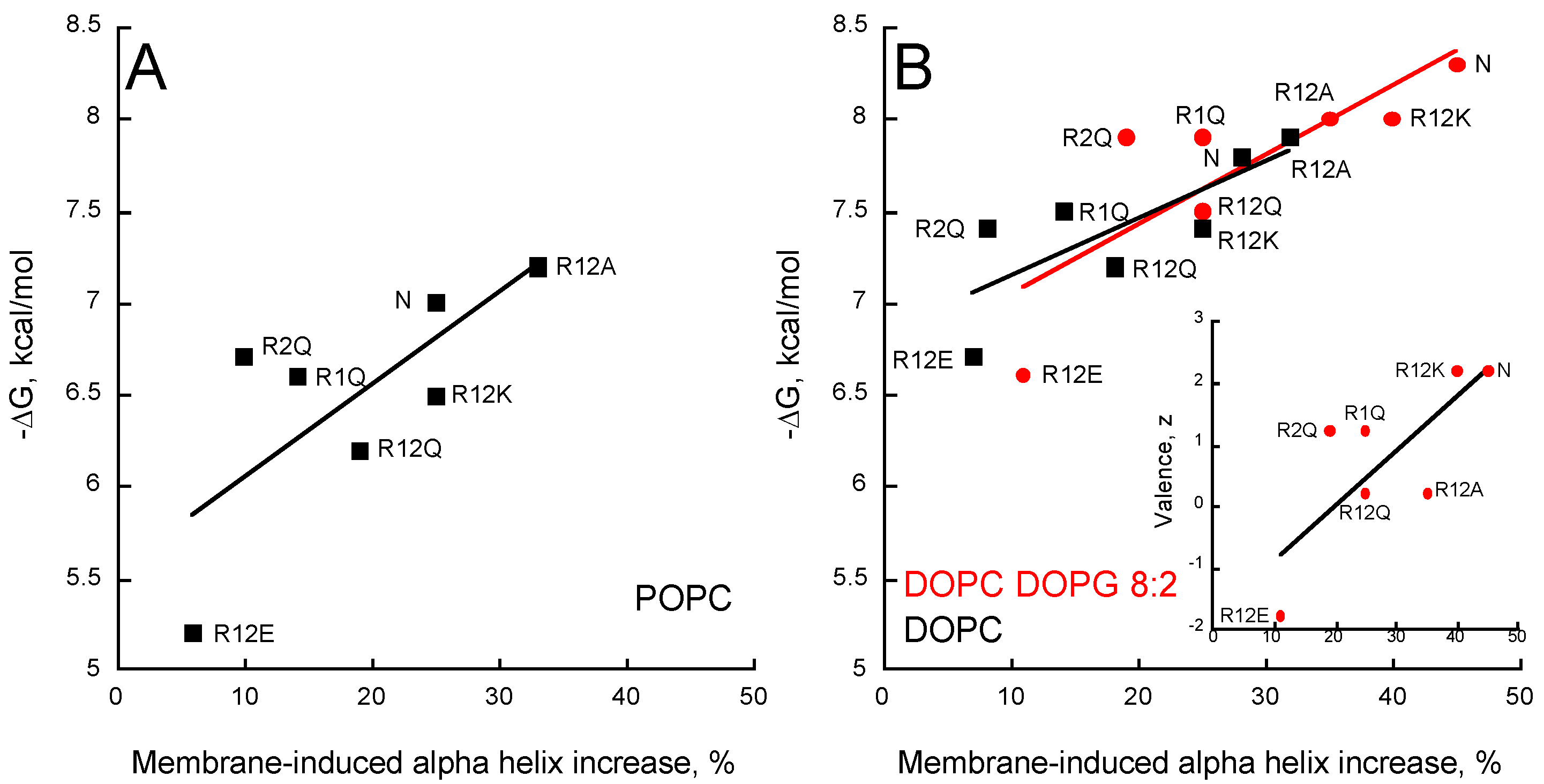
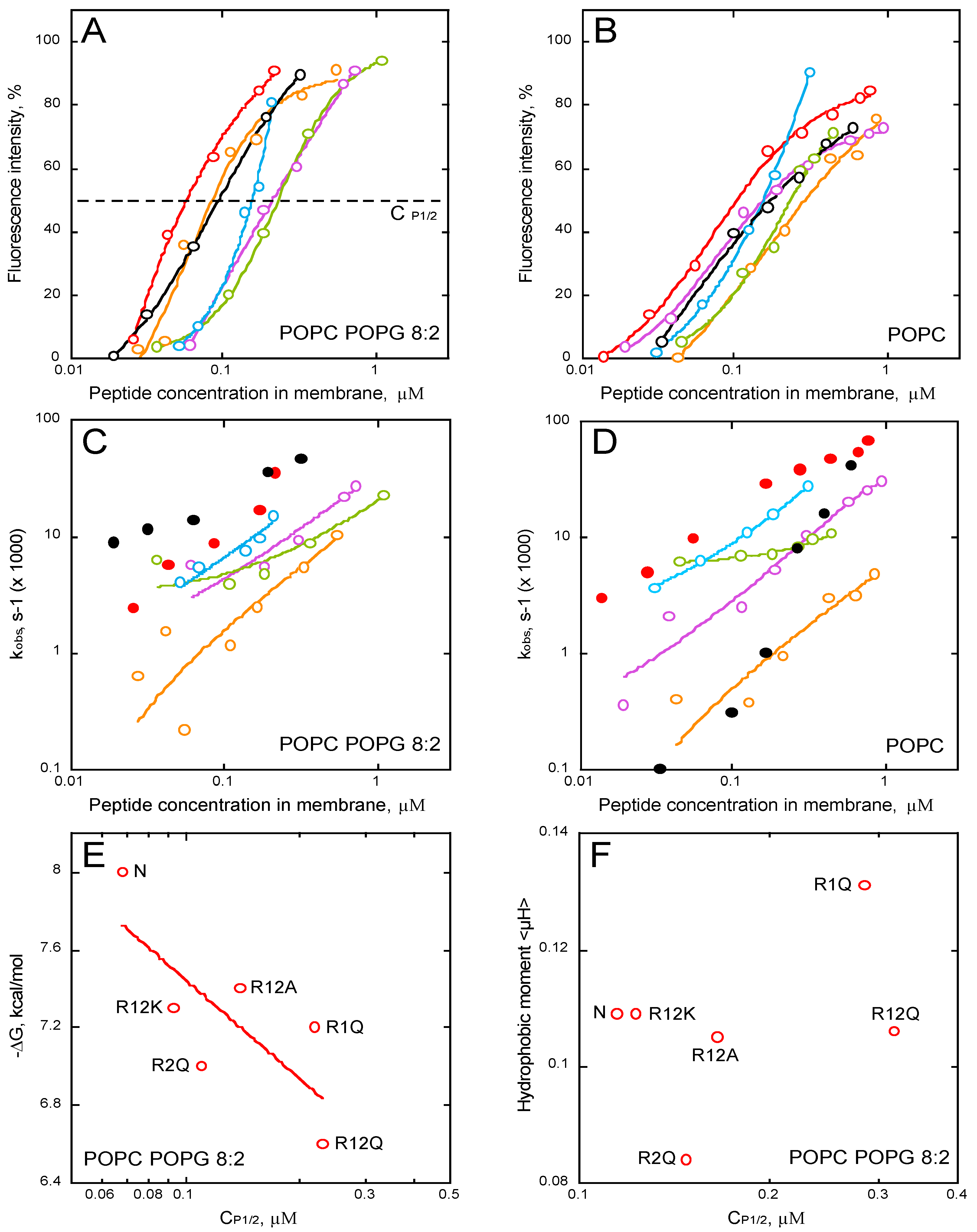
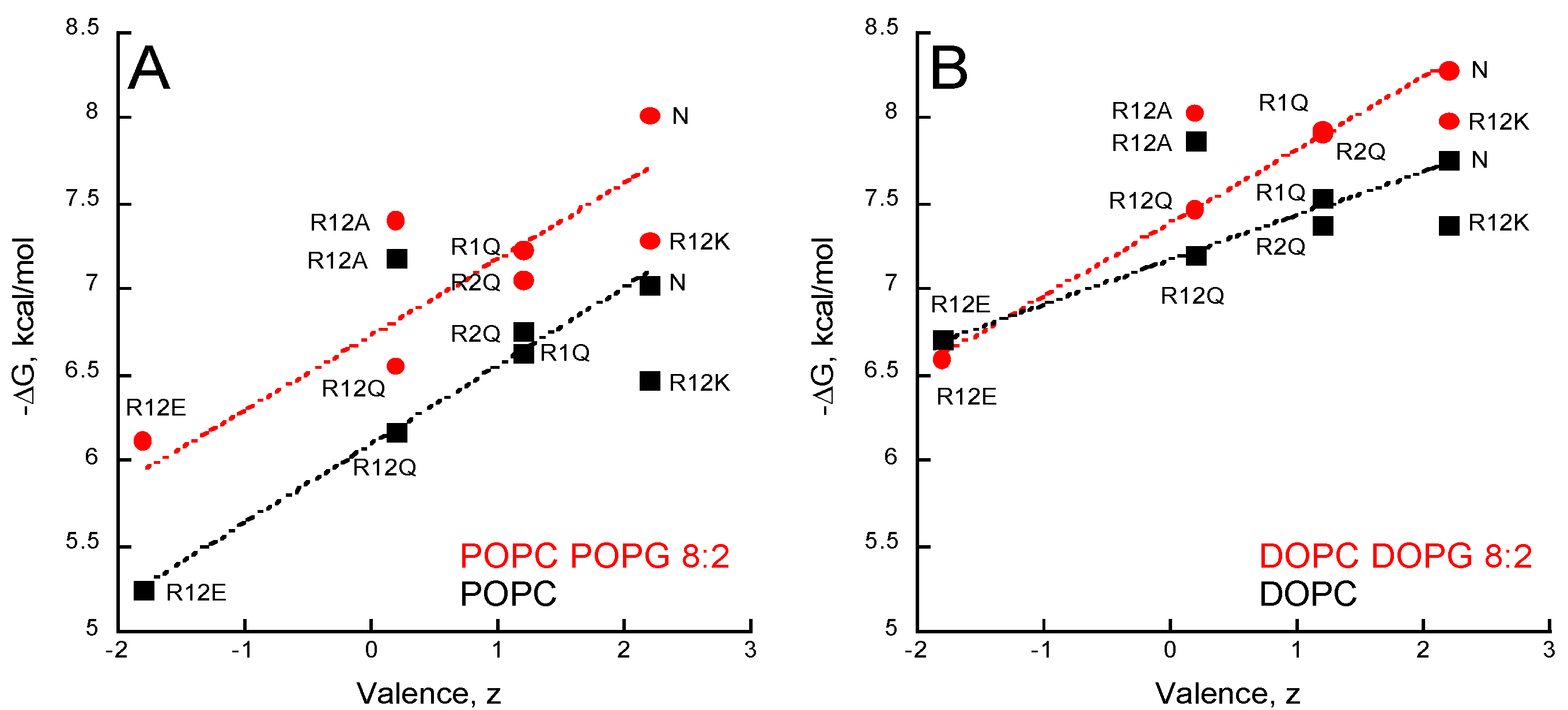
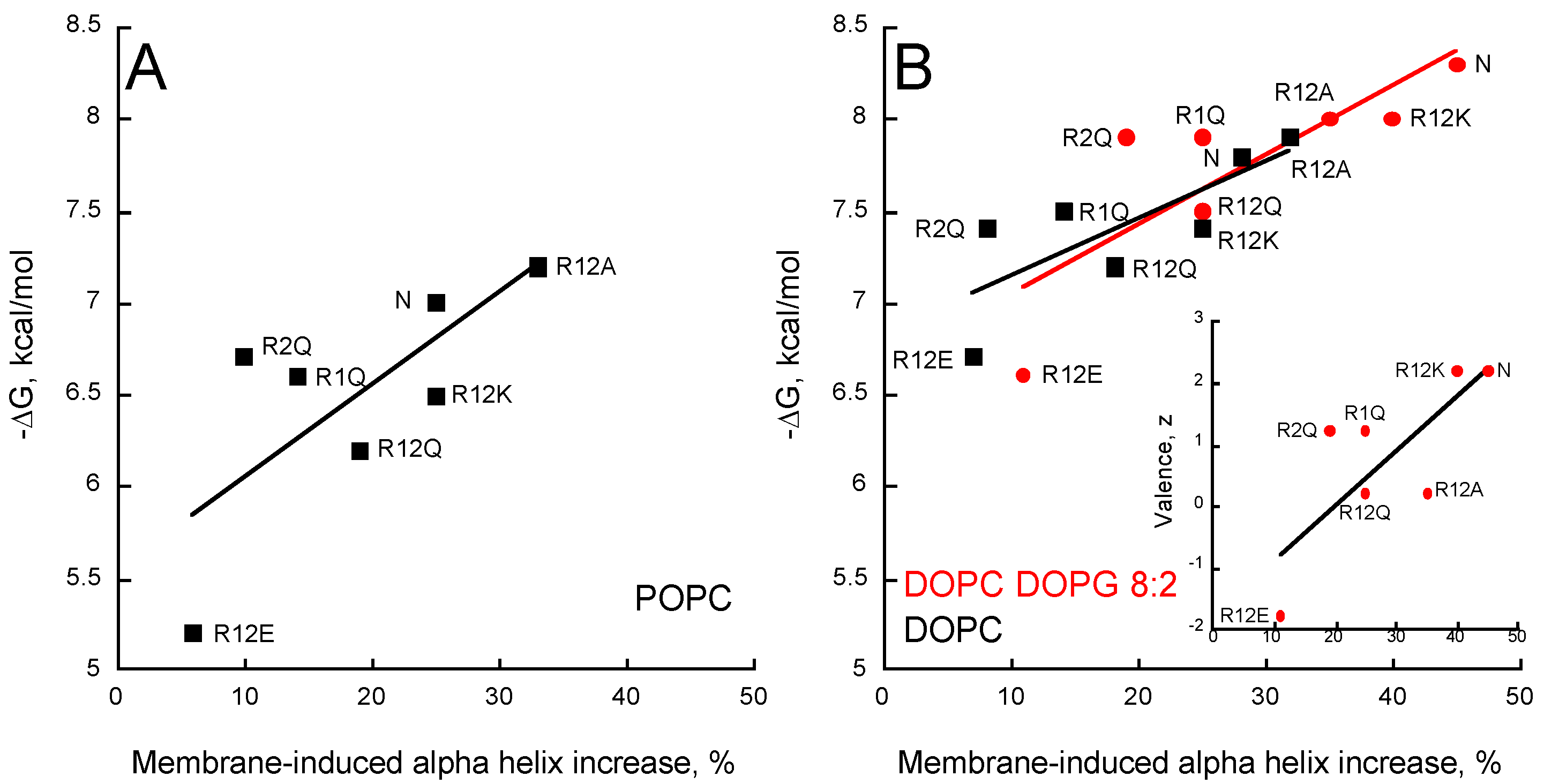
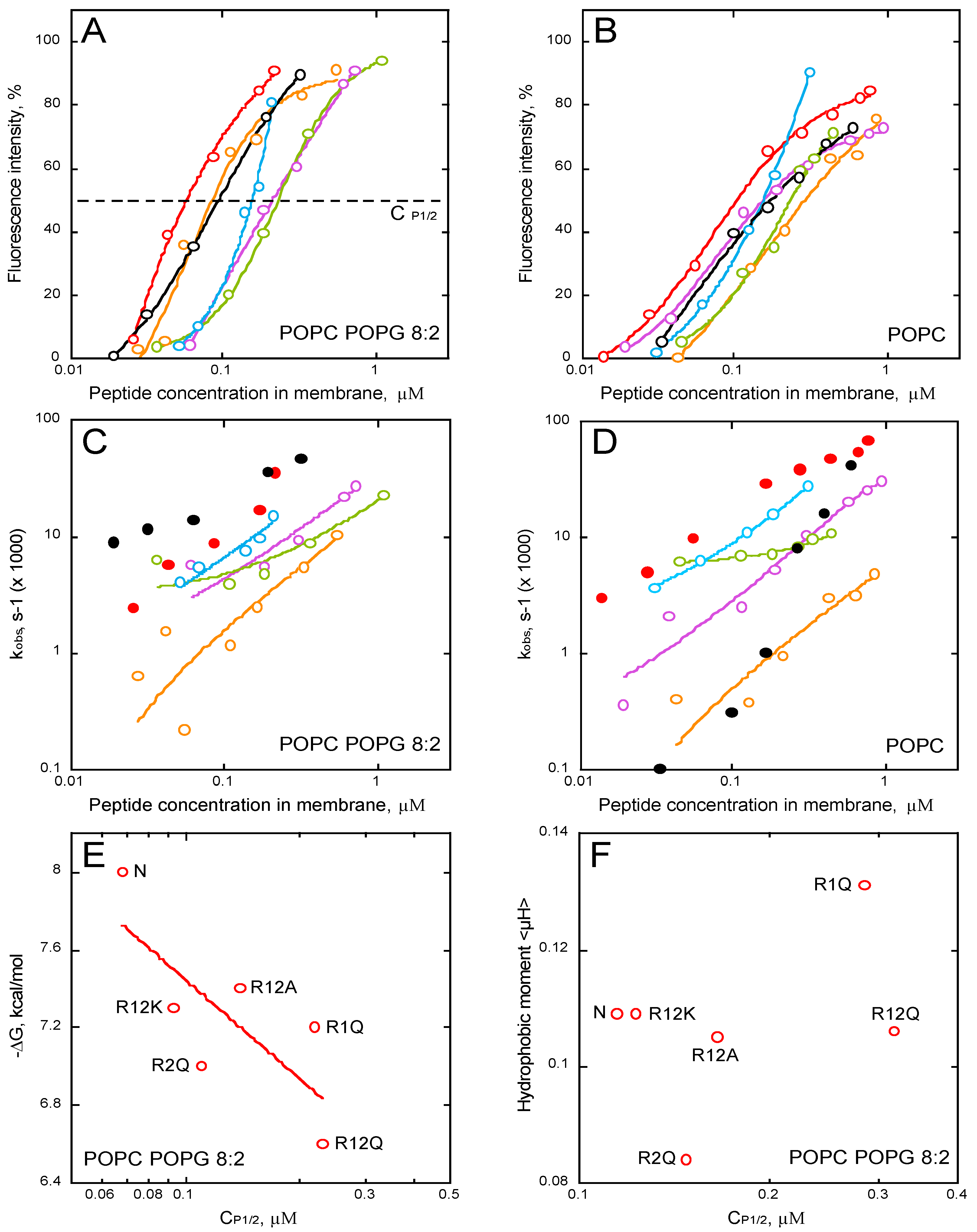
2.5. Membrane Partition of P454-Derived Peptides
2.6. Synchrotron Radiation Circular Dichroism of P454-Derived Peptides
2.7. Membrane Permeabilization
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents
5.2. Peptides
- -
- The P19 peptide corresponds to the B-helix in AC, i.e., residues 19 to 35 of CyaA (IPAAVLDGIKAVAKEKNW), and a tryptophan was added at its C-terminal extremity (underlined).
- -
- The P89 peptide corresponds to the D-helix in AC, i.e., residues 89 to 107 (APEVIARADNDVNSSLAHGW), with a tryptophan added at its C-terminal extremity (underlined).
- -
- The P233 peptide corresponds to the H-helix in AC, i.e., residues 233 to 254 of CyaA (LDRERIDLLWKIARAGARSAVG), and contains the native tryptophan W242.
- -
- The P414 peptide corresponds to residues 414 to 440 of CyaA (SW SLGEVSD- MAAVEAAELEMTRQVLHA), in which Phe-415 was substituted by a tryptophan residue (underlined).
- -
- The P454 peptide corresponds to residues 454 to 484 of CyaA (ASAHWGQRALQGAQAVAAAQRLVHAIALMTQ) and contains the single native tryptophan W458. Several P454-derived peptides were also synthetized, in which the two arginine residues R461 (hereafter referred to as R1) or R474 (referred to as R2), or both of them (referred to R12) were substituted by either lysines (R12K), glutamines (R1Q, R2Q and R12Q), alanines (R12A), or glutamates (R12E).
- -
- The P495 peptide corresponds to residues 495 to 524 of CyaA (QEAASLSAAVFGLGEASSAVAETVSGFFRGW), and a tryptophan was added at its C-terminal extremity (underlined).
- -
- The P509 peptide is a shorter peptide from P495, corresponding to residues 509 to 524 of CyaA (EASSAVAETVSGFFRGW), and a tryptophan was added at its C-terminal extremity (underlined).
- -
- The P626 peptide corresponds to residues 626 to 647 of CyaA (LVQQSHYADQLDKLAQESSAYGW), and a tryptophan was added at its C-terminal extremity (underlined).
- -
- The P717 peptide corresponds to residues 717 to 733 of CyaA (IIEKLANDYARKIDELGW), and a tryptophan was added at its C-terminal extremity (underlined).
- -
- The P737 peptide corresponds to residues 737 to 771 of CyaA (AYFEKNLQARHEQLANSDGLRKMLADLQAGWNASS) and contains the single native tryptophan W767.
- -
- Finally, the P777 peptide corresponds to residues 777 to 793 of CyaA (TTEISKSALELAAITGNW) and a tryptophan was added at its C-terminal extremity (underlined).
5.3. Lipid Vesicles Preparation
5.4. Tryptophan Fluorescence Titrations
5.5. Peptide Partition from Solution to Membranes
5.6. Circular Dichroism
5.7. Membrane Permeabilization
5.8. Molecular Dynamics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ladant, D.; Brezin, C.; Alonso, J.M.; Crenon, I.; Guiso, N. Bordetella pertussis adenylate cyclase. Purification, characterization, and radioimmunoassay. J. Biol. Chem. 1986, 261, 16264–16269. [Google Scholar] [PubMed]
- Bouchez, V.; Guiso, N. Bordetella pertussis, B. parapertussis, vaccines and cycles of whooping cough. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.A.; Hewlett, E.L. Virulence factors of bordetella pertussis. Annu. Rev. Microbiol. 1986, 40, 661–686. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D.; Ullmann, A. Bordatella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Karst, J.C.; Ntsogo Enguene, V.Y.; Cannella, S.E.; Subrini, O.; Hessel, A.; Debard, S.; Ladant, D.; Chenal, A. Calcium, acylation, and molecular confinement favor folding of bordetella pertussis adenylate cyclase CyaA toxin into a monomeric and cytotoxic form. J. Biol. Chem. 2014, 289, 30702–30716. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [PubMed]
- Cannella, S.E.; Ntsogo Enguene, V.Y.; Davi, M.; Malosse, C.; Sotomayor Perez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from bordetella pertussis. Sci. Rep. 2017, 7, 42065. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D. Interaction of bordetella pertussis adenylate cyclase with calmodulin. Identification of two separated calmodulin-binding domains. J. Biol. Chem. 1988, 263, 2612–2618. [Google Scholar] [PubMed]
- Karst, J.C.; Sotomayor Perez, A.C.; Guijarro, J.I.; Raynal, B.; Chenal, A.; Ladant, D. Calmodulin-induced conformational and hydrodynamic changes in the catalytic domain of bordetella pertussis adenylate cyclase toxin. Biochemistry 2010, 49, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Karst, J.C.; Barker, R.; Devi, U.; Swann, M.J.; Davi, M.; Roser, S.J.; Ladant, D.; Chenal, A. Identification of a region that assists membrane insertion and translocation of the catalytic domain of bordetella pertussis CyaA toxin. J. Biol. Chem. 2012, 287, 9200–9212. [Google Scholar] [CrossRef] [PubMed]
- Subrini, O.; Sotomayor-Perez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the bordetella pertussis CyaA toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Sebo, P.; Bellalou, J.; Ladant, D. Interaction of calcium with bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 1995, 270, 26370–26376. [Google Scholar] [CrossRef] [PubMed]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Prongidi-Fix, L.; Perier, A.; Aisenbrey, C.; Vernier, G.; Lambotte, S.; Haertlein, M.; Dauvergne, M.T.; Fragneto, G.; Bechinger, B.; et al. Deciphering membrane insertion of the diphtheria toxin t domain by specular neutron reflectometry and solid-state nmr spectroscopy. J. Mol. Biol. 2009, 391, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Karst, J.C.; Sotomayor Perez, A.C.; Wozniak, A.K.; Baron, B.; England, P.; Ladant, D. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 2010, 99, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor Perez, A.C.; Karst, J.C.; Davi, M.; Guijarro, J.I.; Ladant, D.; Chenal, A. Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 2010, 397, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Perez, A.C.; Ladant, D.; Chenal, A. Calcium-induced folding of intrinsically disordered repeat-in-toxin (RTX) motifs via changes of protein charges and oligomerization states. J. Biol. Chem. 2011, 286, 16997–17004. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Perez, A.C.; Subrini, O.; Hessel, A.; Ladant, D.; Chenal, A. Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 2013, 135, 11929–11934. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, D.P.; Hernandez, B.; Durand, D.; Hourdel, V.; Sotomayor-Perez, A.C.; Vachette, P.; Ghomi, M.; Chamot-Rooke, J.; Ladant, D.; Brier, S.; et al. Structural models of intrinsically disordered and calcium-bound folded states of a protein adapted for secretion. Sci. Rep. 2015, 5, 14223. [Google Scholar] [CrossRef] [PubMed]
- Sotomayor-Perez, A.C.; Ladant, D.; Chenal, A. Disorder-to-order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins 2015, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-driven folding of RTX domain beta-rolls ratchets translocation of RTX proteins through type I secretion ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Rogel, A.; Hanski, E. Distinct steps in the penetration of adenylate cyclase toxin of bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J. Biol. Chem. 1992, 267, 22599–22605. [Google Scholar] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of bordetella pertussis binds to target cells via the alpha(m)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Finetti, F.; Davi, M.; Patrussi, L.; D'Elios, M.M.; Ladant, D.; Baldari, C.T. The bordetella pertussis adenylate cyclase toxin binds to T cells via lFA-1 and induces its disengagement from the immune synapse. J. Exp. Med. 2011, 208, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 2015, 4, e10766. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bullon, D.; Uribe, K.B.; Martin, C.; Ostolaza, H. Phospholipase a activity of adenylate cyclase toxin mediates translocation of its adenylate cyclase domain. Proc. Natl. Acad. Sci. USA 2017, 114, E6784–E6793. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [PubMed]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with hlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [PubMed]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments crucial for membrane translocation and pore-forming activity of bordetella adenylate cyclase toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively charged residues of the segment linking the enzyme and cytolysin moieties restrict the membrane-permeabilizing capacity of adenylate cyclase toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.; Guo, L.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L. Internal lysine palmitoylation in adenylate cyclase toxin from bordetella pertussis. Science 1994, 266, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.; Walker, C.B.; Guo, L.; Gray, M.C.; Van Cuyk, S.; Ullmann, A.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L.; Sebo, P. Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in escherichia coli. J. Biol. Chem. 1995, 270, 20250–20253. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, V.; Higgins, L.; Chen, W.; Halada, P.; Sebo, P.; Sakamoto, H.; Hackett, M. Mass spectrometric analysis of recombinant adenylate cyclase toxin from bordetella pertussis strain 18323/phsp9. J. Mass Spectrom. 2001, 36, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Basar, T.; Havlicek, V.; Bezouskova, S.; Hackett, M.; Sebo, P. Acylation of lysine 983 is sufficient for toxin activity of bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J. Biol. Chem. 2001, 276, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Basler, M.; Knapp, O.; El-Azami-El-Idrissi, M.; Maier, E.; Konopasek, I.; Benz, R.; Leclerc, C.; Sebo, P. Acylation of lysine 860 allows tight binding and cytotoxicity of bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 2005, 44, 12759–12766. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.S.; Yi, X.B.; Gray, M.C.; Szabo, G.; Hewlett, E.L. Membrane depolarization prevents cell invasion by bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 1995, 270, 9695–9697. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, R.; Rossi, C.; Chenal, A.; Devoisselle, J.M.; Ladant, D.; Chopineau, J. Bordetella pertussis adenylate cyclase toxin translocation across a tethered lipid bilayer. Proc. Natl. Acad. Sci. USA 2013, 110, 20473–20478. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Cornell, R.B. Amphitropic proteins: Regulation by reversible membrane interactions (review). Mol. Membr. Biol. 1999, 16, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.B.; Taneva, S.G. Amphipathic helices as mediators of the membrane interaction of amphitropic proteins, and as modulators of bilayer physical properties. Curr. Protein Peptide Sci. 2006, 7, 539–552. [Google Scholar] [CrossRef]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Mihailescu, M.; Soubias, O.; Worcester, D.; White, S.H.; Gawrisch, K. Structure and dynamics of cholesterol-containing polyunsaturated lipid membranes studied by neutron diffraction and NMR. J. Membr. Biol. 2011, 239, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mihailescu, M.; Vaswani, R.G.; Jardon-Valadez, E.; Castro-Roman, F.; Freites, J.A.; Worcester, D.L.; Chamberlin, A.R.; Tobias, D.J.; White, S.H. Acyl-chain methyl distributions of liquid-ordered and -disordered membranes. Biophys. J. 2011, 100, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine is a key regulator of membrane fluidity in eukaryotic cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Detection of hexagonal phase forming propensity in phospholipid bilayers. Biophys. J. 1993, 64, 290–291. [Google Scholar] [CrossRef]
- De Kruijff, B. Lipid polymorphism and biomembrane function. Curr. Opin. Chem. Biol. 1997, 1, 564–569. [Google Scholar] [CrossRef]
- Toombes, G.E.; Finnefrock, A.C.; Tate, M.W.; Gruner, S.M. Determination of L(alpha)-H(II) phase transition temperature for 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine. Biophys. J. 2002, 82, 2504–2510. [Google Scholar] [CrossRef]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Hristova, K.; Wimley, W.C. A look at arginine in membranes. J. Membr. Biol. 2011, 239, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C.; Ladokhin, A.S.; Hristova, K. Protein folding in membranes: Determining energetics of peptide-bilayer interactions. Methods Enzymol. 1998, 295, 62–87. [Google Scholar] [PubMed]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Sani, M.A.; Separovic, F. How membrane-active peptides get into lipid membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Doherty, T.; Waring, A.J.; Ruchala, P.; Hong, M. Roles of arginine and lysine residues in the translocation of a cell-penetrating peptide from 13C, 31P, and 19F solid-state NMR. Biochemistry 2009, 48, 4587–4595. [Google Scholar] [CrossRef] [PubMed]
- Schug, K.A.; Lindner, W. Noncovalent binding between guanidinium and anionic groups: Focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem. Rev. 2005, 105, 67–114. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, S.; Hong, M. Cationic membrane peptides: Atomic-level insight of structure-activity relationships from solid-state NMR. Amino Acids 2013, 44, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Nakase, I.; Fukuda, Y.; Masuda, R.; Oishi, S.; Shimura, K.; Kawaguchi, Y.; Takatani-Nakase, T.; Langel, U.; Graslund, A.; et al. Cxcr4 stimulates macropinocytosis: Implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K.; Blondelle, S.E. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen. 2005, 8, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010, 584, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; Tieleman, D.P. The importance of membrane defects-lessons from simulations. Acc. Chem. Res. 2014, 47, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Nizard, P.; Gillet, D. Structure and function of diphtheria toxin: From pathology to engineering. J. Toxicol. Toxin Rev. 2002, 21, 321–359. [Google Scholar] [CrossRef]
- Galloux, M.; Vitrac, H.; Montagner, C.; Raffestin, S.; Popoff, M.R.; Chenal, A.; Forge, V.; Gillet, D. Membrane interaction of botulinum neurotoxin a translocation (t) domain. The belt region is a regulatory loop for membrane interaction. J. Biol. Chem. 2008, 283, 27668–27676. [Google Scholar] [CrossRef] [PubMed]
- Varela Chavez, C.; Haustant, G.M.; Baron, B.; England, P.; Chenal, A.; Pauillac, S.; Blondel, A.; Popoff, M.R. The tip of the four n-terminal alpha-helices of clostridium sordellii lethal toxin contains the interaction site with membrane phosphatidylserine facilitating small GTPases glucosylation. Toxins 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Varela Chavez, C.; Hoos, S.; Haustant, G.M.; Chenal, A.; England, P.; Blondel, A.; Pauillac, S.; Lacy, D.B.; Popoff, M.R. The catalytic domains of clostridium sordellii lethal toxin and related large clostridial glucosylating toxins specifically recognize the negatively charged phospholipids phosphatidylserine and phosphatidic acid. Cell. Microbiol. 2015, 17, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Araye, A.; Goudet, A.; Barbier, J.; Pichard, S.; Baron, B.; England, P.; Perez, J.; Zinn-Justin, S.; Chenal, A.; Gillet, D. The translocation domain of botulinum neurotoxin a moderates the propensity of the catalytic domain to interact with membranes at acidic pH. PLoS ONE 2016, 11, e0153401. [Google Scholar]
- Reig, N.; van der Goot, F.G. About lipids and toxins. FEBS Lett. 2006, 580, 5572–5579. [Google Scholar] [CrossRef] [PubMed]
- Montagner, C.; Perier, A.; Pichard, S.; Vernier, G.; Menez, A.; Gillet, D.; Forge, V.; Chenal, A. Behavior of the n-terminal helices of the diphtheria toxin t domain during the successive steps of membrane interaction. Biochemistry 2007, 46, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Vernier, G.; Savarin, P.; Bushmarina, N.A.; Geze, A.; Guillain, F.; Gillet, D.; Forge, V. Conformational states and thermodynamics of alpha-lactalbumin bound to membranes: A case study of the effects of pH, calcium, lipid membrane curvature and charge. J. Mol. Biol. 2005, 349, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2003, 108, 750–760. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The martini force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The martini coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101–014107. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An n.Log(n) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 27–28. [Google Scholar] [CrossRef]
- R-Development-Core-Team. R: A Language and Environment for Statistical Computing; R-Development-Core-Team: Vienna, Austria, 2005. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voegele, A.; Subrini, O.; Sapay, N.; Ladant, D.; Chenal, A. Membrane-Active Properties of an Amphitropic Peptide from the CyaA Toxin Translocation Region. Toxins 2017, 9, 369. https://doi.org/10.3390/toxins9110369
Voegele A, Subrini O, Sapay N, Ladant D, Chenal A. Membrane-Active Properties of an Amphitropic Peptide from the CyaA Toxin Translocation Region. Toxins. 2017; 9(11):369. https://doi.org/10.3390/toxins9110369
Chicago/Turabian StyleVoegele, Alexis, Orso Subrini, Nicolas Sapay, Daniel Ladant, and Alexandre Chenal. 2017. "Membrane-Active Properties of an Amphitropic Peptide from the CyaA Toxin Translocation Region" Toxins 9, no. 11: 369. https://doi.org/10.3390/toxins9110369
APA StyleVoegele, A., Subrini, O., Sapay, N., Ladant, D., & Chenal, A. (2017). Membrane-Active Properties of an Amphitropic Peptide from the CyaA Toxin Translocation Region. Toxins, 9(11), 369. https://doi.org/10.3390/toxins9110369