
Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication


 ,
,  and
and 
Abstract
:
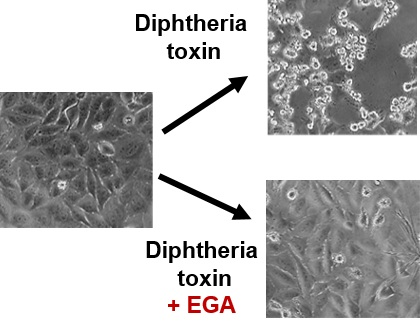{kind=link}
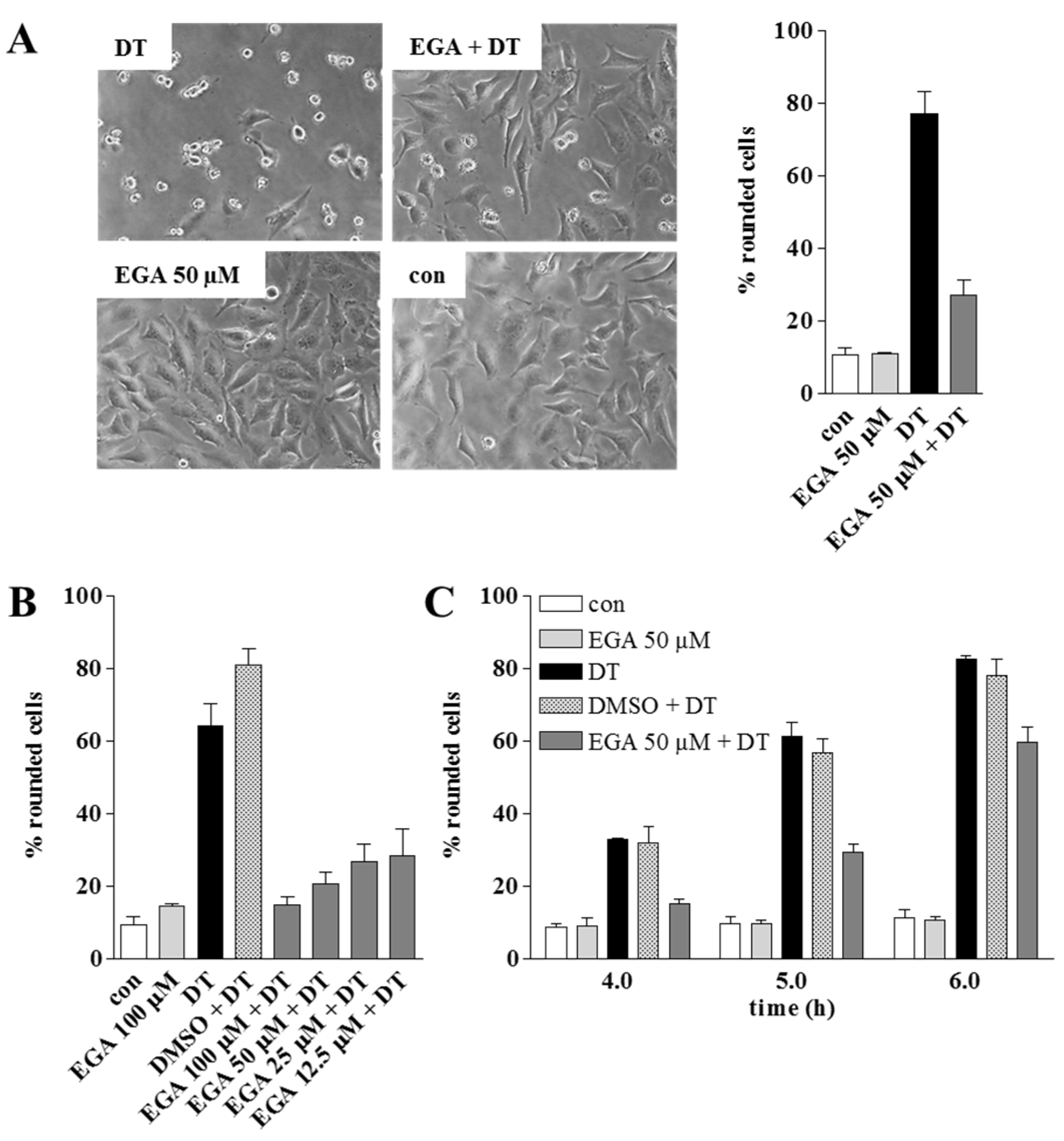{kind=link}
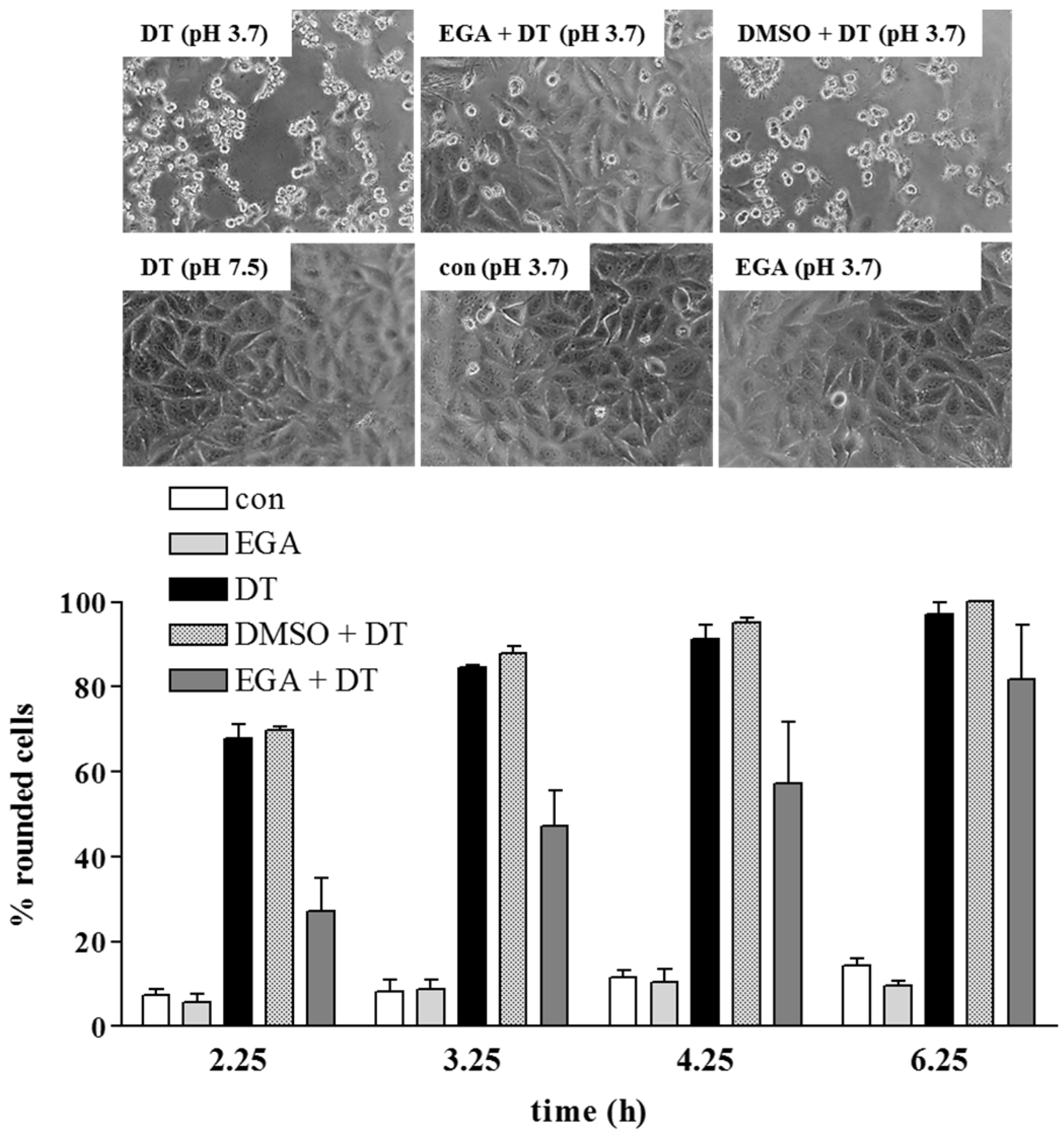{kind=link}
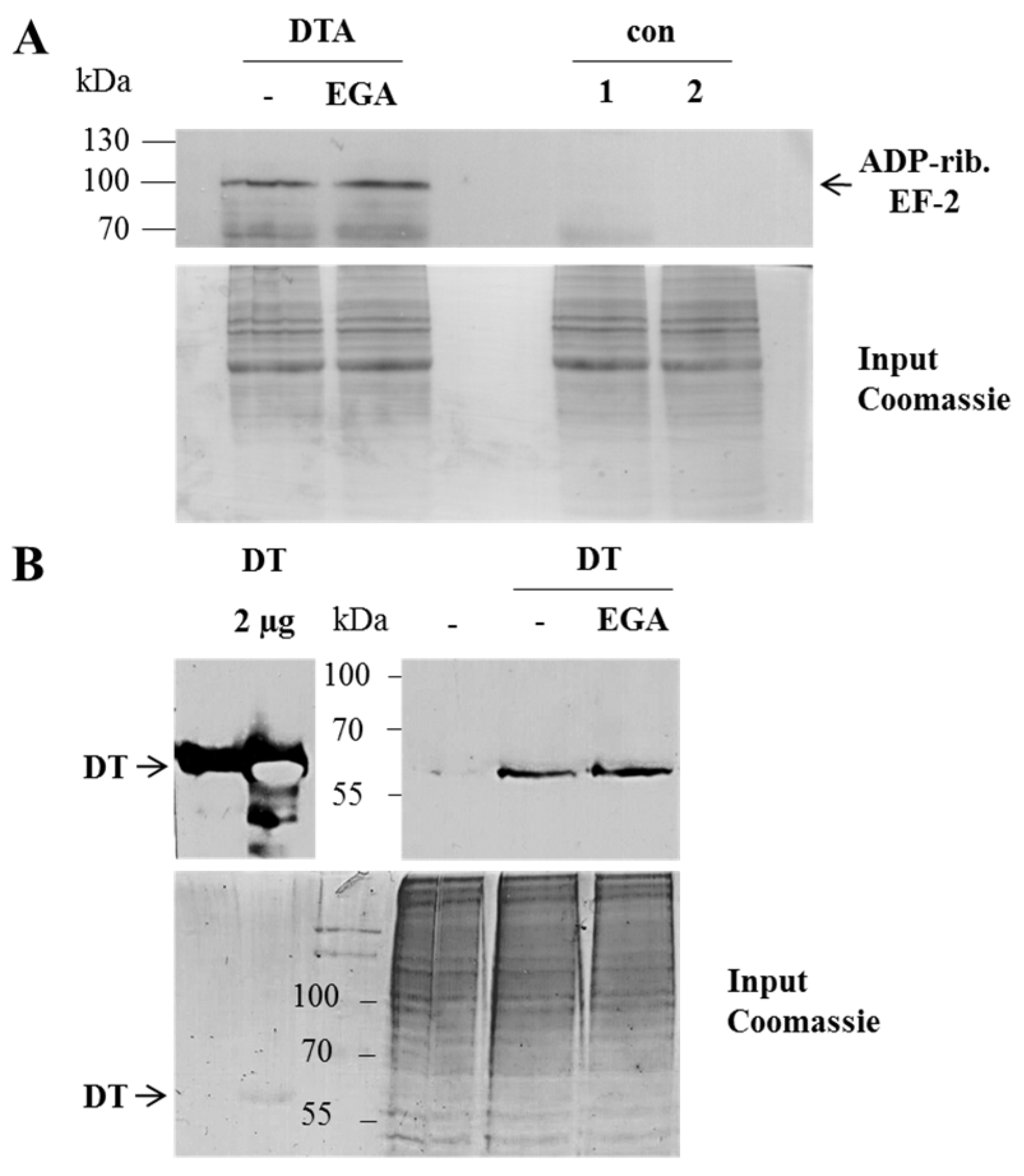{kind=link}
{kind=link}
1. Introduction
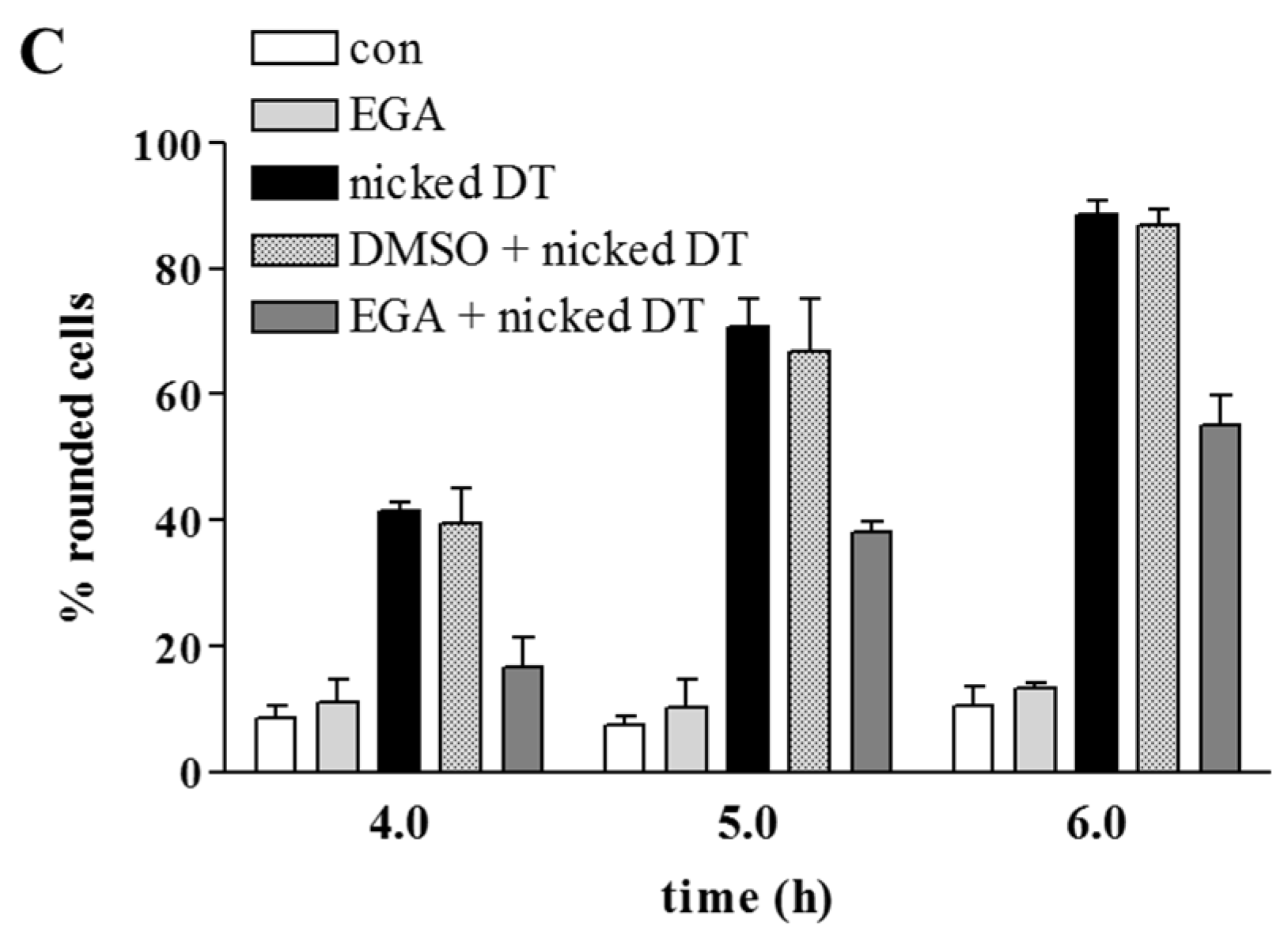
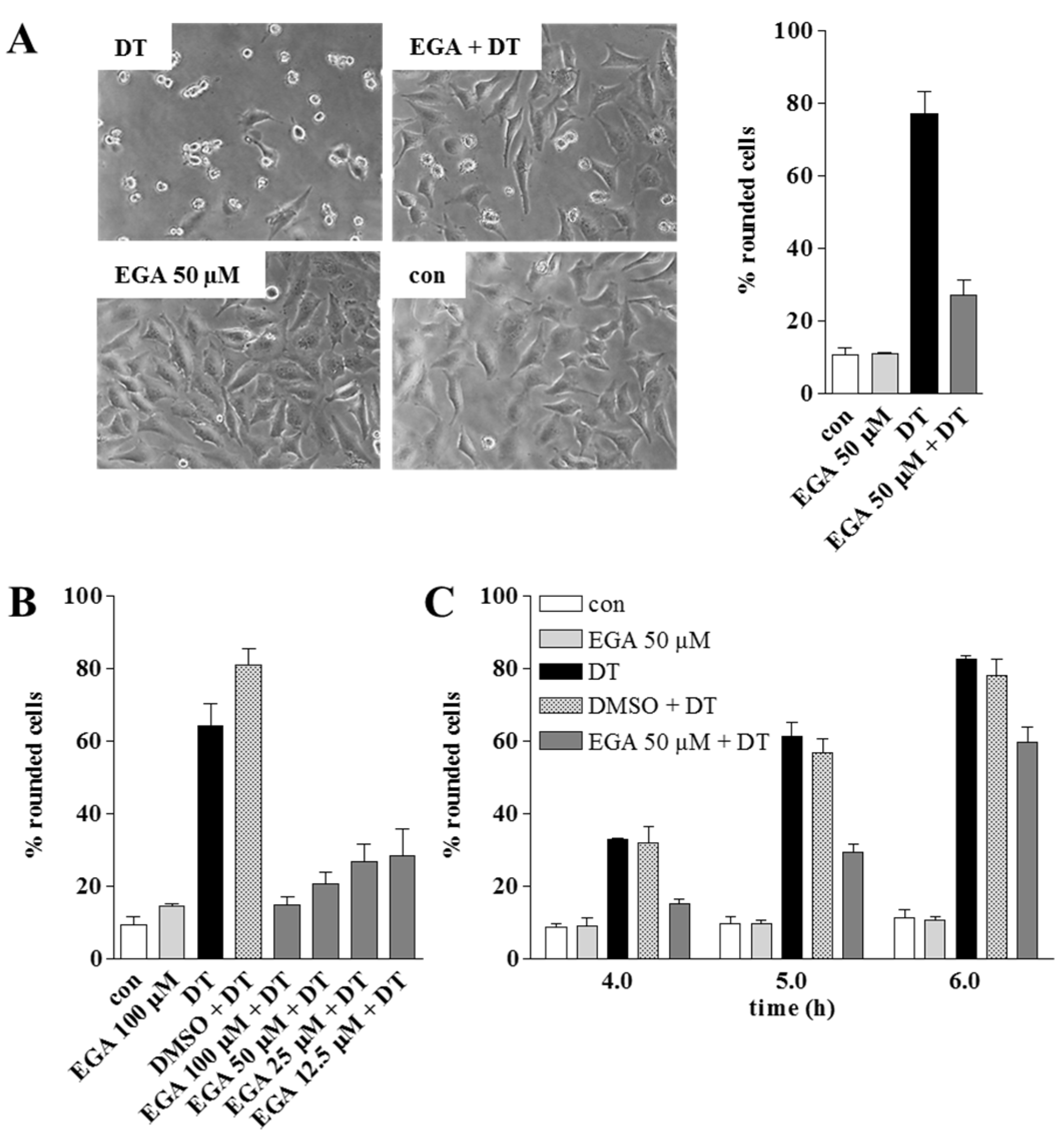
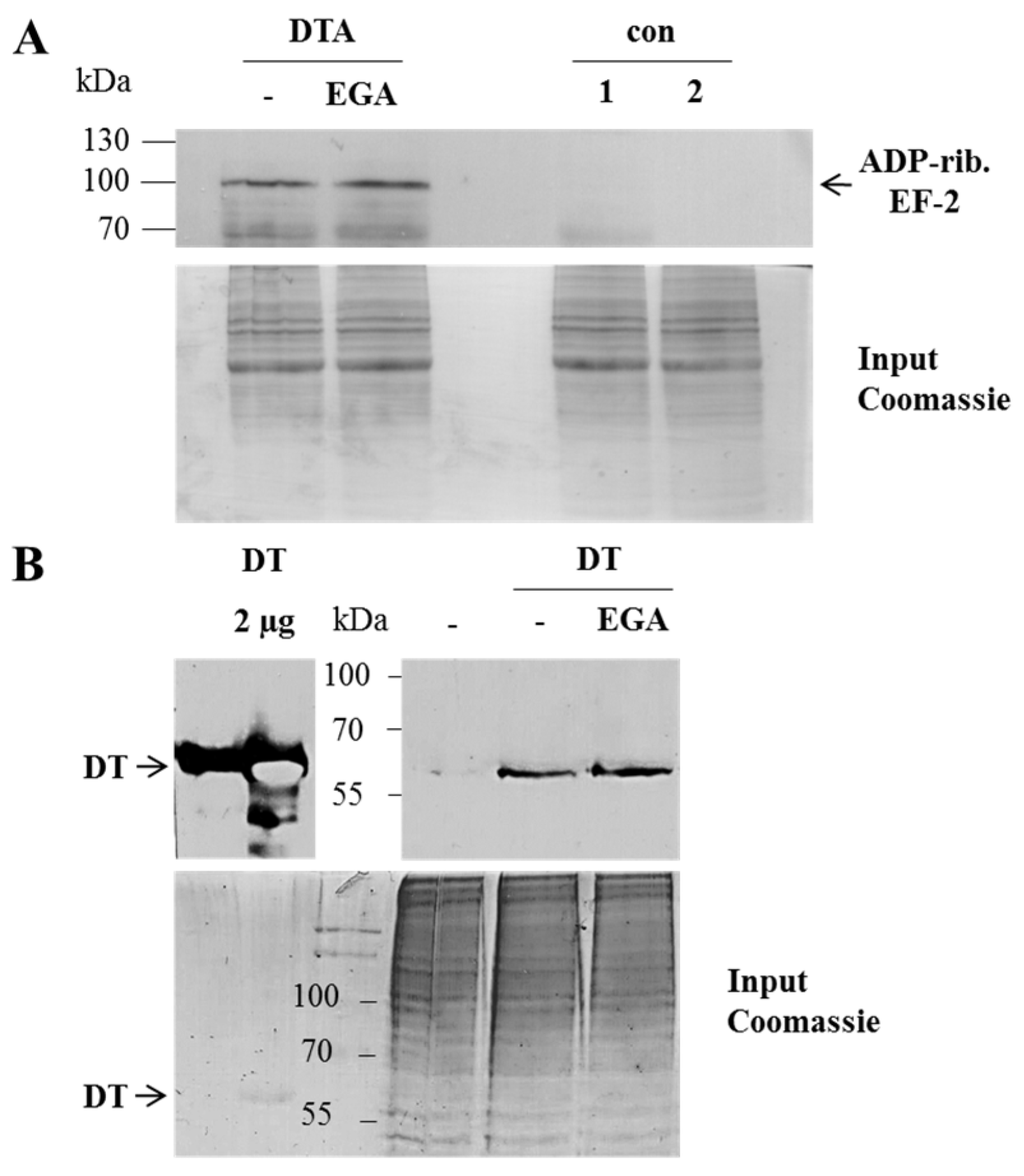
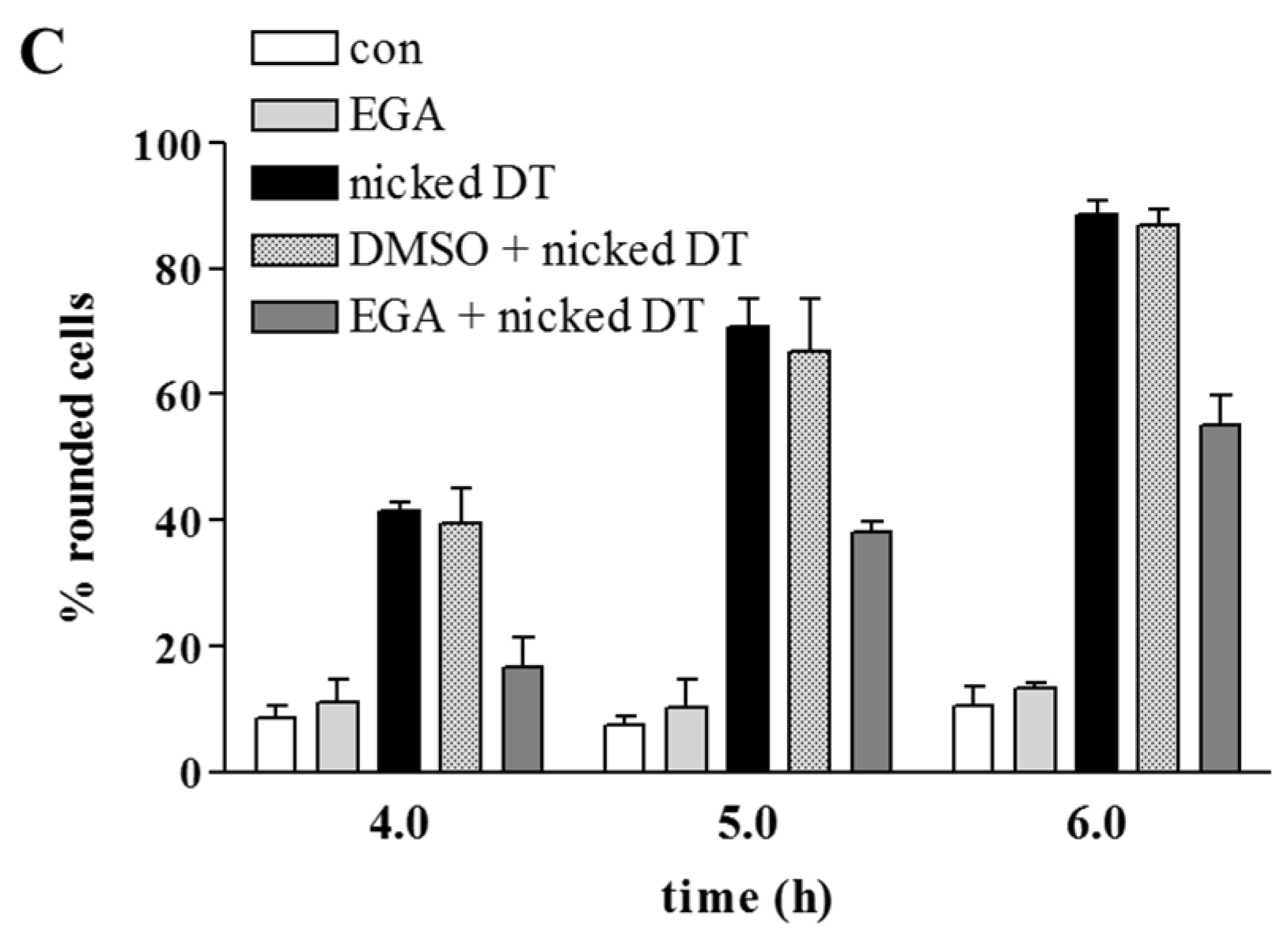
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. Materials and Reagents
4.2. Cell Culture and Cytotoxicity Assays
4.3. SDS-PAGE and Western Blotting
4.4. Proteolytic Activation (Nicking) of DT
4.5. ADP-Ribosylation of EF-2 by DTA in a Cell-Free System
4.6. Binding of DT to Its Cell Surface Receptor
4.7. DTA Translocation Assay across the Cytoplasmic Membrane of Living Cells
4.8. Reproducibility of the Experiments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, A.M. Diphtheria toxin. Annu. Rev. Biochem. 1977, 46, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Cole, H.A. Diphtheria toxin subunit active in vitro. Science 1969, 164, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Schnell, L.; Dmochewitz-Kück, L.; Feigl, P.; Montecucco, C.; Barth, H. Thioredoxin reductase inhibitor auranofin prevents membrane transport of diphtheria toxin into the cytosol and protects human cells from intoxication. Toxicon 2016, 116, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Kandel, J. Structure and activity of diphtheria toxin. I. Thiol-dependent dissociation of a fraction of toxin into enzymically active and inactive fragments. J. Biol. Chem. 1971, 246, 1496–1503. [Google Scholar] [PubMed]
- Iwamoto, R.; Higashiyama, S.; Mitamura, T.; Taniguchi, N.; Klagsbrun, M.; Mekada, E. Heparin-binding EGF-like growth factor, which acts as the diphtheria toxin receptor, forms a complex with membrane protein DRAP27/CD9, which up-regulates functional receptors and diphtheria toxin sensitivity. EMBO J. 1994, 13, 2322–2330. [Google Scholar] [PubMed]
- Naglich, J.G.; Metherall, J.E.; Russell, D.W.; Eidels, L. Expression cloning of a diphtheria toxin receptor: Identity with a heparin-binding EGF-like growth factor precursor. Cell 1992, 69, 1051–1061. [Google Scholar] [CrossRef]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.G.; Nicholls, P.J.; Habig, W.H.; Youle, R.J. The role of proline 345 in diphtheria toxin translocation. J. Biol. Chem. 1993, 268, 3514–3519. [Google Scholar] [PubMed]
- Perier, A.; Chassaing, A.; Raffestin, S.; Pichard, S.; Masella, M.; Ménez, A.; Forge, V.; Chenal, A.; Gillet, D. Concerted protonation of key histidines triggers membrane interaction of the diphtheria toxin T domain. J. Biol. Chem. 2007, 282, 24239–24245. [Google Scholar] [CrossRef] [PubMed]
- Drazin, R.; Kandel, J.; Collier, R.J. Structure and activity of DT II. Attack by trypsin at a specific site within the intact molecule. J. Biol. Chem. 1971, 246, 1504–1510. [Google Scholar] [PubMed]
- Papini, E.; Sandoná, D.; Rappuoli, R.; Montecucco, C. On the membrane translocation of diphtheria toxin: At low pH the toxin induces ion channels on cells. EMBO J. 1988, 7, 3353–3359. [Google Scholar] [PubMed]
- Lemichez, E.; Bomsel, M.; Devilliers, G.; vanderSpek, J.; Murphy, J.R.; Lukianov, E.V.; Olsnes, S.; Boquet, P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 1997, 23, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Wiedlocha, A.; Madshus, I.H.; Mach, H.; Middaugh, C.R.; Olsnes, S. Tight folding of acidic fibroblast growth factor prevents its translocation to the cytosol with diphtheria toxin as vector. EMBO J. 1992, 11, 4835–4842. [Google Scholar] [PubMed]
- Falnes, P.O.; Choe, S.; Madshus, I.H.; Wilson, B.A.; Olsnes, S. Inhibition of membrane translocation of diphtheria toxin A-fragment by internal disulfide bridges. J. Biol. Chem. 1994, 269, 8402–8407. [Google Scholar] [PubMed]
- Stenmark, H.; Olsnes, S.; Madshus, I.H. Elimination of the disulphide bridge in fragment B of diphtheria toxin: Effect on membrane insertion, channel formation, and ATP binding. Mol. Microbiol. 1991, 5, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Leka, O.; Vallese, F.; Pirazzini, M.; Berto, P.; Montecucco, C.; Zanotti, G. Diphtheria toxin conformational switching at acidic pH. FEBS J. 2014, 281, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Umata, T.; Moriyama, Y.; Futai, M.; Mekada, E. The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase. J. Biol. Chem. 1990, 265, 21940–21945. [Google Scholar] [PubMed]
- Sandvig, K.; Olsnes, S. Rapid entry of nicked diphtheria toxin into cells at low pH. Characterization of the entry process and effects of low pH on the toxin molecule. J. Biol. Chem. 1981, 256, 9068–9076. [Google Scholar] [PubMed]
- Sandvig, K.; Olsnes, S. Diphtheria toxin-induced channels in Vero cells selective for monovalent cations. J. Biol. Chem. 1988, 263, 12352–12359. [Google Scholar] [PubMed]
- Moskaug, J.O.; Stenmark, H.; Olsnes, S. Insertion of diphtheria toxin B-fragment into the plasma membrane at low pH. Characterization and topology of inserted regions. J. Biol. Chem. 1991, 266, 2652–2659. [Google Scholar] [PubMed]
- Falnes, P.O.; Madshus, I.H.; Sandvig, K.; Olsnes, S. Replacement of negative by positive charges in the presumed membrane-inserted part of diphtheria toxin B fragment. Effect on membrane translocation and on formation of cation channels. J. Biol. Chem. 1992, 267, 12284–12290. [Google Scholar] [PubMed]
- Ratts, R.; Zeng, H.; Berg, E.A.; Blue, C.; McComb, M.E.; Costello, C.E.; vanderSpek, J.C.; Murphy, R. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J. Cell Biol. 2003, 160, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Dmochewitz, L.; Lillich, M.; Kaiser, E.; Jennings, L.D.; Lang, A.E.; Buchner, J.; Fischer, G.; Aktories, K.; Collier, R.J.; Barth, H. Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell. Microbiol. 2011, 13, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tsuneoka, M.; Nakayama, K.; Hatsuzawa, K.; Komada, M.; Kitamura, N.; Mekada, E. Evidence for the involvement of furin in cleavage and activation of diphtheria toxin. J. Biol. Chem. 1993, 268, 26461–26465. [Google Scholar] [PubMed]
- Gill, D.M.; Pappenheimer, A.M. Structure activity relationships in diphtheria toxin. J. Biol. Chem. 1971, 246, 1485–1491. [Google Scholar] [PubMed]
- Ariansen, S.; Afanasiev, B.N.; Moskaug, J.O.; Stenmark, H.; Madhaus, I.H.; Olsnes, S. Membrane translocation of diphtheria toxin A-fragment: Role of carboxy-terminal region. Biochemistry 1993, 32, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Rappuoli, R.; Murgia, M.; Montecucco, C. Cell penetration of diphtheria toxin. Reduction of the interchain disulfide bridge is the rate-limiting step of translocation in the cytosol. J. Biol. Chem. 1993, 268, 1567–1574. [Google Scholar] [PubMed]
- Madshus, I.H.; Wiedlocha, A.; Sandvig, K. Intermediates in translocation of diphtheria toxin across the plasma membrane. J. Biol. Chem. 1994, 269, 4648–4652. [Google Scholar] [PubMed]
- Moskaug, J.O.; Sandvig, K.; Olsnes, S. Cell-mediated reduction of the interfragment disulfide in nicked DT. A new system to study toxin entry at low pH. J. Biol. Chem. 1987, 262, 10339–10345. [Google Scholar] [PubMed]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, E.J.; Ho, C.L.; Balaji, K.; Clemens, D.L.; Deng, G.; Wang, Y.E.; Elsaesser, H.J.; Tamilselvam, B.; Gargi, A.; Dixon, S.D.; et al. Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E4904–E4912. [Google Scholar] [CrossRef] [PubMed]
- Schnell, L.; Mittler, A.K.; Sadi, M.; Popoff, M.R.; Schwan, C.; Aktories, K.; Mattarei, A.; Tehran, D.A.; Montecucco, C.; Barth, H. EGA protects mammalian cells from Clostridium difficile CDT, Clostridium perfringens iota toxin, and Clostridium botulinum C2 toxin. Toxins 2016, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Tehran, D.A.; Zanetti, G.; Leka, O.; Lista, F.; Fillo, S.; Binz, T.; Shone, C.C.; Rossetto, O.; Montecucco, C.; Paradisi, C.; et al. A Novel inhibitor prevents the peripheral neuroparalysis of botulinum neurotoxins. Sci. Rep. 2015, 5, 17513. [Google Scholar] [CrossRef] [PubMed]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnell, L.; Mittler, A.-K.; Mattarei, A.; Tehran, D.A.; Montecucco, C.; Barth, H. Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication. Toxins 2016, 8, 221. https://doi.org/10.3390/toxins8070221
Schnell L, Mittler A-K, Mattarei A, Tehran DA, Montecucco C, Barth H. Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication. Toxins. 2016; 8(7):221. https://doi.org/10.3390/toxins8070221
Chicago/Turabian StyleSchnell, Leonie, Ann-Katrin Mittler, Andrea Mattarei, Domenico Azarnia Tehran, Cesare Montecucco, and Holger Barth. 2016. "Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication" Toxins 8, no. 7: 221. https://doi.org/10.3390/toxins8070221
APA StyleSchnell, L., Mittler, A.-K., Mattarei, A., Tehran, D. A., Montecucco, C., & Barth, H. (2016). Semicarbazone EGA Inhibits Uptake of Diphtheria Toxin into Human Cells and Protects Cells from Intoxication. Toxins, 8(7), 221. https://doi.org/10.3390/toxins8070221