
Evaluation of the Impact of Mild Steaming and Heat Treatment on the Concentration of Okadaic Acid, Dinophysistoxin-2 and Dinophysistoxin-3 in Mussels



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Influence of Weight in the Processing of Samples
2.2. Influence of Steaming on Weight
2.3. Influence of Steaming on Hydrolysis
2.4. Influence of Steaming and Autoclaving on DTX3 Levels with no Loss of Water
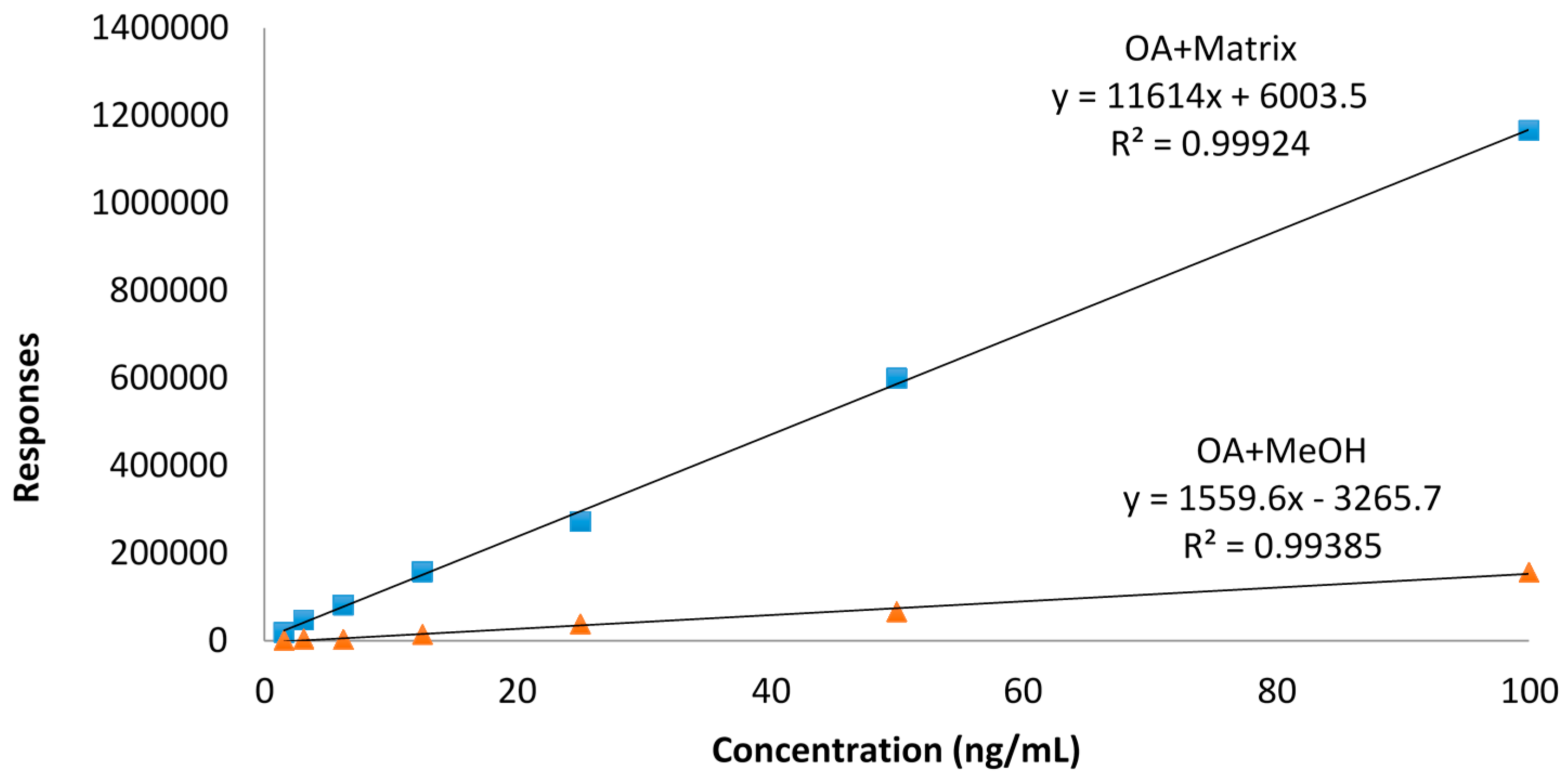
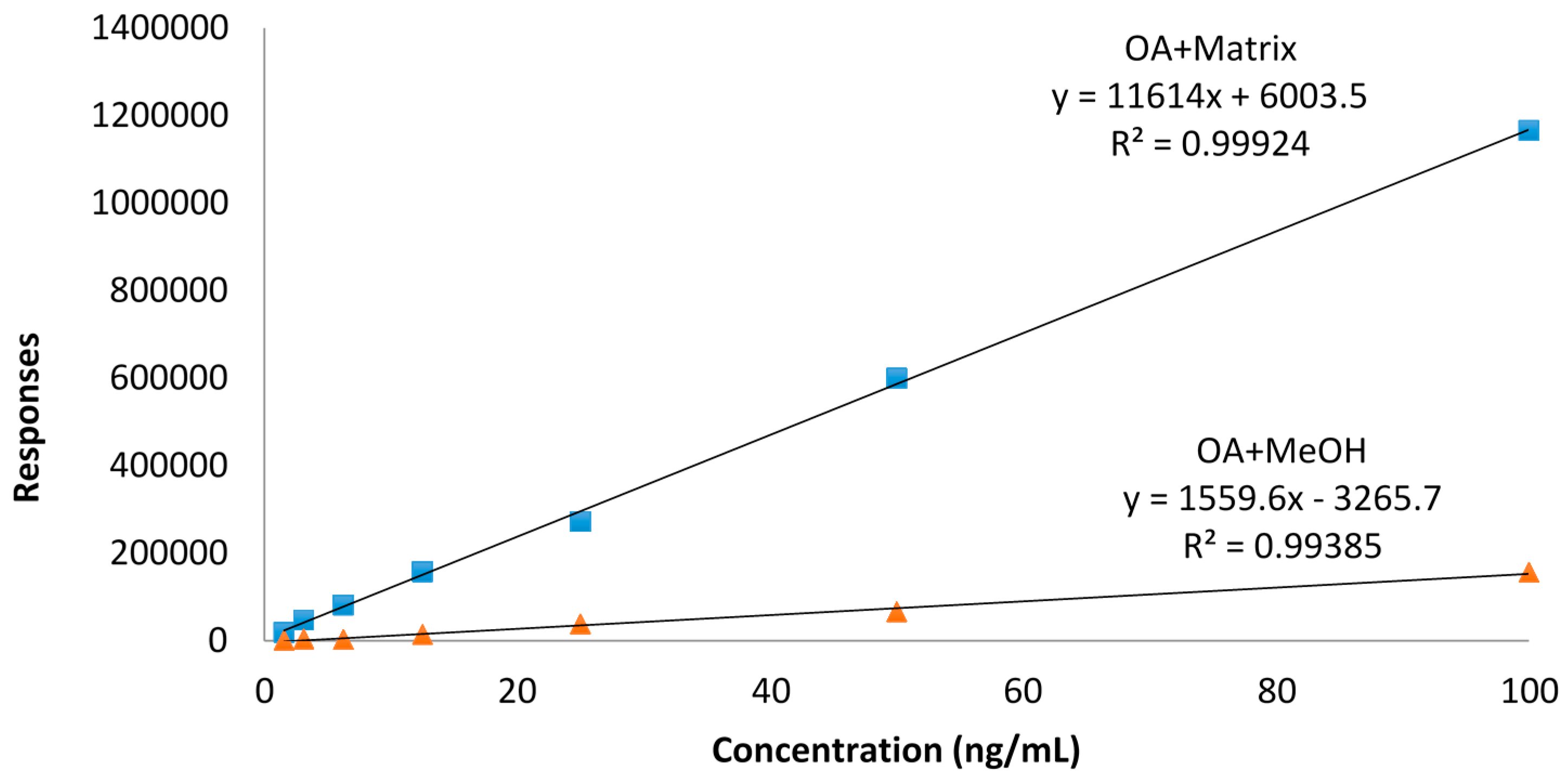
2.5. Influence of Matrix and Steaming on Pure DTX3 Stability with No Loss of Water
3. Conclusions
4. Methods
4.1. Reagents
4.2. Matrix Effect
4.3. Recovery
- Two mussel matrices of known concentration were used, a matrix of certified reference control mussels and a mussel tissue homogenate. The mussel matrices were extracted using the procedure described in the harmonized Standard Operating Procedure (SOP) published by the EU-RL-MB [14].
- The homogenate tissue of mussel was spiked with a known quantity of OA (75 ng/mL final concentration). The matrix was extracted using the procedure described in the EU-harmonized SOP published by the EU-RL-MB [14].
- Three mussel samples were extracted twice with methanol 100% following the procedure described in the EU-harmonized SOP published by the EU-RL-MB [14].
4.4. Dinophysistoxin-3
4.5. Steaming
4.6. Sterilization (Heat)
4.7. Extraction and Hydrolysis Procedure
4.8. LC-MS/MS Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hess, P.; Nguyen, L.; Aasen, J.; Keogh, M.; Kilcoyne, J.; McCarron, P.; Aune, T. Tissue distribution, effects of cooking and parameters affecting the extraction of azaspiracids from mussels, Mytilus edulis, prior to analysis by liquid chromatography coupled to mass spectrometry. Toxicon 2005, 46, 62–71. [Google Scholar] [CrossRef] [PubMed]
- McCarron, P.; Kilcoyne, J.; Hess, P. Effects of cooking and heat treatment on concentration and tissue distribution of okadaic acid and dinophysistoxin-2 in mussels (Mytilus edulis). Toxicon 2008, 51, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- EC. Commission Regulation (EC) No. 2074/2005 of 5 December 2005 laying down implementing measures for certain products under regulation (EC) No. 853/2004. Off. J. Eur. Union 2074, L338, 27–59. [Google Scholar]
- EU. Commission regulation (EU) No. 15/2011 of 10 January 2011 amending Regulation (EC) No. 2074/2005 as regards recognised testing methods for detecting marine biotoxins in live bivalve molluscs. Off. J. Eur. Communities 2011, L6, 3–4. [Google Scholar]
- Panel, E.C. Influence of processing on the levels of lipophilic marine biotoxins in bivalve molluscs. Satement of the Panel on Contaminants in the Food Chain. EFSA J. 2008, 1016, 1–10. [Google Scholar]
- Otero, P.; Alfonso, A.; Alfonso, C.; Rodriguez, P.; Vieytes, M.R.; Botana, L.M. Effect of uncontrolled factors in a validated liquid chromatography-tandem mass spectrometry method question its use as a reference method for marine toxins: Major causes for concern. Anal. Chem. 2011, 83, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- Vale, P.; Sampayo, M.A.D. First confirmation of human diarrhoeic poisonings by okadaic acid esters after ingestion of razor clams (Solen marginatus) and green crabs (Carcinus maenas) in Aveiro lagoon, Portugal and detection of okadaic acid esters in phytoplankton. Toxicon 2002, 40, 989–996. [Google Scholar] [CrossRef]
- Intecmar. Available online: http://www.intecmar.org/pdfs/zonas_2010.pdf (accessed on 10 November 2015).
- Vlamis, A.; Katikou, P. Ecobiology and Geographical Distribution of Potentially Toxic Marine Dinoflagellates, 3rd ed.; CRC Press: Boca Ratón, FL, USA, 2014; pp. 569–625. [Google Scholar]
- Reguera, B.; Pizarro, G. Planktonic dinoflagellates that contain polyether toxins of the old “DSP complex”. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection, 2nd ed.; Botana, L.M., Ed.; CRC Press (Taylor and Francys Group): Boca Raton, FL, USA, 2008; pp. 257–284. [Google Scholar]
- Gonzalez, J.C.; Fontal, O.I.; Vieytes, M.R.; Vieites, J.M.; Botana, L.M. Basis for a new procedure to eliminate diarrheic shellfish toxins from a contaminated matrix. J. Agric. Food Chem. 2002, 50, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Arevalo, F.; Correa, J.; Porro, M.C.; Cabado, A.G.; Vieites, J.M.; Morono, A. Effect of the industrial steaming on the toxicity, estimated by LC-MS/MS, of mussels exposed for a long time to diarrhetic shellfish poisoning (DSP) toxins. Food Chem. 2015, 177, 240–247. [Google Scholar] [CrossRef] [PubMed]
- McCarron, P.; Kilcoyne, J.; Miles, C.O.; Hess, P. Formation of Azaspiracids-3, -4, -6, and -9 via decarboxylation of carboxyazaspiracid metabolites from shellfish. J. Agric. Food Chem. 2009, 57, 160–169. [Google Scholar] [CrossRef] [PubMed]
- EU-RL-MB. Available online: http://aesan.msssi.gob.es/CRLMB/docs/docs/metodos_analiticos_de_desarrollo/EU-Harmonised-SOP-LIPO-LCMSMS_Version5.pdf (accessed on 10 November 2015).

{kind=link}
{kind=link}
| Product Conditions | Sample A: 25 Large Mussels (Bueu) | Sample B: 35 Mussels (Riveira) | Sample C: 23 Mussels (Bueu) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Whole (W) (g) | Flesh (g) | % | Valve Water (mL) % of W | Whole (W) (g) | Flesh (g) | % | Valve Water (mL) % of W | Whole (W) (g) | Flesh (g) | % | Valve Water (mL) % of W | |
| Fresh | 1176 ± 29.5 | 299.6 ± 6.2 | 25.5% ± 0.2 | 367.4 ± 13.5 31.2% | 880 ± 43.1 | 257 ± 12.4 | 29.2% ± 0.3 | 250 ± 16.6 28.4% | 1004 ± 26.3 | 226 ± 7.3 | 22.5% ± 0.5 | 388 ± 4.4 38.6% |
| Lab Steaming | 1140 ± 54.2 | 234 ± 9.9 | 20.6% ± 0.4 | 350 ± 2.8 30.7% | 903.9 ± 46.3 | 195 ± 11.7 | 21.5% ± 0.2 | 283 ± 16.6 31.3% | 986 ± 48.8 | 143 ± 6.5 | 14.6% ± 0.3 | 453 ± 43.5 45.9% |
| Industrial Steaming * | - | 279 ± 6.2 | 19% ± 1 (#) | - | - | 301 ± 8.1 | 22.2% (&) | - | - | 184 ± 2.2 | 13% (**) | - |
| Sample | Fresh Mussels from Farm with Debris and Valve Water (g) | Debris Removed (g) | Valve Water (mL) | Number of Mussels |
|---|---|---|---|---|
| Sample 1 | 5659 | 311 | 400 | 109 |
| Sample 2 | 6690 | 581 | 400 | 122 |
| Sample 3 | 7577 | 635 | 550 | 215 |
| Sample 4 | 7846.5 | 672 | 500 | 142 |
| Product Conditions | Hydrolysis | Sample A | Sample B | Sample C |
|---|---|---|---|---|
| Fresh Product | Before (µg/kg OA/DTX2) | 332 ± 11/104 ± 5.5 | 14.5 ± 1.9/6.7 ± 1.4 | 80 ± 7.6/29.2 ± 1.3 |
| After (µg/kg OA/ DTX2) | 535 ± 36.5/89 ± 2.4 | 76 ± 3.3/13.5 ± 1.5 | 140 ± 19.8/23 ± 0.4 | |
| DTX3 (µg/kg OA-ester/DTX2-ester) | 203/nd | 62/6.8 | 60/nd | |
| % Increased toxin above initial value | 61.14/nd | 427.6/101.5 | 75/nd | |
| Steamed in the Laboratory | Before (µg/kg OA/DTX2) | 467 ± 16.9/108 ± 5.2 | 19.9 ± 0.6/12.7 ± 2.1 | 171 ± 10.4/33 ± 2.7 |
| After (µg/kg OA/ DTX2) | 502 ± 23.2/77.5 ± 3.5 | 58 ± 4.25/11.8 ± 1.6 | 247 ± 13/24.6 ± 2.6 | |
| DTX3 (µg/kg OA-ester/DTX2-ester) | 35/nd | 38/nd | 76/nd | |
| % Increased toxin above initial value | 7.4/nd | 190.9/nd | 44.4/nd | |
| Steamed in the Industry | Before (µg/kg OA/DTX2) | 669.9 ± 33.1/152 ± 2.5 | 20.7 ± 1.3/7.3 ± 1.5 | 187 ± 18.7/51 ± 2.7 |
| After (µg/kg OA/ DTX2) | 588 ± 41/93.6 ± 7 | 39.6 ± 2.6/10 ± 2.5 | 162 ± 12.2/30 ± 2 | |
| DTX3 (µg/kg OA-ester/DTX2-ester) | nd/nd | 18.9/2.7 | nd/nd | |
| % Increased toxin above initial value | nd/nd | 91.3/36.9 | nd/nd |
| A: Standar matrix Mussel-DSP-2 | ||||
| Hydrolysis | at Time 0 | at 10 min | at 20 min | After Autoclave * |
| Before hydrolysis (µg/kg OA) | 369 ± 33 | 417 ± 35 | 442 ± 38 | 197 ± 18 |
| After hydrolysis (µg/kg OA) | 669 ± 59 | 539 ± 46 | 570 ± 49 | 327 ± 13 |
| OA ester (µg/kg OA) | 300 | 122 | 128 | 130 |
| B: Standar matrix Mussel_Control | ||||
| Hydrolysis | at Time 0 | at 10 min | at 20 min | After Autoclave ** |
| Before hydrolysis (µg/kg OA) | 39 ± 14 | 38 ± 11 | 48 ± 13 | - |
| After hydrolysis (µg/kg OA) | 70 ± 28 | 73 ± 18 | 70 ± 18 | - |
| OA ester (µg/kg OA) | 31 | 35 | 22 | - |
| DTX3 (µg/kg OA) | 7-O-palmitoyl Okadaic Ester | |||||
| A | A | A | A | A | A | |
| Time | Control (0’) | Control (0’) | 10´ | 10´ | 20´ | 20´ |
| Water | Matrix | Water | Matrix | Water | Matrix | |
| Before hydrolysis | 0 | 35 ± 8 | 0 | 20 ± 6 | 0 | 22 ± 6 |
| After hydrolysis | 561 ± 73 | 114 ± 16 | 530 ± 69 | 116 ± 17 | 403.7 ± 54 | 110 ± 16 |
| DTX3 (µg/kg OA) | 7-O-palmytoleyl Okadaic Ester | |||||
| B | B | B | B | B | B | |
| Time | Control (0’) | Control (0’) | 10´ | 10´ | 20´ | 20´ |
| Water | Matrix | Water | Matrix | Water | Matrix | |
| Before hydrolysis | 0 | 31 ± 7 | 0 | 36 ± 8 | 0 | 28 ± 7 |
| After hydrolysis | 536 ± 70 | 145 ± 20 | 460 ± 61 | 199 ± 26 | 338 ± 47 | 179 ± 24 |
| DTX3 (µg/kg OA) | Mixture of A and B | |||||
| C | C | C | C | C | C | |
| Time | Control (0’) | Control (0’) | 10´ | 10´ | 20´ | 20´ |
| Water | Matrix | Water | Matrix | Water | Matrix | |
| Before hydrolysis | 0 | 33 ± 7 | 0 | 32 ± 8 | 0 | 27 ± 7 |
| After hydrolysis | 0 | 104 ± 7 | 0 | 96 ± 6 | 0 | 87 ± 6 |
| Compound Name | Precursor Ion | Product Ion | Collision Energy | Polarity |
|---|---|---|---|---|
| DTX1 | 817.5 | 255.2 | 53 | Negative |
| 817.5 | 113 | 66 | - | |
| OA/DTX2 | 803.5 | 255.2 | 52 | Negative |
| 803.5 | 113.2 | 60 | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, I.; Alfonso, A.; Antelo, A.; Alvarez, M.; Botana, L.M. Evaluation of the Impact of Mild Steaming and Heat Treatment on the Concentration of Okadaic Acid, Dinophysistoxin-2 and Dinophysistoxin-3 in Mussels. Toxins 2016, 8, 175. https://doi.org/10.3390/toxins8060175
Rodríguez I, Alfonso A, Antelo A, Alvarez M, Botana LM. Evaluation of the Impact of Mild Steaming and Heat Treatment on the Concentration of Okadaic Acid, Dinophysistoxin-2 and Dinophysistoxin-3 in Mussels. Toxins. 2016; 8(6):175. https://doi.org/10.3390/toxins8060175
Chicago/Turabian StyleRodríguez, Inés, Amparo Alfonso, Alvaro Antelo, Mercedes Alvarez, and Luis M. Botana. 2016. "Evaluation of the Impact of Mild Steaming and Heat Treatment on the Concentration of Okadaic Acid, Dinophysistoxin-2 and Dinophysistoxin-3 in Mussels" Toxins 8, no. 6: 175. https://doi.org/10.3390/toxins8060175
APA StyleRodríguez, I., Alfonso, A., Antelo, A., Alvarez, M., & Botana, L. M. (2016). Evaluation of the Impact of Mild Steaming and Heat Treatment on the Concentration of Okadaic Acid, Dinophysistoxin-2 and Dinophysistoxin-3 in Mussels. Toxins, 8(6), 175. https://doi.org/10.3390/toxins8060175