
CD133, Selectively Targeting the Root of Cancer


Abstract
:
1. Introduction
2. CD133 as Cancer Stem Cell Marker
3. CD133 Expression on Normal Body Cells
4. CD133 Directed Targeted Therapies, Potential and Delivery
5. Potential CD133 Targeting Immunotherapies
5.1. Immunotoxins
5.1.1. C178ABC-CD133MAb
5.1.2. dCD133KDEL
5.1.3. dEpCAMCD133KDEL
5.1.4. Anti-CD133 Conjugated Nanoparticles
5.2. BiTES
5.2.1. MS133
5.2.2. Anti-CD3/CD133 Bispecific Antibody
5.3. BiKES
CD16 × 133
5.4. Trispecific NK Engagers
5.4.1. 133EpCAM16
5.4.2. IL-15 TriKES
5.5. Aptamers
6. Conclusions
Acknowledgement
Author contributions
Conflict of interest
References
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.W.; Clarke, M.F. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev. 2008, 18, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Wicha, M.S. Cancer stem cells: A step toward the cure. J. Clin. Oncol. 2008, 26, 2795–2799. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Petrillo, M.; Bonanno, G.; Scambia, G. Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets 2009, 13, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, S.V.; St Clair, R.; Lyden, D.; Rafii, S. AC133/CD133/Prominin-1. Int. J. Biochem. Cell. Biol. 2005, 37, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.; Fonseca, A.V.; Florek, M.; Freund, D.; Jaszai, J.; Bornhauser, M.; Fargeas, C.A.; Corbeil, D. New insights into the cell biology of hematopoietic progenitors by studying prominin-1 (CD133). Cells Tissues Organs 2008, 188, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, M.; Tian, H.; de Sauvage, F.J. The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 5924–5928. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Fodstad, O.; Lorico, A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells. 2008, 26, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.B.; Nixon, A.M.; Kittanakom, S.; Stewart, J.M.; Chen, G.I.; Curak, J.; Gingras, A.C.; Mazitschek, R.; Neel, B.G.; Stagljar, I.; et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell. Rep. 2012, 2, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Takenobu, H.; Shimozato, O.; Nakamura, T.; Ochiai, H.; Yamaguchi, Y.; Ohira, M.; Nakagawara, A.; Kamijo, T. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 2011, 30, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Wu, P.Y. CD133 as a Marker for Cancer Stem Cells: Progresses and Concerns. Stem Cells Dev. 2009, 18, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Sun, C.; Feng, F.; Ge, M.; Xia, L. Do relevant markers of cancer stem cells CD133 and Nestin indicate a poor prognosis in glioma patients? A systematic review and meta-analysis. J. Exp. Clin. Cancer Res. 2015, 34, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wu, J.D.; Fang, M.M.; Pu, L.Y. Clinicopathological significance and prognostic value of the expression of the cancer stem cell marker CD133 in hepatocellular carcinoma: A meta-analysis. Tumour Biol. 2015, 36, 7623–7630. [Google Scholar] [CrossRef] [PubMed]
- Waldron N, V.D. An Old Idea Tackling a New Problem: Targeted Toxins Specific for Cancer Stem Cells. Antibodies 2013, 2, 82–89. [Google Scholar] [CrossRef]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Pinz, K.; Liu, H.; Golightly, M.; Jares, A.; Lan, F.; Zieve, G.W.; Hagag, N.; Schuster, M.; Firor, A.E.; Jiang, X.; et al. Preclinical targeting of human T cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia 2015, 30, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol. Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Z.; Su, H. Clinical translational research of chimeric antigen receptor-T (CAR-T) cells for the treatment of relapsed and refractory B-cell lymphoma/leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2014, 22, 1137–1141. [Google Scholar] [PubMed]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Watzl, C.; Long, E.O. Exposing tumor cells to killer cell attack. Nat. Med. 2000, 6, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. Stress, NK receptors, and immune surveillance. Science 2001, 294, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Sogn, J.A. Tumor immunology: The glass is half full. Immunity 1998, 9, 757–763. [Google Scholar] [CrossRef]
- Chen, L. Immunological ignorance of silent antigens as an explanation of tumor evasion. Immunol. Today 1998, 19, 27–30. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Lyer, K.S.; Saksena, V.N. A stochastic model of the growth of cancer in cancer. Biometrics 1970, 26, 401–410. [Google Scholar]
- Jackson, E.B.; Brues, A.M. Studies on a Transplantable Embryoma of the Mouse. Cancer Res. Treat. 1941, 1, 494–498. [Google Scholar]
- Makino, S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Ann. N. Y. Acad. Sci. 1956, 63, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.D.; Robson, C.N.; Lang, S.H.; Neal, D.E.; Maitland, N.J.; Collins, A.T. CD133, a novel marker for human prostatic epithelial stem cells. J. Cell. Sci. 2004, 117, 3539–3545. [Google Scholar] [CrossRef] [PubMed]
- Mehra, N.; Penning, M.; Maas, J.; Beerepoot, L.V.; van Daal, N.; van Gils, C.H.; Giles, R.H.; Voest, E.E. Progenitor marker CD133 mRNA is elevated in peripheral blood of cancer patients with bone metastases. Clin. Cancer Res. 2006, 12, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Miki, J.; Furusato, B.; Li, H.; Gu, Y.; Takahashi, H.; Egawa, S.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Rhim, J.S. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007, 67, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Wang, H.L.; Lin, Y.S.; Kumar, K.P.; Lin, H.C.; Chang, C.J.; Lu, C.C.; Huang, T.T.; Martel, J.; Ojcius, D.M.; et al. Identification of CD24 as a cancer stem cell marker in human nasopharyngeal carcinoma. PLoS One 2014, 9, e99412. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.; Kuperwasser, C. Human breast cancer stem cell markers CD44 and CD24: Enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of human pancreatic cancer stem cells. Methods Mol. Biol. 2009, 568, 161–173. [Google Scholar] [PubMed]
- Kucia, M.; Reca, R.; Miekus, K.; Wanzeck, J.; Wojakowski, W.; Janowska-Wieczorek, A.; Ratajczak, J.; Ratajczak, M.Z. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: Pivotal role of the SDF-1-CXCR4 axis. Stem Cells 2005, 23, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Zhao, J. The Role of chemokine receptor CXCR4 in breast cancer metastasis. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar] [PubMed]
- Zheng, X.; Xie, Q.; Li, S.; Zhang, W. CXCR4-positive subset of glioma is enriched for cancer stem cells. Oncol. Res. 2011, 19, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; D’Amico, L.; Moro, M.; Landoni, E.; Perego, P.; Miceli, R.; Gatti, L.; Andriani, F.; Wong, D.; Caserini, R.; et al. Microenvironment-Modulated Metastatic CD133+/CXCR4+/EpCAM—Lung Cancer-Initiating Cells Sustain Tumor Dissemination and Correlate with Poor Prognosis. Cancer Res. 2015, 75, 3636–3649. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J., Jr.; Chang, H.Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, H.; He, L.; Zhang, J.; Ni, B.; Wang, X.; Jin, H.; Cahuzac, N.; Mehrpour, M.; Lu, Y.; et al. CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res. 2008, 14, 6751–6760. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, E.; Lokshin, A.; Levina, V. Lung cancer stem cells as a target for therapy. Anticancer Agents Med. Chem. 2010, 10, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Chen, D.; Yang, J.; Yang, C.; Zhang, Y.; Zhang, H.; Dou, J. Evaluation of characteristics of CD44+ CD117+ ovarian cancer stem cells in three dimensional basement membrane extract scaffold versus two dimensional monocultures. BMC Cell. Biol. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Liao, M.Y.; Lin, W.W.; Wang, Y.P.; Lu, T.Y.; Wu, H.C. Epithelial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial-mesenchymal transition gene expression in colon cancer. J. Biol. Chem. 2012, 287, 39449–39459. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Wicha, M.S. HER2 and breast cancer stem cells: More than meets the eye. Cancer Res. 2013, 73, 3489–3493. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.P.; Seshacharyulu, P.; Vaz, A.; Dey, P.; Batra, S.K. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J. Ovarian Res. 2011, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.; Ortega, F.G.; Salido, M.; Lorente, J.A.; Rodriguez-Rivera, M.; Delgado-Rodriguez, M.; Macia, M.; Fernandez, A.; Corominas, J.M.; Garcia-Puche, J.L.; et al. CD133 expression in circulating tumor cells from breast cancer patients: Potential role in resistance to chemotherapy. Int. J. Cancer 2013, 133, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Di Franco, S.; Iovino, F.; Dieli, F.; Stassi, G.; Todaro, M. CD133 as a target for colon cancer. Expert Opin. Ther. Targets. 2012, 16, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.J.; Rizzo, S.; Ledaki, I.; Davies, M.; Brewer, D.; Attard, G.; de Bono, J.; Hudson, D.L. Expression profiling of CD133+ and CD133− epithelial cells from human prostate. Prostate 2008, 68, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Donnenberg, V.S.; Landreneau, R.J.; Donnenberg, A.D. Tumorigenic stem and progenitor cells: Implications for the therapeutic index of anti-cancer agents. J. Control. Release. 2007, 122, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Damek-Poprawa, M.; Volgina, A.; Korostoff, J.; Sollecito, T.P.; Brose, M.S.; O’Malley, B.W., Jr.; Akintoye, S.O.; DiRienzo, J.M. Targeted inhibition of CD133+ cells in oral cancer cell lines. J. Dent. Res. 2011, 90, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Skubitz, A.P.; Taras, E.P.; Boylan, K.L.; Waldron, N.N.; Oh, S.; Panoskaltsis-Mortari, A.; Vallera, D.A. Targeting CD133 in an in vivo ovarian cancer model reduces ovarian cancer progression. Gynecol. Oncol. 2013, 130, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Immervoll, H.; Hoem, D.; Sakariassen, P.O.; Steffensen, O.J.; Molven, A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Ueno, S.; Arigami, T.; Uchikado, Y.; Setoyama, T.; Arima, H.; Kita, Y.; Kurahara, H.; Okumura, H.; Matsumoto, M.; et al. Prognostic impact of CD133 expression in gastric carcinoma. Anticancer Res. 2010, 30, 2453–2457. [Google Scholar] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Karbanova, J.; Missol-Kolka, E.; Fonseca, A.V.; Lorra, C.; Janich, P.; Hollerova, H.; Jaszai, J.; Ehrmann, J.; Kolar, Z.; Liebers, C.; et al. The stem cell marker CD133 (Prominin-1) is expressed in various human glandular epithelia. J. Histochem. Cytochem. 2008, 56, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Irollo, E.; Pirozzi, G. CD133: To be or not to be, is this the real question? Am. J. Transl. Res. 2013, 5, 563–581. [Google Scholar] [PubMed]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L.A. CD133 as a biomarker for putative cancer stem cells in solid tumours: Limitations, problems and challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Tirino, V.; Camerlingo, R.; Bifulco, K.; Irollo, E.; Montella, R.; Paino, F.; Sessa, G.; Carriero, M.V.; Normanno, N.; Rocco, G.; et al. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell. Death Dis. 2013, 4, e620. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Olin, M.R.; Forster, C.L.; Cruz, K.S.; Panyam, J.; Ohlfest, J.R. Identification of a novel monoclonal antibody recognizing CD133. J. Immunol. Methods 2010, 361, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Waldron, N.N.; Kaufman, D.S.; Oh, S.; Inde, Z.; Hexum, M.K.; Ohlfest, J.R.; Vallera, D.A. Targeting tumor-initiating cancer cells with dCD133KDEL shows impressive tumor reductions in a xenotransplant model of human head and neck cancer. Mol. Cancer Ther. 2011, 10, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Peichev, M.; Naiyer, A.J.; Pereira, D.; Zhu, Z.; Lane, W.J.; Williams, M.; Oz, M.C.; Hicklin, D.J.; Witte, L.; Moore, M.A.; et al. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood 2000, 95, 952–958. [Google Scholar] [PubMed]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Nesterova, A.; Ryan, M.C.; Duniho, S.; Jonas, M.; Anderson, M.; Zabinski, R.F.; Sutherland, M.K.; Gerber, H.P.; Van Orden, K.L.; et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br. J. Cancer. 2008, 99, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Bonanno, G.; Marone, M.; De Ritis, D.; Mariotti, A.; Voso, M.T.; Scambia, G.; Mancuso, S.; Leone, G.; Pierelli, L. Identification of a novel subpopulation of human cord blood CD34− CD133− CD7− CD45+ lineage− cells capable of lymphoid/NK cell differentiation after in vitro exposure to IL-15. J. Immunol. 2003, 171, 2977–2988. [Google Scholar] [CrossRef] [PubMed]
- Suuronen, E.J.; Wong, S.; Kapila, V.; Waghray, G.; Whitman, S.C.; Mesana, T.G.; Ruel, M. Generation of CD133+ cells from CD133− peripheral blood mononuclear cells and their properties. Cardiovasc. Res. 2006, 70, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Glesne, D.; Huberman, E. A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Heeschen, C.; Aicher, A.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation 2003, 108, 2511–2516. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Q.; Wu, X.; Schultz, P.G.; Ding, S. Dedifferentiation of lineage-committed cells by a small molecule. J. Am. Chem. Soc. 2004, 126, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Bouckenooghe, T.; Vandewalle, B.; Moerman, E.; Danze, P.M.; Lukowiak, B.; Muharram, G.; Kerr-Conte, J.; Gmyr, V.; Laine, B.; Pattou, F. Expression of progenitor cell markers during expansion of sorted human pancreatic beta cells. Gene Expr. 2005, 12, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Quirici, N.; Soligo, D.; Caneva, L.; Servida, F.; Bossolasco, P.; Deliliers, G.L. Differentiation and expansion of endothelial cells from human bone marrow CD133+ cells. Br. J. Haematol. 2001, 115, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Donovan, L.K.; Pilkington, G.J. CD133: Holy of grail of neuro-oncology or promiscuous red-herring? Cell. Prolif. 2012, 45, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, V.; Marzesco, A.M.; Corbeil, D.; Huttner, W.B.; Wilsch-Brauninger, M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J. Cell. Biol. 2007, 176, 483–495. [Google Scholar] [CrossRef] [PubMed]
- A.R. Jones, E.V.S. Blood–brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Lichota, J.; Skjorringe, T.; Thomsen, L.B.; Moos, T. Macromolecular drug transport into the brain using targeted therapy. J. Neurochem. 2010, 113, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Watts, R.J. Developing Therapeutic Antibodies for Neurodegenerative Disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Gehling, U.M.; Ergun, S.; Schumacher, U.; Wagener, C.; Pantel, K.; Otte, M.; Schuch, G.; Schafhausen, P.; Mende, T.; Kilic, N.; et al. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood 2000, 95, 3106–3112. [Google Scholar] [CrossRef]
- Adini, A.; Adini, I.; Ghosh, K.; Benny, O.; Pravda, E.; Hu, R.; Luyindula, D.; D’Amato, R.J. The stem cell marker prominin-1/CD133 interacts with vascular endothelial growth factor and potentiates its action. Angiogenesis 2013, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Wientjes, M.G.; Lu, D.; Au, J.L.S. Drug delivery and transport to solid tumors. Pharm. Res. 2003, 20, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, C.; Thomas, X. Potential for bispecific T-cell engagers: Role of blinatumomab in acute lymphoblastic leukemia. Drug Des. Devel. Ther. 2016, 10, 757–765. [Google Scholar] [PubMed]
- Topp, M.S.; Kufer, P.; Gokbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted Therapy With the T-Cell-Engaging Antibody Blinatumomab of Chemotherapy-Refractory Minimal Residual Disease in B-Lineage Acute Lymphoblastic Leukemia Patients Results in High Response Rate and Prolonged Leukemia-Free Survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Ohlfest, J.R.; Zellmer, D.M.; Panyam, J.; Swaminathan, S.K.; Oh, S.; Waldron, N.N.; Toma, S.; Vallera, D.A. Immunotoxin targeting CD133+ breast carcinoma cells. Drug Deliv. Transl. Res. 2012, 3, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Waldron, N.N.; Barsky, S.H.; Dougherty, P.R.; Vallera, D.A. A bispecific EpCAM/CD133-targeted toxin is effective against carcinoma. Target. Oncol. 2014, 9, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target. Oncol. 2015, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yang, Y.; Zhou, P.; Ma, H.; Zhao, X.; He, X.; Wang, T.; Zhang, J.; Liu, Y.; Zhang, T. Targeting CD133high Colorectal Cancer Cells In Vitro and In Vivo With an Asymmetric Bispecific Antibody. J. Immunother. 2015, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.; Wang, Y.; Lv, H.; Guo, Y.; Dai, H.; Wicha, M.S.; Chang, A.E.; Li, Q. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin. Immunol. 2013, 149, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.K.; Niu, L.; Waldron, N.; Kalscheuer, S.; Zellmer, D.M.; Olin, M.R.; Ohlfest, J.R.; Vallera, D.A.; Panyam, J. Identification and characterization of a novel scFv recognizing human and mouse CD133. Drug Deliv. Transl. Res. 2013, 3, 143–151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kreitman, R.J.; Pastan, I. Accumulation of a recombinant immunotoxin in a tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998, 58, 968–975. [Google Scholar] [PubMed]
- Pastan, I.; Chaudhary, V.; FitzGerald, D.J. Recombinant toxins as novel therapeutic agents. Annu. Rev. Biochem. 1992, 61, 331–354. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ohlfest, J.R.; Zellmer, D.M.; Panyam, J.; Swaminathan, S.K.; Oh, S.; Waldron, N.N.; Toma, S.; Vallera, D.A. Immunotoxin targeting CD133+ breast carcinoma cells. Drug Deliv. Transl. Res. 2013, 3, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Trzpis, M.; McLaughlin, P.M.; de Leij, L.M.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [PubMed]
- van der Gun, B.T.; Melchers, L.J.; Ruiters, M.H.; de Leij, L.F.; McLaughlin, P.M.; Rots, M.G. EpCAM in carcinogenesis: The good, the bad or the ugly. Carcinogenesis 2010, 31, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer. 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia. 2012, 17, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell. Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gokbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Edwards, R.P.; Parker, L.P.; DeMars, L.R.; Herzog, T.J.; Lentz, S.S.; Morris, R.T.; Akerley, W.L.; Holloway, R.W.; Method, M.W.; et al. Catumaxomab for the treatment of malignant ascites in patients with chemotherapy-refractory ovarian cancer: A phase II study. Int. J. Gynecological cancer 2014, 24, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P promotes pancreatic cancer growth, survival, and invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hu, H.; Tong, X.; Jiang, Q.; Zhu, H.; Zhang, S. Calcium-binding protein S100P and cancer: Mechanisms and clinical relevance. J. Cancer Res. Clin. Oncol. 2012, 138, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, K.A.; Prehn, J.L.; Landers, C.; Han, Q.; Luo, X.; Cha, S.C.; Wei, P.; Targan, S.R. TL1A synergizes with IL-12 and IL-18 to enhance IFN-gamma production in human T cells and NK cells. J. Immunol. 2004, 172, 7002–7007. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Zhang, B.; Gleason, M.K.; Oh, S.; Weiner, L.M.; Kaufman, D.S.; McCullar, V.; Miller, J.S.; Verneris, M.R. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother. Radiopharm. 2013, 28, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Ranson, T.; Vosshenrich, C.A.; Corcuff, E.; Richard, O.; Muller, W.; Di, S. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood 2003, 101, 4887–4893. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Legrand, N.; Alves, N.L.; Jaron, B.; Weijer, K.; Plet, A.; Corcuff, E.; Mortier, E.; Jacques, Y.; Spits, H.; et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009, 206, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Interleukin-15 in the treatment of cancer. Expert Rev. Clin. Immunol. 2014, 10, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Basak, G.W.; Zapala, L.; Wysocki, P.J.; Mackiewicz, A.; Jakobisiak, M.; Lasek, W. Interleukin 15 augments antitumor activity of cytokine gene-modified melanoma cell vaccines in a murine model. Oncol. Rep. 2008, 19, 1173–1179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK cell activation of an anti-carcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol. Ther. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Munger, W.; DeJoy, S.Q.; Jeyaseelan, R., Sr.; Torley, L.W.; Grabstein, K.H.; Eisenmann, J.; Paxton, R.; Cox, T.; Wick, M.M.; Kerwar, S.S. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: Comparison with interleukin-2. Cell. Immunol. 1995, 165, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.T.; McCullar, V.; Zhou, X.; Schmohl, J.; Zhang, B.; Lenvik, A.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL-15 Trispecific Killer Engagers (TriKEs) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Ward, A.C.; De, A.; Yang, C.J.; Wei, M.; Duan, W. Clinical applications of aptamers and nucleic acid therapeutics in haematological malignancies. Br. J. Haematol. 2011, 155, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Orava, E.W.; Cicmil, N.; Gariepy, J. Delivering cargoes into cancer cells using DNA aptamers targeting internalized surface portals. Biochim. Biophys. Acta 2010, 1798, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef] [PubMed]
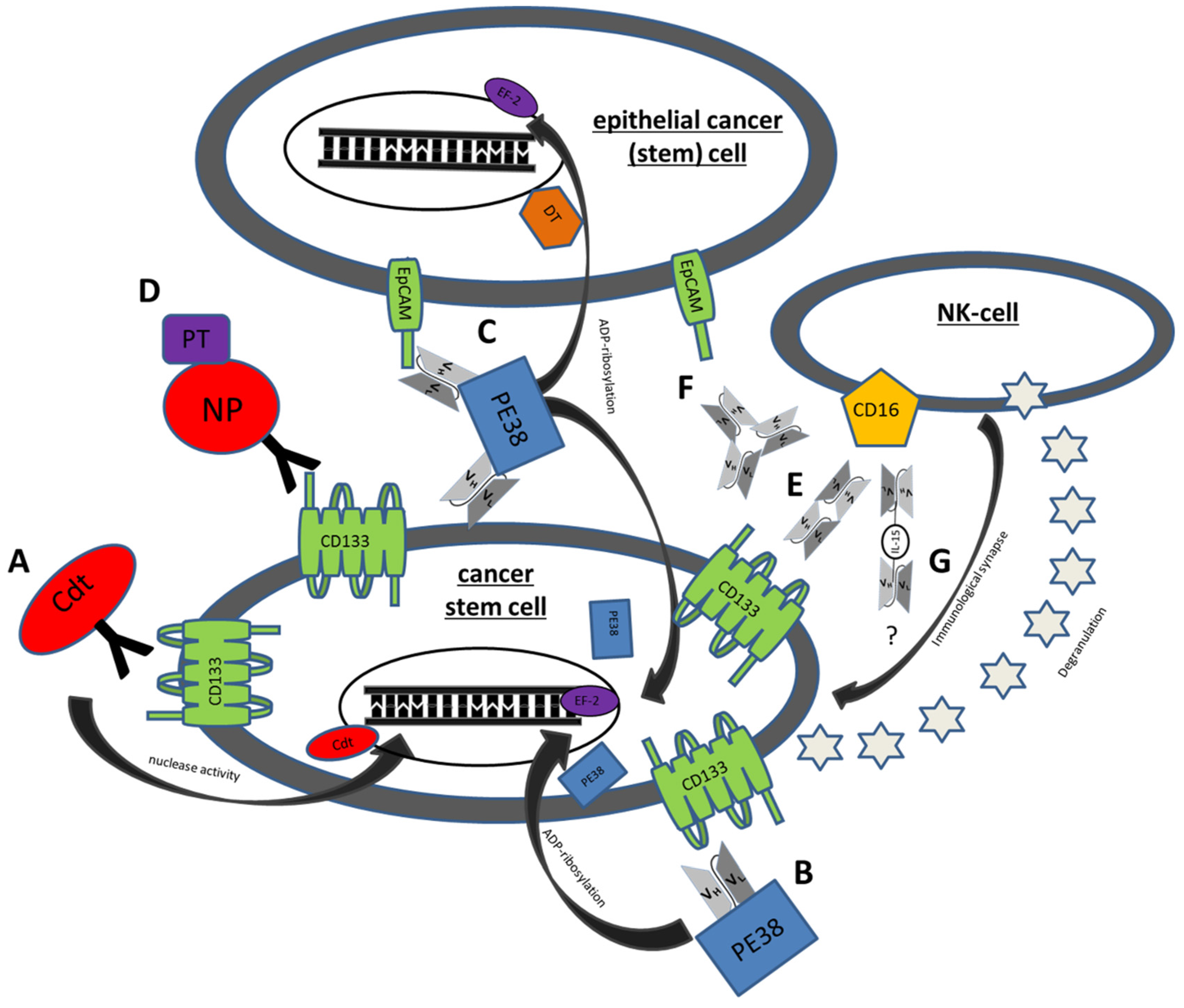

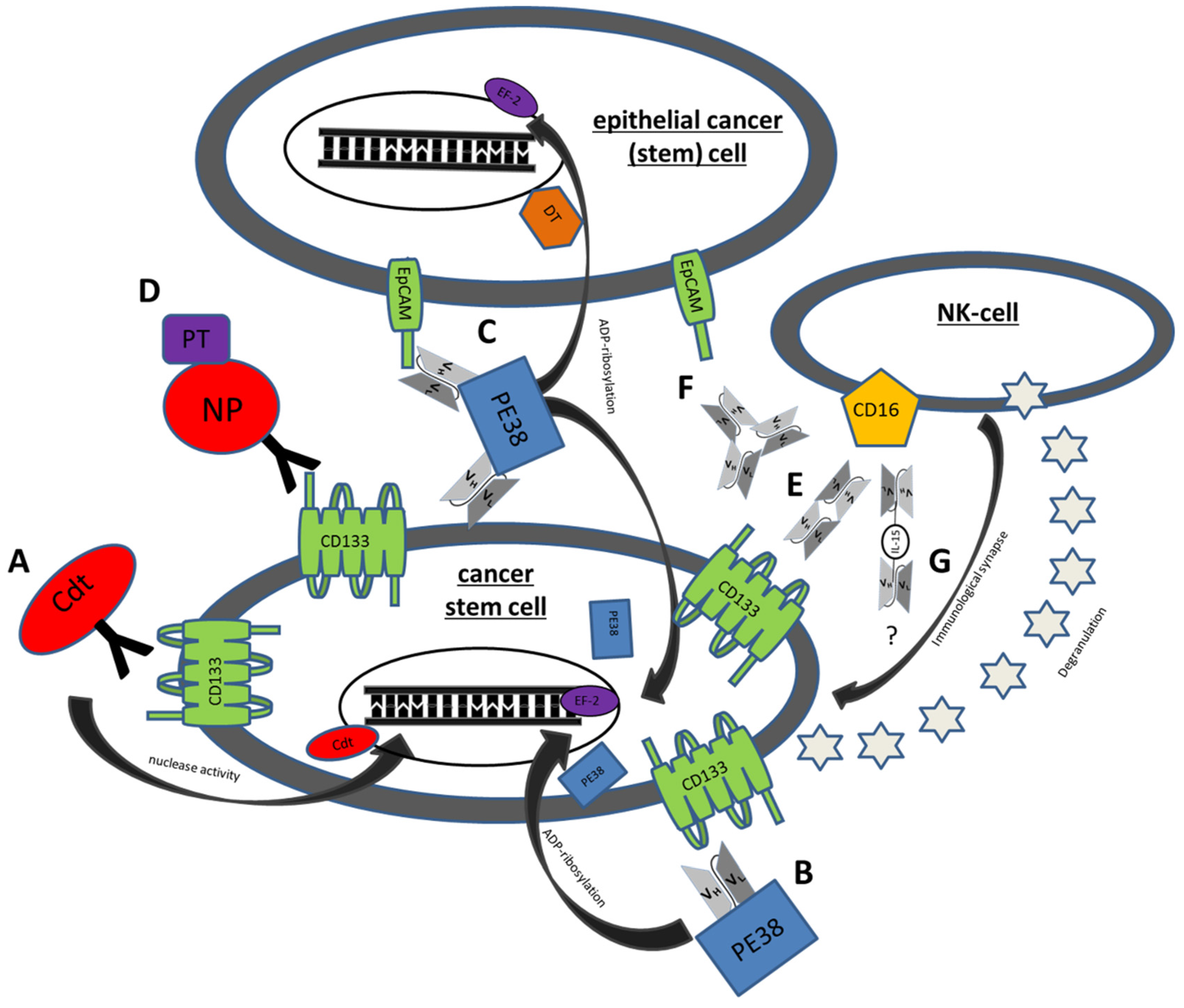
{kind=link}
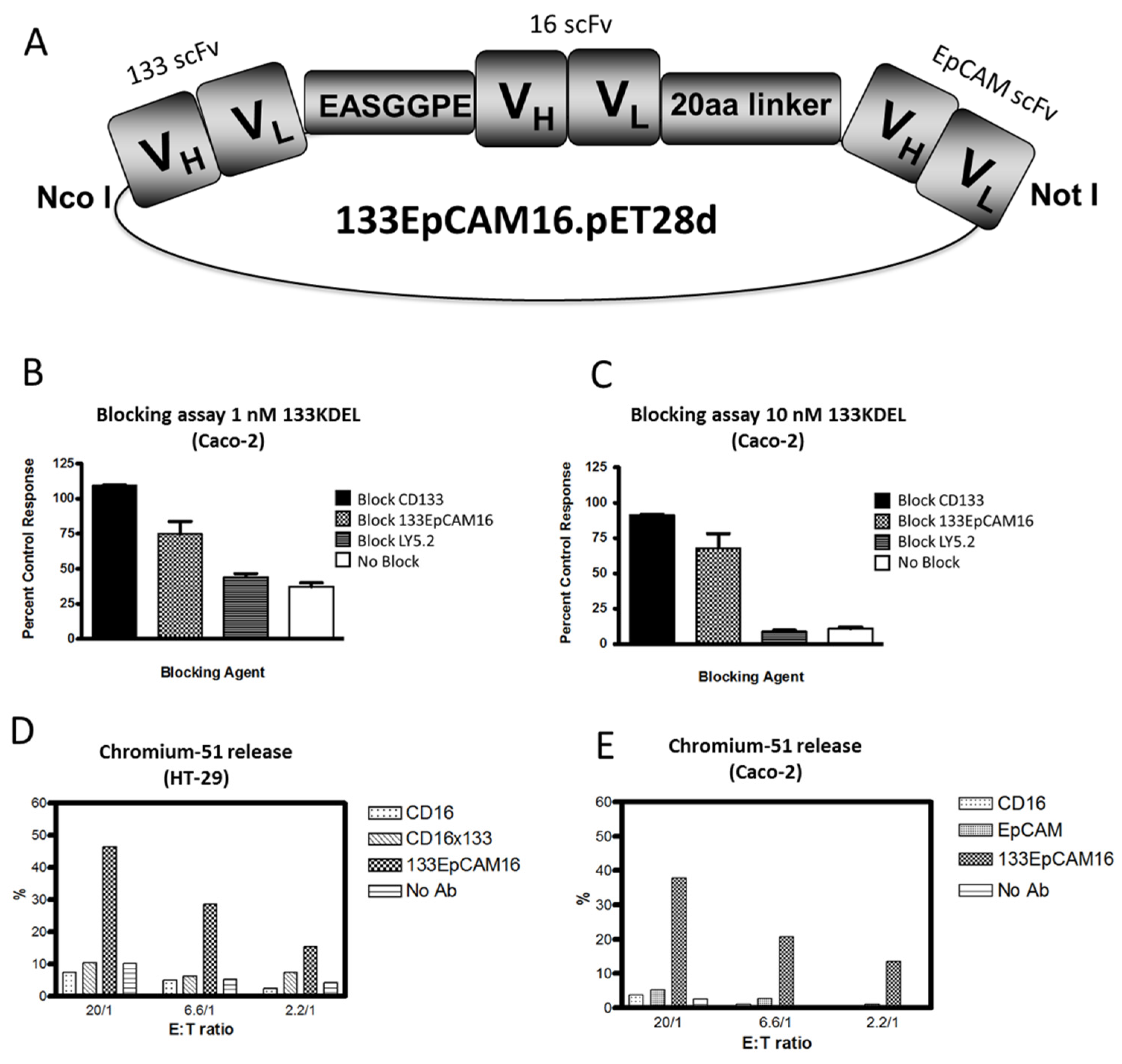
{kind=link}
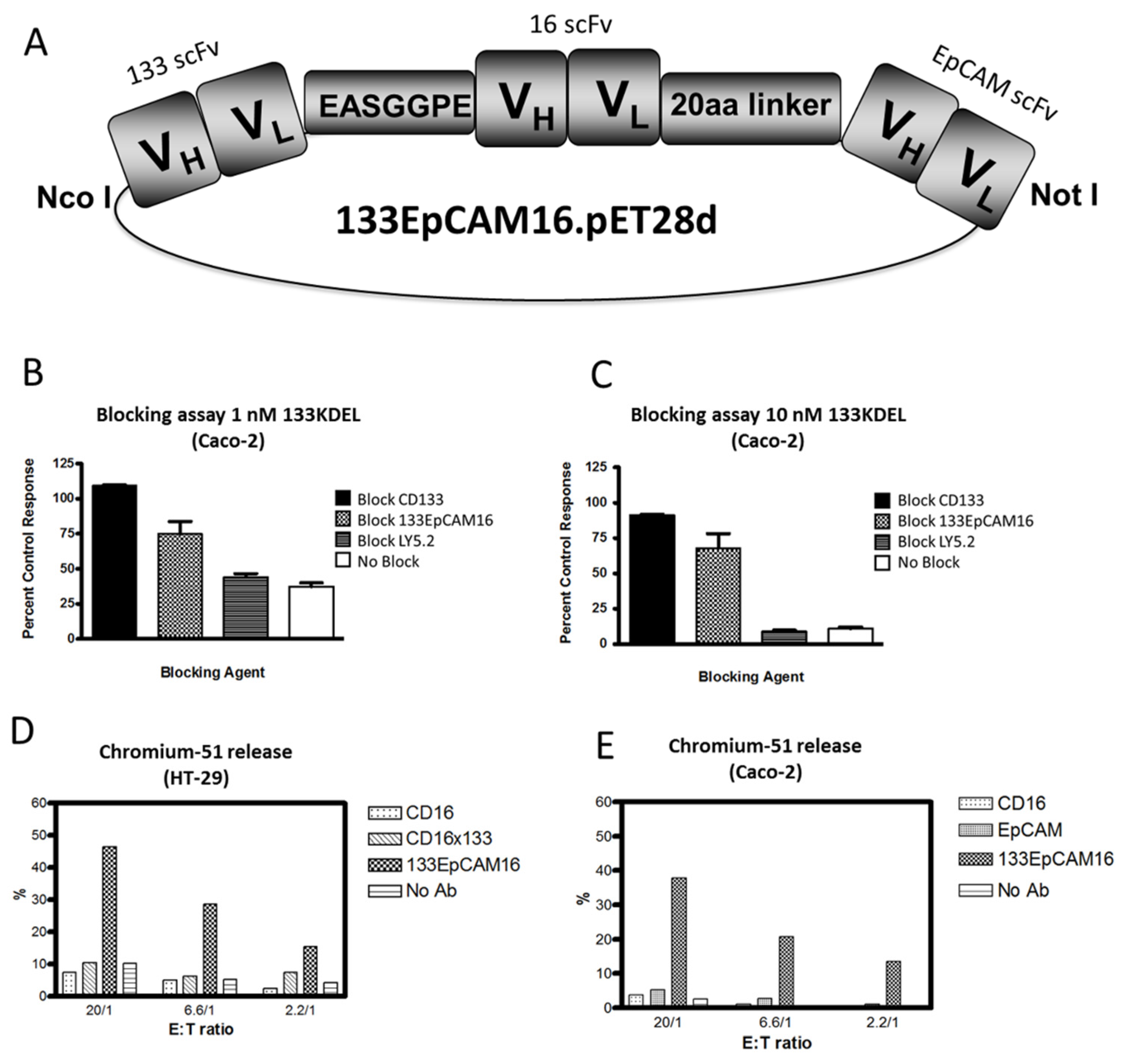{kind=link}
| Marker | Source of Malignancy | Ref. |
|---|---|---|
| CD20 | Melanoma | [39] |
| CD24 | Nasopharyngeal | [40] |
| Breast | [41] | |
| Pancreatic | [42] | |
| CXCR4 | Breast | [43,44] |
| Glioma | [45] | |
| Lung | [46] | |
| CD47 | Bladder | [47] |
| Breast | [48] | |
| CD44 | Bladder | [47] |
| Colon | [49] | |
| Gastric | [50] | |
| Ovarian | [51] | |
| Pancreatic | [42] | |
| CD117 | Lung | [52] |
| Ovarian | [53] | |
| EpCAM | Colon | [54] |
| Breast | [55] | |
| HER2/ERBB2 | Breast | [56] |
| Ovarian | [57] | |
| CD34 | Acute myeloid leukemia | [1] |
| Tissue Source | CD133+ Cell Group | Ref. | CD133+ Tumor Cells in Tumor Tissue |
|---|---|---|---|
| Breast | Cancer inducing subpopulation | [58,59] | Unknown |
| Colon | Cancer inducing subpopulation | [37,60] | 2.5% |
| Prostate | Subpopulation | [38,61] | 0.5% |
| Melanoma | Cancer inducing subpopulation | [62] | 1% |
| Lung | Cancer inducing subpopulation | [63] | 10% |
| HNSCC | Subpopulation | [64] | 18% |
| Ovarian | Cancer inducing subpopulation | [65] | 5.6%–16% |
| Pancreatic | Subpopulation | [66]/[67] | >1%/>15% |
| Gastric | Subpopulation | [68] | >1% |
| Hepathocellular | Subpopulation | [69] | 1%–3% |
| Drug | Cancer Subtype | Cell Line | Ref. | Result |
|---|---|---|---|---|
| C178ABC-CD133MAb | HNSCC | CAL-27 | [64] | Inhibition of proliferation (in vitro) |
| dCD133KDEL | HNSCC | UMSCC-11B | [62] | Inhibition of proliferation, degradation (in vitro) |
| NA-SCC | Inhibition of proliferation (in vitro) | |||
| Ovarian | NIH:OVCAR5 | [65] | Inhibition of growth (in vivo) | |
| Breast | MDA-MB-231 | [102] | Inhibition of proliferation (in vivo) | |
| DTEpCAMCD133KDEL | HNSCC | UMSCC-11B | [103] | Inhibition of proliferation and CR (in vivo) |
| Colorectal | Caco-2 | Inhibition of proliferation (in vitro) | ||
| HT-29 | Inhibition of proliferation (in vitro) | |||
| Breast | BT-474 | Inhibition of proliferation (in vitro) | ||
| SK-BR3 | Inhibition of proliferation (in vitro) | |||
| Glioma | U87 | No effect | ||
| Lymphoma | Raji | No effect | ||
| CD133 NP | Colorectal | Caco-2 | [104] | Particle uptake (in vitro) |
| Breast | mammospheres | Cell elimination (in vitro) | ||
| MDA-MB-231 | Tumor decline (in vivo) | |||
| CD16 × 133 | Colorectal | Caco-2 | [105] | Cell elimination (in vitro) |
| Lymphoma | Daudi | Cell elimination (in vitro) | ||
| MS133 | Colorectal | HCT 116 | [106] | Cell elimination (in vitro)/repression of tumor initiation (in vivo) |
| CD133CD3 bispecific antibody | Pancreatic | SW1990 | [107] | Cell elimination/inhibition of tumor growth (in vivo) |
| Hepatic | Hep3B | Cell elimination |
| Cell Line | Dose (µg) | Anti-CD133-FITC (%) | Anti-EpCAM-FITC (%) | Anti-CD16-FITC (%) |
|---|---|---|---|---|
| Caco-2 | 0.5 | 40.0 | 99.6 | - |
| 1 | 41.5 | 99.7 | - | |
| 2 | 48.0 | 99.7 | 0.6 | |
| HT-29 | 1 | 0.8 | 98.2 | - |
| 2 | 1.4 | 99.2 | 0.8 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmohl, J.U.; Vallera, D.A. CD133, Selectively Targeting the Root of Cancer. Toxins 2016, 8, 165. https://doi.org/10.3390/toxins8060165
Schmohl JU, Vallera DA. CD133, Selectively Targeting the Root of Cancer. Toxins. 2016; 8(6):165. https://doi.org/10.3390/toxins8060165
Chicago/Turabian StyleSchmohl, Jörg U., and Daniel A. Vallera. 2016. "CD133, Selectively Targeting the Root of Cancer" Toxins 8, no. 6: 165. https://doi.org/10.3390/toxins8060165
APA StyleSchmohl, J. U., & Vallera, D. A. (2016). CD133, Selectively Targeting the Root of Cancer. Toxins, 8(6), 165. https://doi.org/10.3390/toxins8060165