
Paulistine—The Functional Duality of a Wasp Venom Peptide Toxin


Abstract
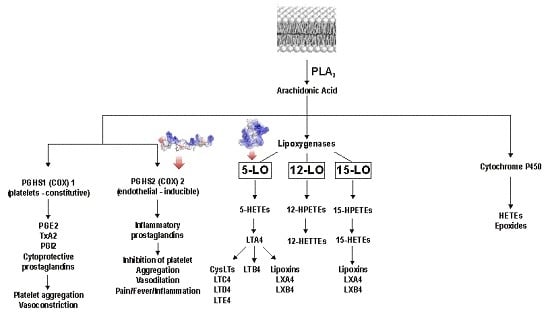
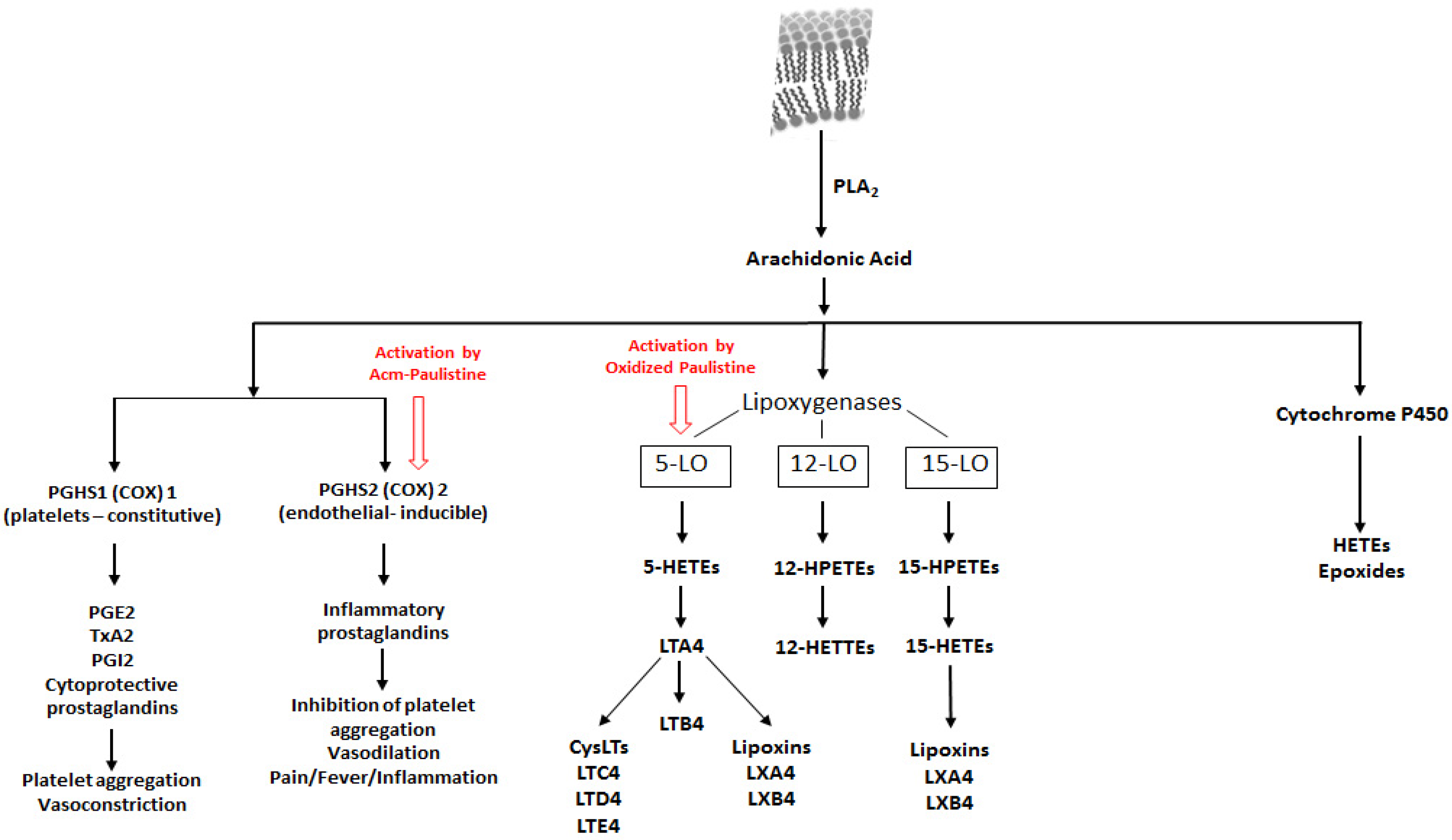
:
1. Introduction
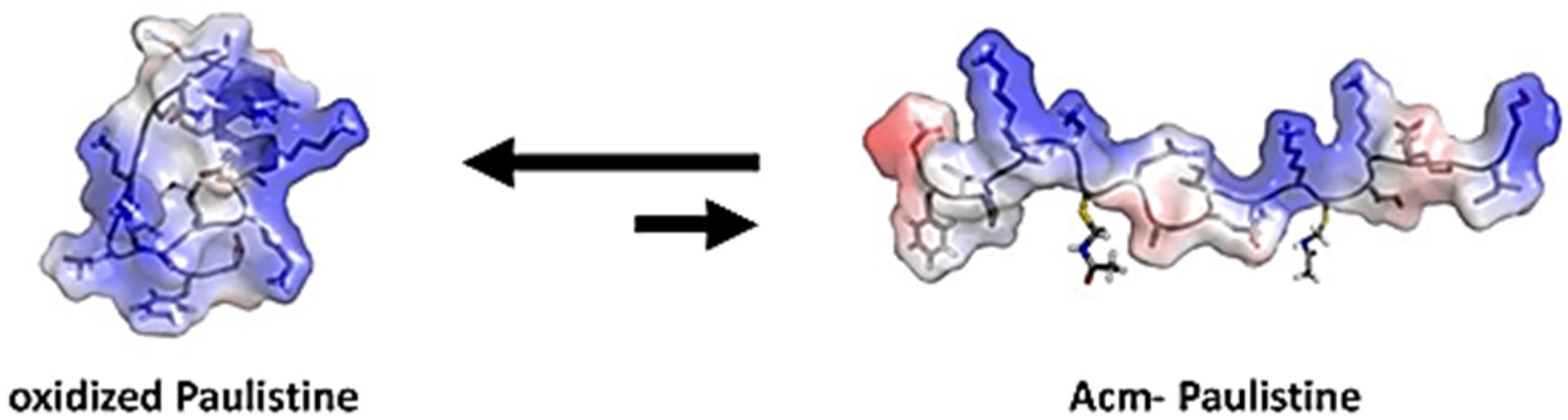
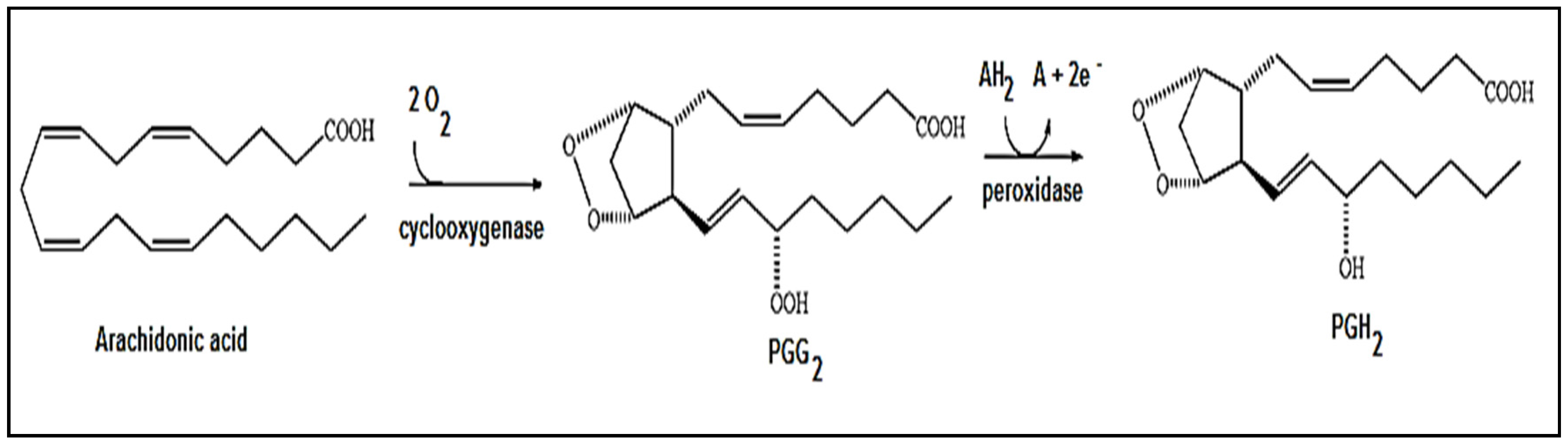
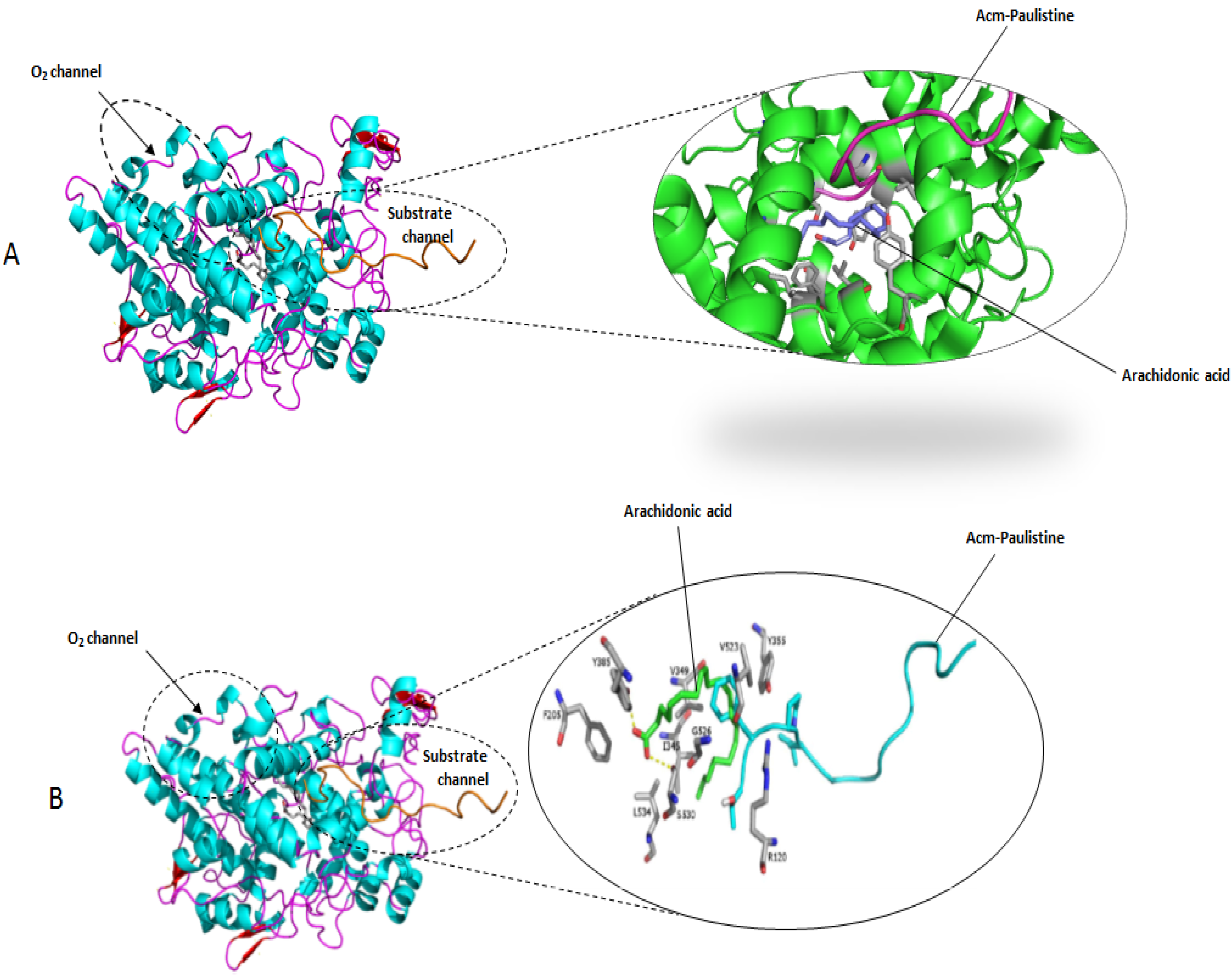
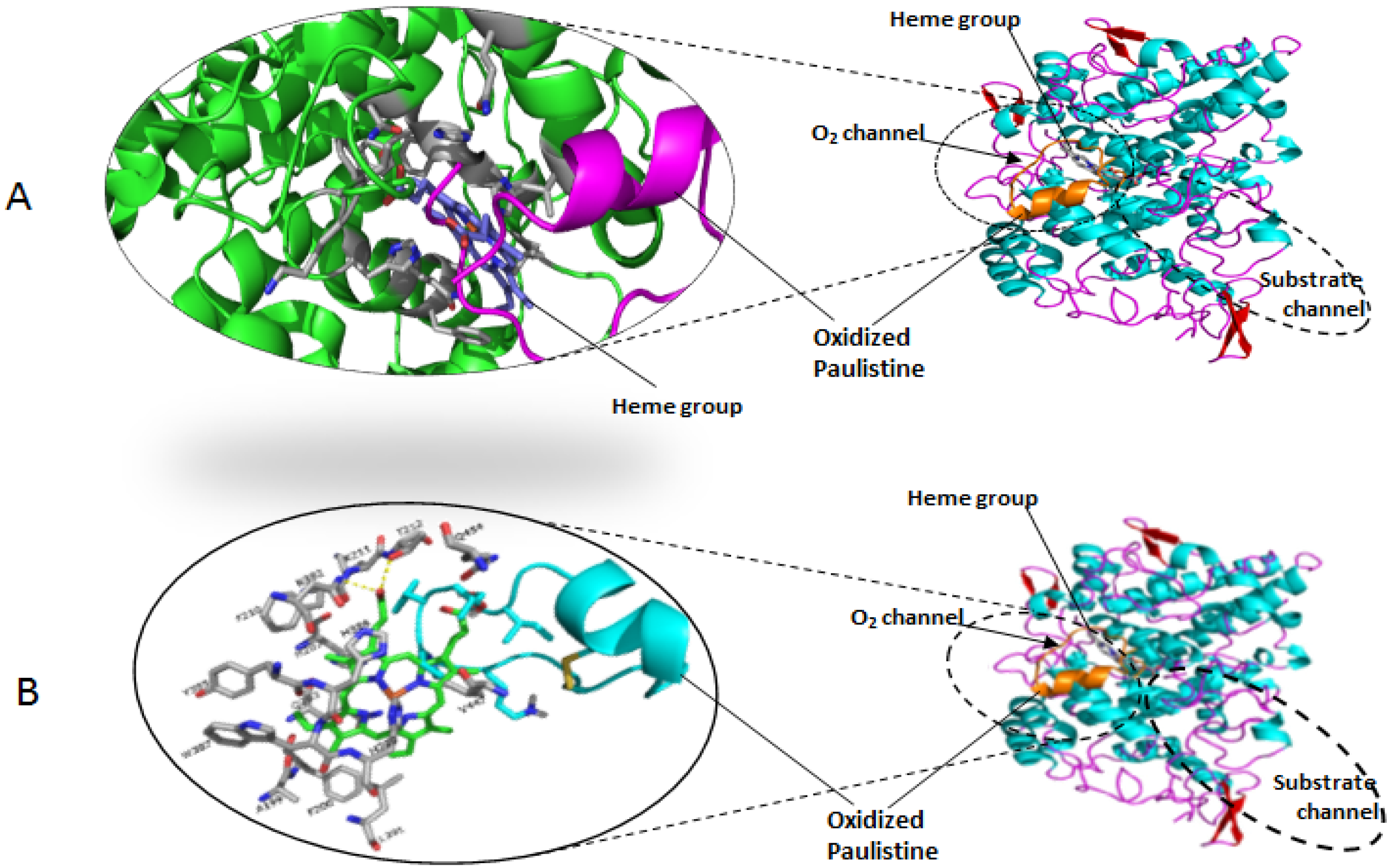
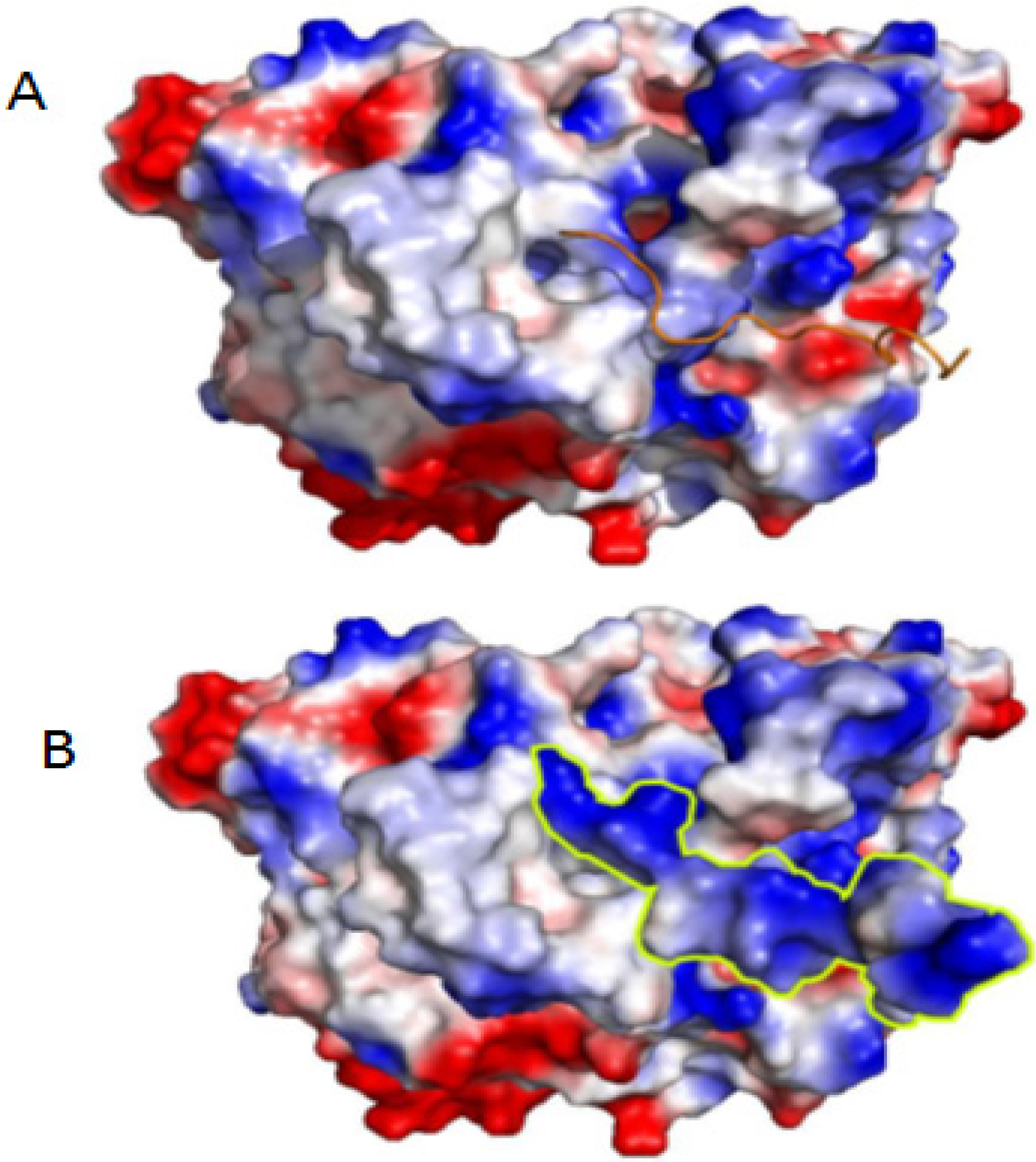
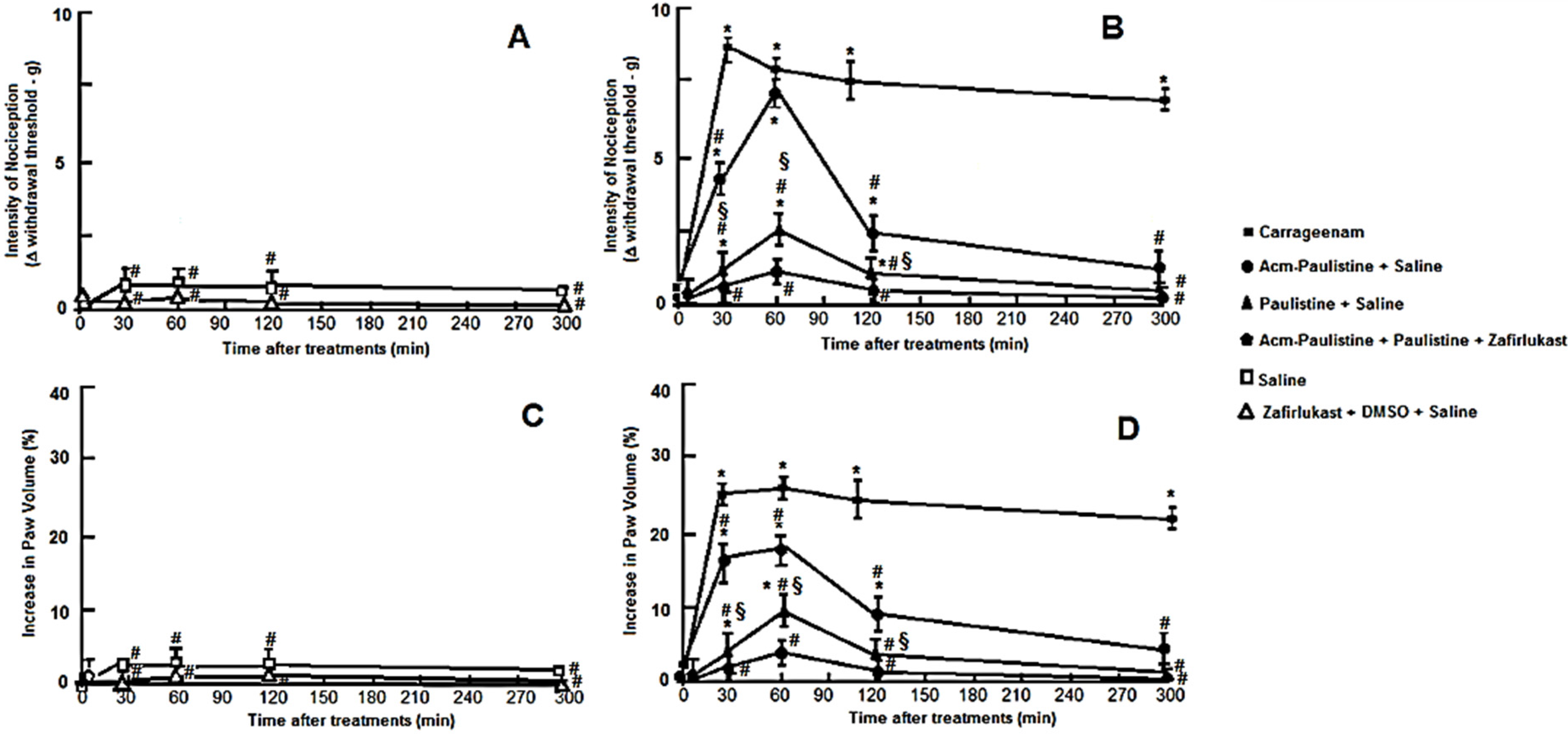
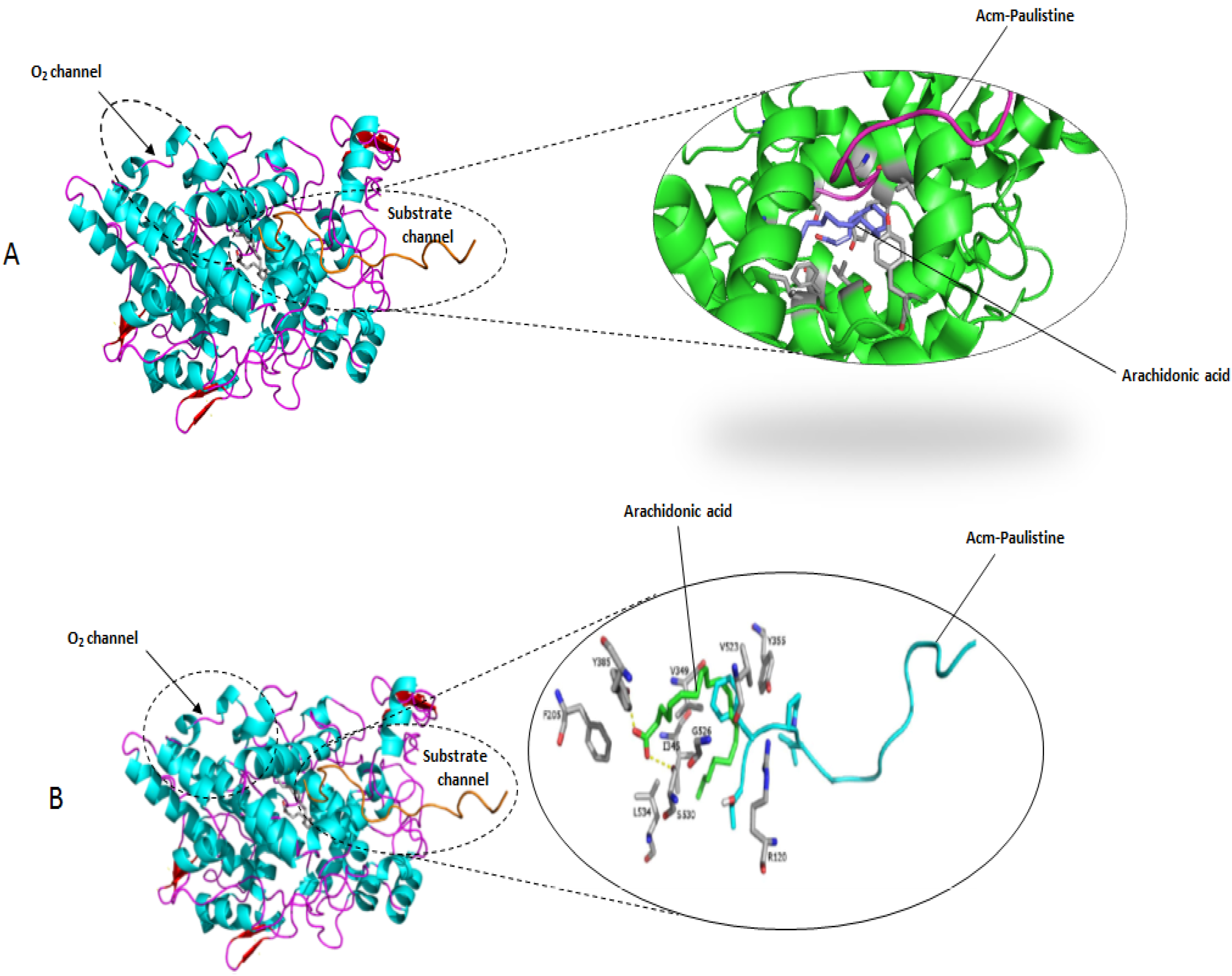
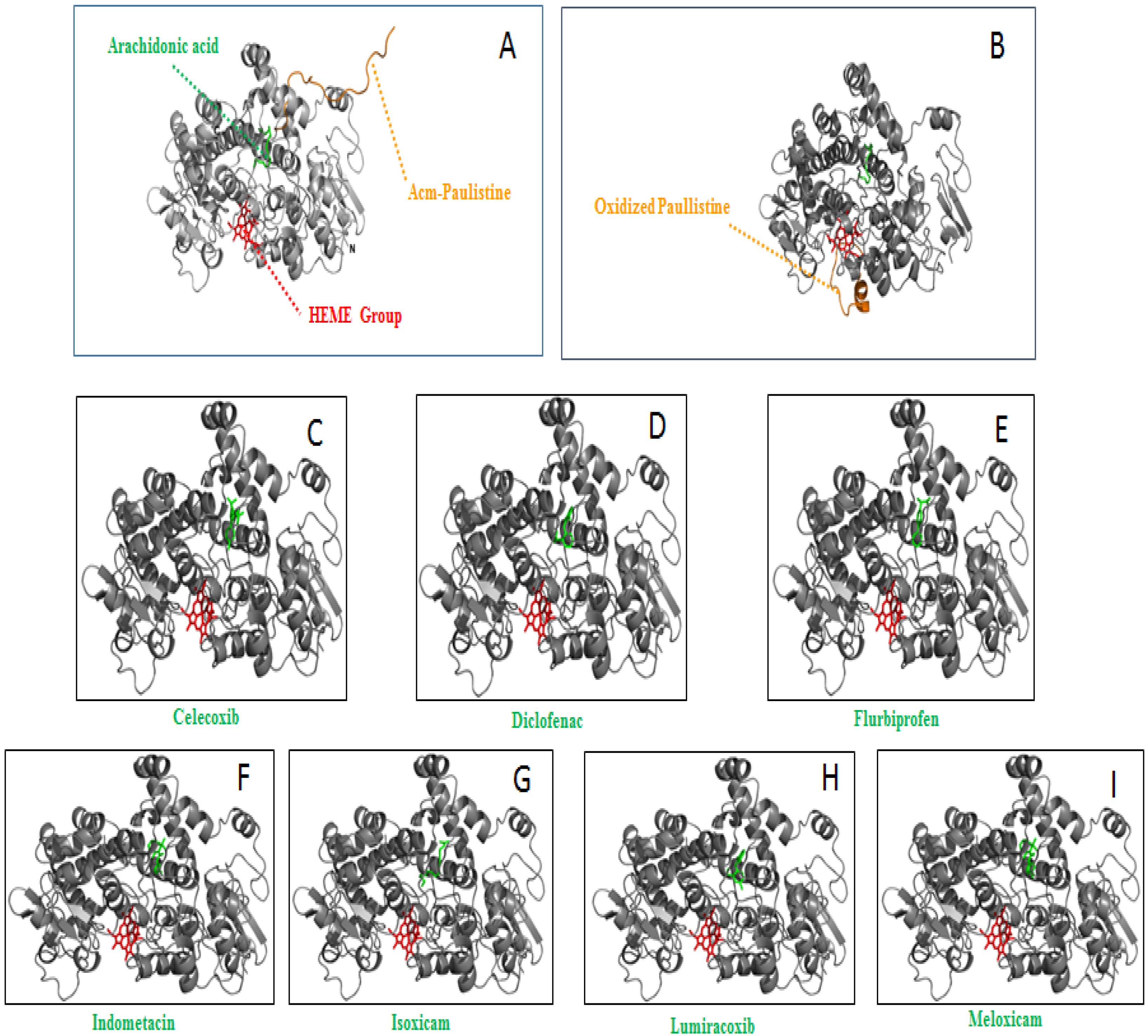
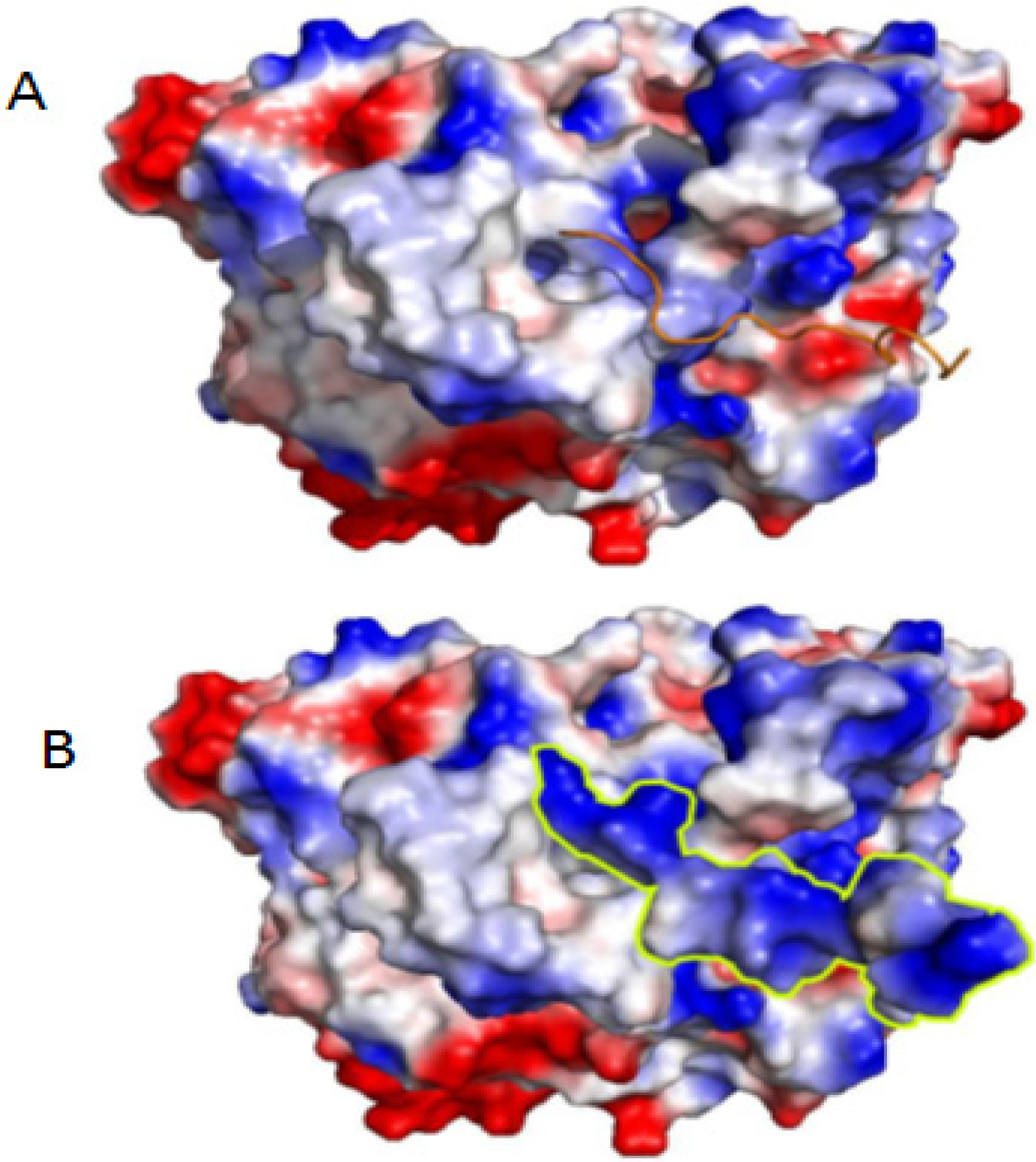
2. Results and Discussion
3. Experimental Section
3.1. Peptide Synthesis
3.2. Mass Spectrometry
3.3. Molecular Modeling
3.4. Molecular Docking Simulations
3.5. Biological Assays
3.5.1. Hyperalgesic and Edematogenic Effects
3.5.2. Von Frey Electronic Pressure Meter Paw Tests for Mice
3.5.3. Evaluation of Edema
3.5.4. Evaluation of Nociceptivity
3.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Souza, B.M.; Palma, M.S. Monitoring the positioning of short Polycationic peptides in model lipid bilayers by combining hydrogen/deuterium exchange and electrospray ionization mass spectrometry. Biochim. Biophys. Acta 2008, 1778, 2797–2805. [Google Scholar] [CrossRef] [PubMed]
- Turillazzi, S.; Mastrobuoni, G.; Dani, F.R.; Moneti, G.; Pieraccini, G.; La Marca, G.; Bartolucci, B.; Perito, D.; Lambardi, V.; Cavallini, L.; et al. Dominulin A and B: Two new antibacterial peptides identified on the cuticle and in the venom of the social paper wasp Polistes dominulus using MALDI-TOF, MALDI-TOF/TOF, and ESI-Ion Trap. J. Am. Soc. Mass Spectrom. 2006, 17, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.S. Chapter 58: Hymenoptera Venom Peptides. In Handbook of Biologically Active Peptides, 2nd ed.; Kastin, A., Ed.; Academic Press: San Diego, CA, USA, 2013; pp. 416–422. [Google Scholar]
- Santos, L.D.; Santos-Pinto, J.R.A.; Menegasso, A.R.S.; Saidemberg, D.M.; Garcia, A.M.C.; Palma, M.S. Proteomic profiling of the molecular targets of interactions of the mastoparan Polybia MP-III at the level of endosomal membranes from rat mast cells. Proteomics 2012, 12, 2682–2693. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.S.; Brochetto-Braga, M.R. Biochemical variability between venoms from different honey bees (Apis mellifera) races. Comp. Biochem. Physiol. C 1993, 106, 423–427. [Google Scholar]
- Baptista-Saidemberg, N.B.; Saidemberg, D.M.; Palma, M.S. Chemometric analysis of Hymenoptera toxins and defensins: Model for predicting the biological activity of novel peptides from venoms and hemolymph. Peptides 2011, 32, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Baptista-Saidemberg, N.B.; Saidemberg, D.M.; Palma, M.S. Profiling the peptidome of the venom from the social wasp Agelaia pallipes pallipes. J. Proteom. 2011, 74, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
- Banks, B.E.C.; Shipolini, R.A. Chemistry and pharmacology of honeybee venoms. In Venoms of Hymenoptera: Biochemical, Pharmacological and Behavioral Aspects; Piek, T., Ed.; Academic Press: London, UK, 1986; pp. 329–416. [Google Scholar]
- Dohtsu, K.; Hagiwara, K.; Palma, M.S.; Nakajima, T. Isolation and sequence analysis of peptides from the venom of Protonectarina sylveirae (Hymenoptera, Vespidae) (I). Nat. Toxins 1993, 1, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.C.; Souza, B.M.; Dias, N.B.; Brigatte, P.; Mourelle, D.; Arcuri, H.A.; Dos Santos Cabrera, M.P.; Stabeli, R.G.; Ruggiero Neto, J.; Palma, M.S. Study of structure-function relationship of peptide Paulistine: A novel class toxin from the venom of the social wasps Polybia paulista. Biochim. Biophys. Acta 2014, 1840, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Nelson, K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7384–7388. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Vecchio, A.J.; Sharma, N.P.; Jurban, B.J.; Malkowski, M.G.; Smith, W.L. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 2011, 286, 19035–19046. [Google Scholar] [CrossRef] [PubMed]
- O'Banion, M.K. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit. Rev. Neurobiol. 1999, 13, 45–82. [Google Scholar] [PubMed]
- Prusakiewicz, J.J.; Duggan, K.C.; Rouzer, C.A.; Marnett, L.J. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 2009, 48, 7353–7355. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Kiefer, J.R.; Marnett, L.J. Cyclooxygenase enzymes: Catalysis and inhibition. Curr. Opin. Struct. Biol. 2001, 11, 752–760. [Google Scholar] [CrossRef]
- Seibert, K.; Masferrer, J.L. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994, 4, 17–23. [Google Scholar] [PubMed]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef] [PubMed]
- Kermode, J.; Butt, W.; Shann, F. Comparison between prostaglandin E1 and epoprostenol (prostacyclin) in infants after heart surgery. Br. Heart J. 1991, 66, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, H. Role of thromboxane derived from COX-1 and -2 in hepatic microcirculatory dysfunction during endotoxemia in mice. Hepatology 2004, 39, 139–150. [Google Scholar] [CrossRef] [PubMed]
- DuBois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.A.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [PubMed]
- Yamaki, T.; Endoh, K.; Miyahara, M.; Nagamine, I.; Huong, N.T.H.; Sakurai, H.; Pokorny, J.; Yano, T. Prostaglandin E2 activates Src signaling in lung adenocarcinoma cell via EP3. Cancer Lett. 2004, 214, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Green, G.A. Understanding NSAIDs: From aspirin to COX-2. Clin. Cornerstone 2001, 3, 50–59. [Google Scholar] [CrossRef]
- Cao, Y.; Prescott, S.M. Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J. Cell. Physiol. 2002, 190, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Loll, P.J.; Picot, D.; Garavito, R.M. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat. Struct. Biol. 1995, 2, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef] [PubMed]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated h-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- De Souza, B.M.; Palma, M.S. Peptides from hymenoptera venoms: Biochemistry, pharmacology and potential applications in health and biotechnology. In Animal Toxins: The State of Art. Perspectives on Health and Biotechnology; UFMG Press: Belo Horizonte, Brazil, 2009; pp. 273–297. [Google Scholar]
- Nelson, D.L.; Cox, M.M. Lehninger's Principles of Biochemistry, 5th ed.; W.H. Freeman and Co.: New York, NY, USA, 2008; p. 359. [Google Scholar]
- Diaz, J.H. Non-steroidal anti-inflammatory drugs (NSAIDs). In Color Atlas of Human Poisoning and Envenoming; CRC Press: Boca Raton, FL, USA, 2007; p. 82. [Google Scholar]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In Algorithms in Bioinformatics; Springer Berlin Heidelberg: Berlin, Germany, 2002; Volume 2452, pp. 185–200. [Google Scholar]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Bailey, R.A. Design of Comparative Experiments; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Bound Surface (Å2) | ΔGbind (kcal/mol) * | TΔS (kcal/mol) * |
|---|---|---|---|
| Oxidized Paulistine | 1764 | −16.5 | 9.6 |
| Arachidonic acid | 940 | 2.0 | 4.5 |
| Celecoxibe | 1909 | 2.2 | 4.8 |
| Diclofenaco | 1816 | −8.4 | 3.9 |
| Flurbiprofeno | 1739 | 4.1 | 3.6 |
| Indometacin | 1957 | −7.7 | 4.6 |
| Isoxicam | 1842 | −6.8 | 4.2 |
| Lumiracoxibe | 692 | −11.8 | 4.0 |
| Meloxicam | 1875 | −9.4 | 4.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcuri, H.A.; Gomes, P.C.; De Souza, B.M.; Dias, N.B.; Brigatte, P.; Stabeli, R.G.; Palma, M.S. Paulistine—The Functional Duality of a Wasp Venom Peptide Toxin. Toxins 2016, 8, 61. https://doi.org/10.3390/toxins8030061
Arcuri HA, Gomes PC, De Souza BM, Dias NB, Brigatte P, Stabeli RG, Palma MS. Paulistine—The Functional Duality of a Wasp Venom Peptide Toxin. Toxins. 2016; 8(3):61. https://doi.org/10.3390/toxins8030061
Chicago/Turabian StyleArcuri, Helen Andrade, Paulo Cesar Gomes, Bibiana Monson De Souza, Nathalia Baptista Dias, Patrícia Brigatte, Rodrigo Guerino Stabeli, and Mario Sergio Palma. 2016. "Paulistine—The Functional Duality of a Wasp Venom Peptide Toxin" Toxins 8, no. 3: 61. https://doi.org/10.3390/toxins8030061
APA StyleArcuri, H. A., Gomes, P. C., De Souza, B. M., Dias, N. B., Brigatte, P., Stabeli, R. G., & Palma, M. S. (2016). Paulistine—The Functional Duality of a Wasp Venom Peptide Toxin. Toxins, 8(3), 61. https://doi.org/10.3390/toxins8030061